

Abstract
In recent years it has became evident that the transcriptome of most species has little protein-coding capacity and that the abundance of non-coding RNA was previously overlooked. Non-coding RNAs were initially thought to be transcriptional noise, however, a growing number of studies is showing that many of these RNAs have important regulatory functions. Here, we review the progress done in apicomplexan parasites in this rapidly growing field.
In recent years, genome sequencing has revealed that the genomes of all eukaryotes studied to date are nearly totally transcribed, generating a vast number of non-coding RNAs (ncRNAs) [1]. Whether the vast majority of ncRNAs are functional or simply transcriptional noise has been controversial. However, recent studies are revealing that a substantial number of ncRNAs are indeed functional [2]. A fraction of these RNAs with short open reading frames (ORFs) may encode peptides, but on the other hand some currently annotated ORFs may well be false (i.e. some ncRNAs contain ORFs but do not encode proteins). NcRNAs are classified as long ncRNAs (>200 nucleotides); [1], or short ncRNAs (<200 nt and typically ~20–30 nt long). There are 3 main classes of well studied short ncRNAs: short interfering RNAs (siRNAs), microRNAs (miRNAs), and PIWI-interacting RNAs (piRNAs) [3]. Short ncRNAs have been classically implicated in gene silencing pathways directing translational repression or messenger RNA (mRNA) degradation [3], and they have been linked to heterochromatin formation [4]. Although the literature is dominated by short ncRNAs, there is increasing evidence for the presence of functional long ncRNAs in many organisms. Long ncRNAs are commonly associated with cellular differentiation and the development of complex organisms [4,5], it has been proposed that the epigenetic trajectories of differentiation are primarily programmed by RNA regulatory networks [5].
When comparing different species through evolution, it has been shown that the amount of ncRNA increases with the morphological complexity and vertebrate species contain more ncRNA than all other species studied to date [5]. In contrast, the number of protein-coding genes does not scale-up consistently with morphological complexity [6]. Apicompexan parasites are single cell lower eukaryotes, and their morphology is simple compared to higher eukaryotes. However, most of these parasites have very complex life cycles, and the malaria parasite, Plasmodium falciparum, contains more ncRNA than either the multicellular organism Phanerochaete chrysosporium or Dictyostelium discoideum, which is either single cell or multicellular depending on the lifecycle stage [5].
Bearing in mind that ncRNAs have central regulatory roles, and apicomplexan parasites are important pathogens, it is very likely that many of these RNAs regulate progression through the infectious cycle of these parasites or regulate the expression of virulence factors. This has been shown for other microbial pathogens, for example, in several pathogenic bacteria, regulatory RNAs are involved in controlling virulence related genes. NcRNAs from Salmonella pathogenicity island are suggested to be involved in regulating virulence mechanisms and antibiotic resistance [7]. In Vibrio cholerae and Pseudomonas aeruginosa small RNAs regulate quorum sensing, genes encoding virulence factors and biofilm formation [8,9].
This review is focused on Plasmodium and Toxoplasma, but since members of the phylum apicomplexa share many biological characteristics and the genomes of other apicomplexans are now available, most of the methods presented here to identify ncRNAs and future directions discussed, are also applicable to other apicomplexan parasites.
Functions of ncRNAs
In this section, I describe the main established functions of ncRNAs in eukaryotes. Their function in apicomplexan parasites is discussed in the next section.
Short ncRNA
The 3 main classes of small RNAs (siRNA, miRNA and piRNA) interact with members of the Argonaute/Piwi protein family which functions as the core of a diverse set of complexes called RNA-induced silencing complexes, RISCs [3]. RISCs use the short RNAs as guides for sequence specific silencing. They either induce the degradation of mRNAs or repress mRNA translation, a process called RNA interference (RNAi).
MiRNAs were first discovered in a genetic screen designed to identify regulators of development [10]. A couple of well studied miRNAs, lin-4 and let-7, regulate developmental timing in the worm C. elegans [11]. Although miRNAs were originally considered to be restricted to multicellular eukaryotes, they are also present in lower eukaryotes such as the ciliated protozoan Tetrahymena. In Tetrahymena massive DNA rearrangements are involved in the differentiation of macronuclei from micronuclei and small RNAs are essential in this process [12,13]. Components of the RNAi machinery have been found in many eukaryotes, suggesting that miRNA regulation is an ancestral feature of eukaryotic cells [14].
In many cases, individual miRNAs are involved in targeting gene batteries, repressing multiple mRNAs that are not needed at a particular developmental stage [15]. For instance, miR-124 controls a gene regulatory network involved in neuronal differentiation [16]. MiRNAs can also regulate transcription and alternative splicing. The transcription factor lin-14 is repressed by the miRNA lin-4 [17]. Repressors of alternative splicing, PTBP1 and PTBP2, are regulated by miRNAs [18].
Long ncRNA
The diversity of long ncRNAs with correspondingly diverse functionality makes simple generalizations about ncRNA function difficult [1]. Long ncRNAs are defined as longer that 200 bp, but their sizes vary from ~300 bps to ~20 kbs and they are usually bigger that 1 Kb [10]. Some are spliced or alternatively spliced, and some are polyadenylated.
