

Abstract
Major life events involving social rejection are strongly associated with onset of depression. To account for this relation, we propose a psychobiological model in which rejection-related stressors elicit a distinct and integrated set of cognitive, emotional, and biological changes that may evoke depression. In this model, social rejection events activate brain regions involved in processing negative affect and rejection-related distress (e.g., anterior insula, dorsal anterior cingulate cortex). They also elicit negative self-referential cognitions (e.g., “I’m undesirable,” “Other people don’t like me”) and related self-conscious emotions (e.g., shame, humiliation). Downstream biological consequences include upregulation of the hypothalamic-pituitary-adrenal axis, sympathetic-adrenal-medullary axis, and inflammatory response. Pro-inflammatory cytokines play an important role in this process because they induce a constellation of depressotypic behaviors called sickness behaviors. Although these changes can be short-lived, sustained inflammation may occur via glucocorticoid resistance, catecholamines, sympathetic innervation of immune organs, and immune cell aging. This response also may be moderated by several factors, including prior life stress, prior depression, and genes implicated in stress reactivity.
Keywords: Life stress, Social rejection, Depression, Dorsal anterior cingulate cortex, Cortisol, Inflammation, Glucocorticoid resistance, Immune cell aging, Stress sensitization, 5-HTTLPR
Depression is a serious psychiatric condition that approximately 20% of people experience in their lifetime (Kessler et al., 2010). Although many questions remain regarding its etiology, risk for the disorder substantially increases following an acute major life event (Monroe et al., 2009). Stressors of this type typically involve serious threats to an important relationship or job, or significant changes in health, housing, or financial status. Depressed individuals are 2.5- to 10-times more likely than nondepressed persons to have experienced a recent major life event (Kendler et al., 1995; Shrout et al., 1989), and up to 82% of depressive episodes appear to be precipitated by such stress (Mazure, 1998). Major life events, therefore, are one of the best predictors of an impending onset of depression.
Given the consistency with which depression follows major life stress, there can be little doubt that such events play a role in bringing about at least some forms of depression. At the same time, little is known about what makes particular stressors depressogenic. A fundamental question along these lines is whether some life events increase risk for depression more than others and, if so, why? This question has been underappreciated in part because early theories postulated that organismic responses to stress are uniform and similar regardless of the eliciting condition (see Selye, 1956). This view is not uncommon but rather pervades the literature on stress and depression. For example, very few studies have looked beyond stressor severity to investigate how the characteristics of different life events might predict clinical aspects of depression (Hammen, 2005). This has occurred despite evidence that biological and behavioral responses may not in fact be uniform across stressors, but may be differentiated and specific (Denson et al., 2009; Kemeny, 2003; Weiner, 1992).
The present review explores an instance of this stressor specificity by examining the psychobiology of social rejection and depression. We focus on social rejection because rejection-related life events increase risk for depression to a greater degree than any other type of stress (Kendler et al., 2003). Our overarching goal is twofold: first, identify the attributes that make social rejection particularly depressogenic; and second, propose a model to explain how rejection events elicit a distinct and integrated set of cognitive, emotional, and biological changes that may evoke depression. We argue that while such changes can be adaptive, several factors may prolong these reactions, leading to sustained inflammatory processes that precipitate depression for vulnerable individuals.
1. Interpersonal loss, social rejection, and depression
Interpersonal loss has long been regarded as central to depression. This focus can be traced back to Freud’s (1917/1957) Mourning and Melancholia, and is represented in many contemporary psychodynamic (Blatt, 2004), cognitive (Beck, 1967), and social (Brown and Harris, 1978) accounts of the disorder. Generally speaking, these theories posit that close social bonds confer several important benefits, such as nurturance, social connection, physical protection, and the potential for reproductive success. Consequently, stressors that disrupt such bonds are hypothesized to produce immediate and intense distress (Bowlby, 1980; Gilbert, 1992). Life events research has supported this prediction. For example, individuals who experience a key interpersonal loss are nearly twice as likely to develop depression as individuals without a recent loss (Monroe et al., 1999).
All interpersonal loss events, however, are not “created equal.” For instance, an intimate relationship can end when you break up with your partner but also when your partner breaks up with you. This difference may seem subtle, but it is crucial because different types of interpersonal loss have different attributes and pose very different risks for depression. As summarized in Table 1, individuals who break up with their partner are 10.2-times more likely to develop depression than those without such stress. However, individuals who are broken-up with are 21.6-times more likely to develop depression (Kendler et al., 2003). Thus, risk for depression more than doubles based on this distinction alone. And importantly, because the life events included in these comparisons were all rated as having high threat, these differential risks by event type are not likely due to differences in stressor severity.
Table 1.