Many long ncRNAs mediate epigenetic changes recruiting chromatin remodeling complexes. Recently, 3,300 large intergenic ncRNAs (lincRNAs) were analyzed using chromatin state maps and ~20% of these RNAs are bound to polycomb repressive complex PCR2 [19]. PCR2 is a methyltransferase that trimethylates H3K27, repressing transcription. Another example is the fbp1+ locus of S. Pombe. The chromatin at this locus is progressively converted to an open configuration by a cascade of ncRNAs [20].
An increasing number of long ncRNAs have been shown to play a role regulating transcription. In some cases, promoters can be transcribed into long ncRNAs. For example, the human DHFR gene has an alternative promoter that is transcribed into a ncRNA that forms a stable complex with the major promoter, directs the interaction with the transcription factor IIB, and induces the dissociation of the preinitiation complex, thereby repressing transcription [21]. There is also a growing number of long ncRNAs involved in post-transcriptional regulation, including splicing and translation [1].
Like short ncRNAs, long ncRNAs are not restricted to multicellular eukaryotes. In unicellular eukaryotes, long ncRNAs play a role regulating different processes that control gene expression and cell differentiation [10]. In Oxytricha trifallax, maternal RNA templates guide correct and precise DNA rearrangements at a specific stage during development [22]. In Leishmania, developmentally regulated RNAs are transcribed from subtelomeric repeats generating sense and antisense ncRNAs that are exported to the cytosol and processed by trans-splicing [23].
NcRNA in apicomplexan parasites
In Plasmodium there are some reports showing that double-stranded RNA (dsRNA) mediates gene silencing [24–27]. Although the level of mRNA was reduced in dsRNA treated parasites, there is no direct evidence that the downregulation was due to the RNAi pathway and database mining failed to identify RNAi gene candidates in any of the Plasmodium species [28]. Baum et al., used RNA-based and comparative genomic approaches to determine if RNAi is functional in malaria parasites [29]. These authors concluded that RNAi is not functional and previous reports of RNAi could be explained by general toxicity or an antisense mechanism [29]. Therefore, the existence of a “classical” RNAi pathway appears not likely.
Two research groups carried out a systematic search and analysis of short RNAs in P. falciparum coming to the conclusion that there are no miRNAs [30,31]. Again, these results suggest that “classical” miRNAs are not present in Plasmodium, but it is possible that other types of miRNA do exist. In fact, new classes of small RNA are constantly being discovered in other organisms. For example, whole-genome tiling arrays identified a new class named PASR (promoter-associated short RNA) which map to transcription start sites and TASR (termini-associated short RNA) which map to the transcription termination sites [32]. Also, with the current available protocols it is not possible to clone 5′ or 3′ chemically modified small RNAs, which is likely to impair the discovery of novel families. While miRNAs are elusive in Plasmodium, translational repression (TR) is not. TR is essential for sexual development and mediated by an RNA helicase that is a member of the DDX6 family of DEAD-box RNA helicases [33].
The ribosomal RNA (rRNA) of Plasmodium has a unique biology, two different developmentally regulated genes encode the small subunit, one gene is mainly expressed in the asexual blood stages and the other is mainly transcribed in the sporozoite forms [34]. Chakrabarti et al., reported that the organization of the rRNA multigene family is unusual, there are only a few genes dispersed on different chromosomes [35]
An exceptionally high frequency of antisense RNAs was observed in the P. falciparum genome [36]. These antisense transcripts are distributed across all 14 chromosomes and strikingly, an inverse relationship was observed between sense and antisense transcript abundance across the entire parasite transcriptome. This observation raised the possibility that some loci can be regulated by antisense RNAs [36]. In a following report, it was demonstrated that the antisense RNA is synthesized by RNA polymerase II in the nuclear fractions and is more common than originally expected [37]. The antisense loci analyzed by Militello et al.,., are not predicted to contain ORFs and it is not likely that they encode proteins. The authors suggested a regulatory role for the antisense RNA; in particular, they proposed the hypothesis that antisense RNA regulates stage-specific gene expression since the levels of most sense transcripts are tightly regulated through the parasite life cycle. In addition, two reports show that antisense RNA can be used as a tool to silence gene expression [38,27].
Upadhyay et al., took advantage of the A-T richness of the P. falciparum genome, using a bioinformatics approach to identify ncRNAs in intergenic regions with high G-C content, followed by the analysis of conservation of these regions between malaria parasite species [39]. Using this strategy they successfully identified small nuclear RNAs predicted to be involved in splicing, such as U1, U2, U4, U5 and U6, and 10 ncRNAs of unknown function but which are highly conserved among Plasmodium species [39].
As in other eukaryotes, a recent study showed that the ncRNAs in P. falciparum are likely to be associated with chromosome dynamics and higher-level nuclear organization [40]. These authors showed that the centromeres contain bidirectional promoters that generate small ncRNAs. These ncRNAs localize to the nucleus and associate with the centromeres themselves, suggesting that these RNAs play a role in the maintenance and function of centromeric chromatin. Interestingly, the authors point out that although their data suggest that these RNAs are processed into short, discrete bands of similar sizes (75 and 175 nucleotides), the genome does not contain homologues of the genes involved in processing short RNAs or RNAi machinery. This observation raises the question of whether Plasmodium has a different but analogous system to process small ncRNA.
Chromatin remodeling is important for silencing of var genes. The virulence var genes are responsible for antigenic variation which results from switches in expression between the members of this multigene family. Recently, it was demonstrated that the chromatin surrounding the var genes generates long sense and antisense ncRNAs [41]. This transcripts are generated by a bidirectional promoter located within the conserved intron of the var genes and they associate with the chromatin, suggesting that these ncRNAs may be components of the chromatin structure that regulates antigenic variation; in addition to the epigenetic marks previously described [41].
NcRNA is heavily dependent on its secondary structure which is usually required to accomplish its function. Two research groups carried out an extensive analysis of Plasmodium structural RNAs. Chakrabarti et al., used a comparative genomics approach to identify numerous RNAs such as telomerase RNA, snoRNAs, spliceosomal snRNAs, SRP RNA, MRP RNA, RNAse P RNA, and 6 new RNAs of unknown function [35]. The expression of telomerase RNA was confirmed by Northern-blots and it is believed to be involved in maintaining telomere integrity. Since the multigene families implicated in antigenic variation are located in the subtelomeric regions, maintaining the telomere structure may be critical for the parasites. In addition, one of the RNAs of unkwon function is arranged in clusters flanking the rifin or var surface antigens. This observation led the authors to hypothesize that this particular ncRNA plays a role in the expression or maintenance of these surface antigens [35]. The snoRNA is located in intergenic regions or introns, similar to other protozoa, and it is predicted to be involved in the modification of rRNA and snRNA. They also found candidate genes for each of the snRNAs in most of the malaria genomes and verified some of them by Northern. The snRNA is predicted to fold into similar conformations of metazoan and fungal snRNAs. RNase P and MRP RNAs, which are responsible for tRNA and rRNA processing, were verified by Northern and align well with secondary structures of other organisms [35].
The other research group that analyzed structural RNAs used the RNA gene-finding tool RNAz to identify conserved ncRNAs across eight Plasmodium genomes [42]. They identified 604 novel ncRNA structures and, combining microarray and Northern data, they provided evidence for the expression of 33 RNAs, some of which were expressed stage-specifically during the asexual life cycle. Importantly, the authors show that these RNAs are subject to purifying selection. This criterion has been widely used in many other eukaryotic species where it has helped to show that particular ncRNAs are functional.
Raabe et al., made two small size cDNA libraries from P. falciparum and carried out an extensive analysis of small ncRNAs [43]. They identified 630 novel ncRNAs including snoRNAs, telomere- and subtelomere-associated ncRNAs, antisense RNAs, and other intergenic ncRNAs. The chromosome ends are organized in telomere repeats followed by a subtelomeric domain containing 6 different repetitive elements, followed by members of the var, rif and stevor families. This group found one ncRNA associated with the telomeres and and 70 ncRNAs associated with the subtelomeric region, and they proposed that these RNAs may form part of an RNA regulatory network involved in gene silencing or may form DNA/RNA duplexes which could contribute to high recombination at the chromosome ends. They also detected 328 antisense RNAs complementary to protein coding genes encoding a broad range of proteins. The large number of antisense RNAs to different biochemical pathways suggests they may participate in the regulation of gene expression. Interestingly, this group also reported antisense RNAs to members of almost all var genes.
As discussed above, different research groups demonstrated that the var genes and the chromatin surrounding the var genes, express a large number of distinct types of ncRNAs. This suggests that ncRNAs may play a critical role in the virulence of malaria parasites.
In contrast to Plasmodium species, Toxoplasma gondii’s genome contains good RNAi gene candidates [28]. Nonetheless, efficient and widespread use of RNAi for specific gene silencing remains elusive in Toxoplasma. Many laboratories have attempted to use this tool to downregulate gene expression but there are very few reports that show successful double-stranded RNA induced gene silencing [44–46]. Al-Riyahi et al., showed that a T. gondii homolog of argonaute (TgAgo), with highest amino acid similarity to Arabidopsis Ago1, is indeed expressed in the two asexual stages, tachyzoites and bradyzoites [46]. The authors showed that a transgenic parasite line harboring a knockdown of the endogenous TgAgo gene is impaired in its ability to induce specific dsRNA gene silencing. Although Al-Riyahi et al., were not able to show the presence of specific siRNAs, these remain the most convincing data showing a functional RNAi-like pathway in T. gondii. Of note, the ToxoDB gene prediction used by Ullu et al., shows that TgAgo contains both a PAZ and a PIWI domain [28]. However, the experimental data obtained by Al-Riyahi et al., RT-PCR, 5′-RACE and Northern blots, do not support the ToxoDB gene prediction; this group showed that TgAgo is a much shorter protein than the argonaute proteins of higher eukaryotes [46]. This group reported that TgAgo contains a conserved PIWI domain but the N-terminal peptide is different from the conserved PAZ domain of other eukaryotes [58]. Taken together with the paucity of double-stranded RNA induced gene silencing reports, it may suggest that this pathway has its unique characteristics in T. gondii.
Like in P. falciparum, a high frequency of antisense RNAs was observed in T. gondii [47]. Radke et al., used serial analysis of gene expression (SAGE), to examine the T. gondii transcriptome. This group analyzed SAGE tag frequency and orientation in different sets of predicted genes, and they reported ~21% of antisense transcription [47]. As shown for P. falciparum, T. gondii also shows an inverse relationship between the frequency of antisense transcripts and the level of sense transcription [47].
Genome-wide discovery and validation of ncRNAs remains to be done in T. gondii. Gissot et al., used a tiling microarray covering 650 kb of chromosome 1b, to study the epigenetic organization and transcription pattern of tachyzoites [48]. Most of the modified histone peaks are located close to the 5′ end of predicted genes and correlate with gene expression. Using this approach, they discovered one transcript which does not have an ORF and appears to represent a ncRNA [48]. The microarray used by this group contains 1% of the T. gondii genome, but this approach could provide a good overall picture of ncRNAs if applied to the entire genome.
Different labs reported (at meetings) the presence of many T. gondii ESTs with no obvious ORFs, some of which lie in intergenic regions. It is likely that a number of these ESTs correspond to long ncRNAs, but additional bioinformatic or experimental evidence is needed to confirm this. Given that long ncRNAs are associated with cellular differentiation and development in many species [4,5,10], it is tempting to speculate that these RNAs may play important roles in the transition from tachyzoites to bradyzoites. Bradyzoite differentiation mutants have been generated in several laboratories [49–52]. In very few cases have the loci disrupted in these mutants been identified and linked to the mutant phenotype. Some of the bradyzoite differentiation mutants have insertions in places that do not contain any gene prediction, it is possible that part of these loci correspond to ncRNAs. Two of the bradyzoite differentiation mutants we generated [49] have insertions inside putative ncRNAs. One of these mutants has an insertion within a 2.6 Kb transcript with no obvious ORF, which is up-regulated 24 fold in bradyzoites. The other mutant has an insertion within a locus that encodes two overlapping transcripts, one predicted to be a splicing factor and the other is a 5 Kb antisense transcript with no obvious ORF [Matrajt lab, manuscript in preparation].
Histone-modifying complexes have been linked to differentiation in T. gondii. Behnke et al., showed that, similar to other eukaryotes, conventional promoter mechanisms work with the chromatin-remodeling machinery to regulate bradyzoite gene expression [53]. However, how the chromatin-remodeling machinery is recruited to specific genomic loci is a major unanswered question. To this end, it was recently shown that a member of the ApiAP2 family of transcription factors is involved in the heterochromatin formation at the chromosome ends of Plasmodium falciparum [54]. In other eukaryotes, there are a growing number of studies showing that ncRNAs can mediate epigenetic changes by recruiting chromatin-remodeling complexes [19]. It is believed that ncRNAs can provide specificity to individual genomic loci, and it would not be surprising if ncRNAs work in concert with chromatin remodelers to re-program the expressed genome during the transition from tachyzoites to bradyzoites. The main families of histone-modifying enzymes involved in chromatin-remodeling have been identified in the genomes of most apicomplexan parasites [55]. NcRNAs could be involved in any process that requires reprogramming of the expressed genome in apicomplexan parasites.
Distinguishing ncRNAs from protein-coding RNAs is not trivial
Since in the last few years it has become clear that the majority of eukaryotic transcriptomes corresponds to ncRNA, the correct annotation of ncRNAs as well as the discrimination between protein-coding and ncRNA has become critical. Unlike protein-coding RNA where sequence motifs and the primary sequence are helpful to infer functionality, ncRNA function is often heavily dependent on its secondary structure. Therefore, computational methods developed for protein-coding genes cannot be used to search for ncRNAs, and it is becoming apparent that the identification of ncRNAs requires novel strategies and poses new challenges. Indeed, correct genome annotation will require techniques to distinguish between protein-coding RNA, functional ncRNA, and transcriptional noise. In this section, I discuss the main approaches currently used to discriminate between ncRNA and mRNA.
The first obvious place to start is to look at the ORFs. More than 95% of the proteins deposited in public databases are more than a 100 amino acids in length. This length is also 2 standard deviations above the average length of ORFs that occur in a random sequence of 1 Kilobase [56]. The Functional Annotation of the Mammalian Genome, FANTOM consortium, used a cutoff of 300 nt (100 aa) to identify putative mRNAs. Using this criterion alone will misclassify some ncRNAs, specially the long ones, that by chance contain long ORFs. For example, the ncRNAs Xist, H19, and Mirg are functional ncRNAs although they consist of ORFs longer than 100 aa [57].
Another criterion that has been used is ORF conservation, since ORF similarity to known proteins provides indirect evidence of function as an mRNA [56]. The program CSTminer has exploited the tendency of protein-coding sequences to favor synonymous base changes versus non-synonymous ones to distinguish ncRNA from mRNA [58]. The limitation of this method is the number of genomes available for comparison and even the absence of ORF conservation with related species does not guarantee the absence of function [56]. In individual cases, in vitro translation assays or assessing association with polysomes can be informative, but these methods are far from definitive [56].
Because ncRNAs frequently require specific secondary structures to accomplish their function, a number of studies use structural approaches to identify ncRNA. In addition, the predicted structure of an RNA under study can be compared with a database of functionally annotated structural signatures [59]. Therefore, ncRNA identification can involve secondary structure prediction and/or secondary structure comparison. Three main approaches are used: thermodynamic, probabilistic and covariation-based [59]. Since these approaches are complimentary, they are typically used in combination to decrease their individual limitations.
Thermodynamic approaches are based on the hypothesis that the RNA is folded in the most stable structure, i.e. the one having the minimum free energy (MFE). Several programs based on the MFE are available, such as RNAfold or RNAKinetics, which also takes into account the fact that RNA molecules may start folding during transcription [60, 61]. The probabilistic approaches are built by estimating parameters from a set of known examples, called training samples, using data from RNA structure databases [62]. Covariation methods are based on the assumption that homologous RNAs have common ancestry and function. These RNAs are expected to have similar structures and modest similarity in sequence. Covariation methods are comparative; the input sequences can be: (a) aligned and unfolded sequences; (b) unaligned and individually folded sequences; or (c) unaligned and unfolded sequences [63]. A method to improve current RNA structural alignments on incomplete sequence fragments was recently developed [64].
Chakrabarti et al., applied covariation methods to several Plasmodium species to identify structural RNAs [35]. This is the first genome-wide study on structural RNAs in malaria parasites. Similar studies remain to be done with all other apicomplexan parasites.
The development of ncRNA gene finder algorithms is still a challenge, but important progress was made recently with Support Vector Machine technology (SVM) at the forefront [65]. This technology uses supervised learning algorithms designed to distinguish ncRNA from mRNA. The algorithm CONC (for coding – non-coding) uses multiple distinct features to distinguish mRNA from ncRNA, such as peptide length, homology with known proteins, amino acid composition, secondary structure, and sequence entropy [65]. Another algorithm, CPC (for Coding Potential Calculator), accomplished comparable performance in less time [66]. A web-based interface is available for CPC at: http://cpc.cbi.pku.edu.cn/.
Mourier et al., used the ncRNA gene finder RNAz and identified 604 novel RNA structures in P. falciparum [42]. RNAz is a Support Vector Machine classifier that uses multiple alignments. This is first study using the SVM technology in malaria parasites. It is interesting to note that this study is largely complimentary to the study done by Chakrabarti et al., which shows that the use of different bioinformatic tools could be advantageous. Using the SVM technology with other apicomplexan parasites will be an important step to advance this field.
Using the SVM technology in apicomplexan parasites will have its challenges. For instance, the algorithm CONC requires to train the software with a set of ncRNAs and a set of coding RNAs, and then feed the classifier with real sequence data. This could be a challenge with apicomplexan genomes where the number of ncRNAs is limited. However, there is a growing number of databases specialized in helping the annotation of ncRNAs, such as NONCODE that collects a wide variety of ncRNAs (small and long) from 861 organisms covering all kingdoms of life [67].
Conclusions
The research on ncRNAs is clearly in its infancy in parasitic organisms. One place to start this effort is to take advantage of the constant improvements in the ncRNA gene finders, such as the SVM technology, and apply them to the apicomplexan genomes. It is reasonable to start by looking at the conserved ncRNAs. However, ncRNAs have generally low sequence conservation. To this end, new techniques such as next-generation sequencing will facilitate the discovery of novel classes of ncRNA, as well as their annotation in the genomes.
Once ncRNAs are identified, the next obvious challenge is to elucidate their function and mechanism of action. An overview of the ncRNA regulatory functions studied in other eukaryotes is shown in Figure 1. This figure shows all the research areas that are yet to be explored in these parasites; ncRNAs are involved in the regulation of gene expression at all levels.
Fig. 1.
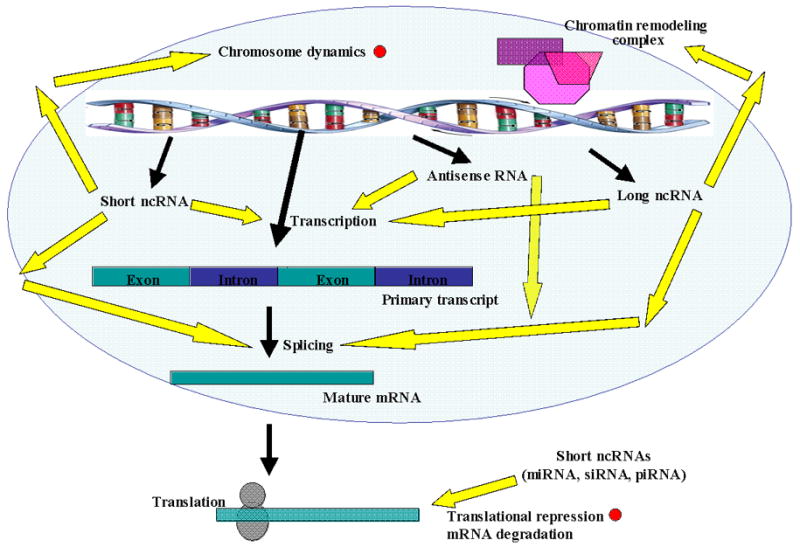
Main function of ncRNAs in eukaryotes. In apicomplexan parasites, the expression of many ncRNAs has been confirmed but their function was studied in a few cases (red dots), see text for details.
In other eukaryotes, several approaches to study ncRNA function have been developed. For example, Guttman et al., identified over a thousand large ncRNAs [68]. In order to examine their possible function, they made a custom array containing the ncRNAs identified plus protein coding genes. Then they clustered the ncRNAs together with the protein coding genes into sets of co-expressed genes. This approach allowed the authors to associate groups of ncRNAs with distinct biological processes [68]. A similar approach could be useful in apicomplexan parasites.
Having the ncRNAs annotated in the genomes of these parasites can also help to infer putative functions. For example, any laboratory that is running a forward genetic screen will be able to check if the transcript they are working on is predicted to be an ncRNA. This will aid to link ncRNAs to phenotypes and possible function. Also, until recently, the gene expression field was dominated by the concept that most important tasks are carried out by proteins. If investigators working in any aspect of gene expression have in mind that ncRNAs could be involved in these processes, they will not discard transcripts that do not have an ORF, or they will include ncRNAs in their working models.
Apicomplexan parasites are of medical significance. NcRNAs may play a critical role in the virulence of these parasites. Novel ncRNAs could be explored for drug targeting. This will also involve the intersection with other fields, such as RNA structural biology.
Acknowledgments
I would like to thank Dr. Gary Ward (University of Vermont) and Dr. Sergio Angel (IIB-INTECH, CONICET, Argentina) for critically reviewing the manuscript. The Matrajt lab is supported by the Vermont Genetics Network (VGN) through NIH grant 1 P20 RR16462 from the BRIN program of the NCRR; The American Heart Association AHA 0535047N; and the NIH P20 RR021905.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mercer TR, Dinger ME, et al. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–9. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 2.Carninci P, Kasukawa T, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–63. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 3.Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457(7228):405–12. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]
- 4.Amaral PP, Dinger ME, et al. The eukaryotic genome as an RNA machine. Science. 2008;319(5871):1787–9. doi: 10.1126/science.1155472. [DOI] [PubMed] [Google Scholar]
- 5.Mattick JS. A new paradigm for developmental biology. J Exp Biol. 2007;210:1526–47. doi: 10.1242/jeb.005017. [DOI] [PubMed] [Google Scholar]
- 6.Taft RJ, Pheasant M, et al. The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays. 2007;29(3):288–99. doi: 10.1002/bies.20544. [DOI] [PubMed] [Google Scholar]
- 7.Chinni SV, Raabe CA, et al. Experimental identification and characterization of 97 novel npcRNA candidates in Salmonella enterica serovar Typhi. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bordi C, Lamy MC, et al. Regulatory RNAs and the HptB/RetS signalling pathways fine-tune Pseudomonas aeruginosa pathogenesis. Mol Microbiol. 2010 doi: 10.1111/j.1365-2958.2010.07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Svenningsen SL, Tu KC, et al. Gene dosage compensation calibrates four regulatory RNAs to control Vibrio cholerae quorum sensing. Embo J. 2009;28(4):429–39. doi: 10.1038/emboj.2008.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amaral PP, Mattick JS. Noncoding RNA in development. Mamm Genome. 2008;19(7–8):454–92. doi: 10.1007/s00335-008-9136-7. [DOI] [PubMed] [Google Scholar]
- 11.Reinhart BJ, Slack FJ, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403(6772):901–6. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 12.Malone CD, Anderson AM, et al. Germ line transcripts are processed by a Dicer-like protein that is essential for developmentally programmed genome rearrangements of Tetrahymena thermophila. Mol Cell Biol. 2005;25(20):9151–64. doi: 10.1128/MCB.25.20.9151-9164.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Taverna SD, et al. RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev. 2007;21(12):1530–45. doi: 10.1101/gad.1544207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerutti H, Casas-Mollano JA. On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet. 2006;50(2):81–99. doi: 10.1007/s00294-006-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giraldez AJ, Mishima Y, et al. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312(5770):75–9. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- 16.Makeyev EV, Maniatis T. Multilevel regulation of gene expression by microRNAs. Science. 2008;319(5871):1789–90. doi: 10.1126/science.1152326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee RC, Feinbaum RL, et al. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 18.Boutz PL, Chawla G, et al. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev. 2007;21(1):71–84. doi: 10.1101/gad.1500707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khalil AM, Guttman M, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106(28):11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirota K, Miyoshi T, et al. Stepwise chromatin remodelling by a cascade of transcription initiation of non-coding RNAs. Nature. 2008;456(7218):130–4. doi: 10.1038/nature07348. [DOI] [PubMed] [Google Scholar]
- 21.Martianov I, Ramadass A, et al. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. 2007;445(7128):666–70. doi: 10.1038/nature05519. [DOI] [PubMed] [Google Scholar]
- 22.Nowacki M, Vijayan V, et al. RNA-mediated epigenetic programming of a genome-rearrangement pathway. Nature. 2008;451(7175):153–8. doi: 10.1038/nature06452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dumas C, Chow C, et al. A novel class of developmentally regulated noncoding RNAs in Leishmania. Eukaryot Cell. 2006;5(12):2033–46. doi: 10.1128/EC.00147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McRobert L, McConkey GA. RNA interference (RNAi) inhibits growth of Plasmodium falciparum. Mol Biochem Parasitol. 2002;119(2):273–8. doi: 10.1016/s0166-6851(01)00429-7. [DOI] [PubMed] [Google Scholar]
- 25.Malhotra P, Dasaradhi PV, et al. Double-stranded RNA-mediated gene silencing of cysteine proteases (falcipain-1 and -2) of Plasmodium falciparum. Mol Microbiol. 2002;45(5):1245–54. doi: 10.1046/j.1365-2958.2002.03105.x. [DOI] [PubMed] [Google Scholar]
- 26.Mohmmed A, Dasaradhi PV, et al. In vivo gene silencing in Plasmodium berghei--a mouse malaria model. Biochem Biophys Res Commun. 2003;309(3):506–11. doi: 10.1016/j.bbrc.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 27.Crooke A, Diez A, et al. Transient silencing of Plasmodium falciparum bifunctional glucose-6-phosphate dehydrogenase- 6-phosphogluconolactonase. Febs J. 2006;273(7):1537–46. doi: 10.1111/j.1742-4658.2006.05174.x. [DOI] [PubMed] [Google Scholar]
- 28.Ullu E, Tschudi C, et al. RNA interference in protozoan parasites. Cell Microbiol. 2004;6(6):509–19. doi: 10.1111/j.1462-5822.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- 29.Baum J, Papenfuss AT, et al. Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res. 2009;37(11):3788–98. doi: 10.1093/nar/gkp239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rathjen T, Nicol C, et al. Analysis of short RNAs in the malaria parasite and its red blood cell host. FEBS Lett. 2006;580(22):5185–8. doi: 10.1016/j.febslet.2006.08.063. [DOI] [PubMed] [Google Scholar]
- 31.Xue X, Zhang Q, et al. No miRNA were found in Plasmodium and the ones identified in erythrocytes could not be correlated with infection. Malar J. 2008;7:47. doi: 10.1186/1475-2875-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapranov P, Cheng J, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316(5830):1484–8. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 33.Mair GR, Braks JA, et al. Regulation of sexual development of Plasmodium by translational repression. Science. 2006;313(5787):667–9. doi: 10.1126/science.1125129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waters AP, Syin C, et al. Developmental regulation of stage-specific ribosome populations in Plasmodium. Nature. 1989;342(6248):438–40. doi: 10.1038/342438a0. [DOI] [PubMed] [Google Scholar]
- 35.Chakrabarti K, Pearson M, et al. Structural RNAs of known and unknown function identified in malaria parasites by comparative genomics and RNA analysis. Rna. 2007;13(11):1923–39. doi: 10.1261/rna.751807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gunasekera AM, Patankar S, et al. Widespread distribution of antisense transcripts in the Plasmodium falciparum genome. Mol Biochem Parasitol. 2004;136(1):35–42. doi: 10.1016/j.molbiopara.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Militello KT, Patel V, et al. RNA polymerase II synthesizes antisense RNA in Plasmodium falciparum. Rna. 2005;11(4):365–70. doi: 10.1261/rna.7940705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardiner DL, Holt DC, et al. Inhibition of Plasmodium falciparum clag9 gene function by antisense RNA. Mol Biochem Parasitol. 2000;110(1):33–41. doi: 10.1016/s0166-6851(00)00254-1. [DOI] [PubMed] [Google Scholar]
- 39.Upadhyay R, Bawankar P, et al. A screen for conserved sequences with biased base composition identifies noncoding RNAs in the A-T rich genome of Plasmodium falciparum. Mol Biochem Parasitol. 2005;144(2):149–58. doi: 10.1016/j.molbiopara.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 40.Li F, Sonbuchner L, et al. Nuclear non-coding RNAs are transcribed from the centromeres of Plasmodium falciparum and are associated with centromeric chromatin. J Biol Chem. 2008;283(9):5692–8. doi: 10.1074/jbc.M707344200. [DOI] [PubMed] [Google Scholar]
- 41.Epp C, Li F, et al. Chromatin associated sense and antisense noncoding RNAs are transcribed from the var gene family of virulence genes of the malaria parasite Plasmodium falciparum. Rna. 2009;15(1):116–27. doi: 10.1261/rna.1080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mourier T, Carret C, et al. Genome-wide discovery and verification of novel structured RNAs in Plasmodium falciparum. Genome Res. 2008;18(2):281–92. doi: 10.1101/gr.6836108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raabe CA, Sanchez CP, et al. A global view of the nonprotein-coding transcriptome in Plasmodium falciparum. Nucleic Acids Res. 2010;38(2):608–17. doi: 10.1093/nar/gkp895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Anouti F, Ananvoranich S. Comparative analysis of antisense RNA, double-stranded RNA, and delta ribozyme-mediated gene regulation in Toxoplasma gondii. Antisense Nucleic Acid Drug Dev. 2002;12(4):275–81. doi: 10.1089/108729002320351593. [DOI] [PubMed] [Google Scholar]
- 45.Al-Anouti F, Quach T, et al. Double-stranded RNA can mediate the suppression of uracil phosphoribosyltransferase expression in Toxoplasma gondii. Biochem Biophys Res Commun. 2003;302(2):316–23. doi: 10.1016/s0006-291x(03)00172-4. [DOI] [PubMed] [Google Scholar]
- 46.Al Riyahi A, Al-Anouti F, et al. Single argonaute protein from Toxoplasma gondii is involved in the double-stranded RNA induced gene silencing. Int J Parasitol. 2006;36(9):1003–14. doi: 10.1016/j.ijpara.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 47.Radke JR, Behnke MS, et al. The transcriptome of Toxoplasma gondii. BMC Biol. 2005;3:26. doi: 10.1186/1741-7007-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gissot M, Kelly KA, et al. Epigenomic modifications predict active promoters and gene structure in Toxoplasma gondii. PLoS Pathog. 2007;3(6):e77. doi: 10.1371/journal.ppat.0030077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matrajt M, Donald RG, et al. Identification and characterization of differentiation mutants in the protozoan parasite Toxoplasma gondii. Mol Microbiol. 2002;44(3):735–47. doi: 10.1046/j.1365-2958.2002.02904.x. [DOI] [PubMed] [Google Scholar]
- 50.Singh U, Brewer JL, et al. Genetic analysis of tachyzoite to bradyzoite differentiation mutants in Toxoplasma gondii reveals a hierarchy of gene induction. Mol Microbiol. 2002;44(3):721–33. doi: 10.1046/j.1365-2958.2002.02903.x. [DOI] [PubMed] [Google Scholar]
- 51.Anderson MZ, Brewer J, et al. A pseudouridine synthase homologue is critical to cellular differentiation in Toxoplasma gondii. Eukaryot Cell. 2009;8(3):398–409. doi: 10.1128/EC.00329-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanchinathan P, Brewer JL, et al. Disruption of a locus encoding a nucleolar zinc finger protein decreases tachyzoite-to-bradyzoite differentiation in Toxoplasma gondii. Infect Immun. 2005;73(10):6680–8. doi: 10.1128/IAI.73.10.6680-6688.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Behnke MS, Radke JB, et al. The transcription of bradyzoite genes in Toxoplasma gondii is controlled by autonomous promoter elements. Mol Microbiol. 2008;68(6):1502–18. doi: 10.1111/j.1365-2958.2008.06249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flueck C, Bartfai R, et al. A major role for the Plasmodium falciparum ApiAP2 protein PfSIP2 in chromosome end biology. PLoS Pathog. 6(2):e1000784. doi: 10.1371/journal.ppat.1000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hakimi MA, Deitsch KW. Epigenetics in Apicomplexa: control of gene expression during cell cycle progression, differentiation and antigenic variation. Curr Opin Microbiol. 2007;10(4):357–62. doi: 10.1016/j.mib.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 56.Dinger ME, Pang KC, et al. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;4(11):e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prasanth KV, Spector DL. Eukaryotic regulatory RNAs: an answer to the ‘genome complexity’ conundrum. Genes Dev. 2007;21(1):11–42. doi: 10.1101/gad.1484207. [DOI] [PubMed] [Google Scholar]
- 58.Castrignano T, Canali A, et al. CSTminer: a web tool for the identification of coding and noncoding conserved sequence tags through cross-species genome comparison. Nucleic Acids Res. 2004;32(Web Server issue):W624–7. doi: 10.1093/nar/gkh486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Machado-Lima A, del Portillo HA, et al. Computational methods in noncoding RNA research. J Math Biol. 2008;56(1–2):15–49. doi: 10.1007/s00285-007-0122-6. [DOI] [PubMed] [Google Scholar]
- 60.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31(13):3429–31. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Danilova LV, Pervouchine DD, et al. RNAKinetics: a web server that models secondary structure kinetics of an elongating RNA. J Bioinform Comput Biol. 2006;4(2):589–96. doi: 10.1142/s0219720006001904. [DOI] [PubMed] [Google Scholar]
- 62.Do CB, Woods DA, et al. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006;22(14):e90–8. doi: 10.1093/bioinformatics/btl246. [DOI] [PubMed] [Google Scholar]
- 63.Tabaska JE, Cary RB, et al. An RNA folding method capable of identifying pseudoknots and base triples. Bioinformatics. 1998;14(8):691–9. doi: 10.1093/bioinformatics/14.8.691. [DOI] [PubMed] [Google Scholar]
- 64.Kolbe DL, Eddy SR. Local RNA structure alignment with incomplete sequence. Bioinformatics. 2009;25(10):1236–43. doi: 10.1093/bioinformatics/btp154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu J, Gough J, et al. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genet. 2006;2(4):e29. doi: 10.1371/journal.pgen.0020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kong L, Zhang Y, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(Web Server issue):W345–9. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Solda G, I, Makunin V, et al. An Ariadne’s thread to the identification and annotation of noncoding RNAs in eukaryotes. Brief Bioinform. 2009;10(5):475–89. doi: 10.1093/bib/bbp022. [DOI] [PubMed] [Google Scholar]
- 68.Guttman M, Amit I, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]