Different types of interpersonal loss, their key attributes, and associated risk for depression
| Event Type | Definition | Key Attributes | Risk for Onset of Depression* |
|---|---|---|---|
| Death | Death of Subject’s close tie, such as a spouse, parent, or child |
Loss of significant other/confidant; unpremeditated; low on social-evaluative threat; low on controllability; no social demotion, or loss in social status, value, or regard |
9.9 ± 0.4 |
| Self-initiated separation |
Significant separation, falling- out, or rift in a relationship involving a close tie, where the separation is mutual or initiated by Subject |
Loss of significant other/confidant; premeditated; low on social-evaluative threat; high on controllability; no social demotion, or loss in social status, value, or regard |
10.2 ± 0.6 |
| Other-initiated separation |
Same as above, except the separation is initiated by Subject’s partner |
Loss of significant other/confidant; premeditated; high on social-evaluative threat; low on controllability; intentional social demotion, and loss in social status, value, and regard |
21.6 ± 0.9 |
Estimates of risk are from Kendler et al., 2003, and are given as mean risk for onset of depression in the month of event ± SE percentage.
Social rejection events not only greatly increase risk for depression, they also appear to bring about depression more quickly than non-rejection events. For example, Slavich and colleagues examined the effects of a specific type of rejection called targeted rejection (TR), which involves the exclusive, active, and intentional rejection of an individual by others (Slavich et al., 2009). Individuals who experienced a recent severe TR event became depressed approximately three times faster than those who experienced a severe life event that was not defined as TR. Again, because these events were all rated severe using a state-of-the-art, interview-based measure of stress, the relatively quicker onset of depression following TR is likely due to the specific characteristics that define TR events, as opposed to differences in the severity of TR versus non-TR life events.
2. Depressogenic attributes of social rejection
So, why is social rejection so depressogenic? One possibility concerns the adaptive value of social connection and the poignant manner in which rejection events disrupt social bonds. Because of the critical benefits that social relationships provide, humans possess a fundamental drive to maintain positive social status, value, and regard, all of which increase likelihood of continued social inclusion (Baumeister and Leary, 1995). In fact, because of their common goal to promote survival, we may strive to preserve social status as strongly as we strive to preserve physical well-being, a process that has been called social self-preservation (Dickerson et al., 2004; Gruenewald et al., 2004).
Social rejection events threaten social self-preservation because they involve high degrees of negative social evaluation, or social-evaluative threat. They also imply devaluation of the person by others, and forecast potential or actual involuntary social exclusion (Gilbert, 1992; Kemeny, 2009). As a result, rejection events may elicit negative self-referential cognitions concerning one’s social worth and esteem, such as “I’m undesirable,” “I’m unlovable,” and “Other people don’t like me” (Monroe et al., 2007a). These cognitions, which are hallmarks of many depressive episodes, may in turn give rise to self-conscious emotions like shame and humiliation, which are associated with biological processes (e.g., inflammation) that support behavioral disengagement and withdrawal (Kemeny, 2009). Although these outcomes may be evoked by other types of stress, we argue that rejection-related stressors possess the greatest propensity for initiating this particular set of negative cognitions and emotions.
In sum, social rejection events are relatively unique insofar as they include elements of social-evaluative threat, social demotion, and social exclusion. These attributes threaten social self-preservation and are more likely to characterize social rejection events (e.g., being broken up with, getting fired) than non-rejection events (e.g., initiating a break up, quitting a job). As a result, rejection may elicit negative self-referential cognitions and self-conscious emotions, which are associated with biological processes that promote depressotypic behaviors. These responses may be short-lived for many individuals, but sustained for persons who are at risk for depression.
3. Neural and peripheral mediators of the social rejection response
We turn now to our second goal, which is to evaluate how rejection “gets under the skin” to evoke depression. Our basic formulation is that rejection activates a coordinated and distinct psychobiological response that possesses short-term adaptive value in preparing the organism to deal with potential threats. In some instances, however, these changes may become prolonged, conferring increased risk for depression.
3.1. Social rejection and the biological stress response
Psychobiological responses to rejection begin with the perception of social threat. A growing body of research suggests that stressors involving social rejection and exclusion activate neural regions involved in processing negative affect and the distress associated with physical pain. These regions include the anterior insula and dorsal anterior cingulate cortex (dACC; Lieberman and Eisenberger, 2009). Although activation in multiple biological systems may follow, the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic-adrenal-medullary (SAM) axis are particularly responsive. Degree of reactivity differs based on subtle features of the social environment, such as availability of social support (O’Donovan and Hughes, 2008). Moreover, this reactivity may be indexed by two biomarkers: the glucocorticoid cortisol, which is released by the HPA axis, and the catecholamines epinephrine and norepinephrine, which are released by the SAM axis.
A variety of stressful events can activate the HPA and SAM axes. However, a meta-analytic review of 208 studies revealed that rejection-related stressors are particularly activating (Dickerson and Kemeny, 2004). Specifically, stressful conditions characterized by low controllability and high social-evaluative threat were associated with the greatest cortisol responses and slowest recovery of cortisol to baseline levels. These findings were subsequently confirmed in a laboratory study. Compared to participants in a condition involving no social-evaluative threat, those in the social-evaluative threat condition exhibited greater increases in shame and greater decrements in social self-esteem; they also showed greater HPA axis activation, as indexed by cortisol. Moreover, increases in shame and decrements in self-esteem were both associated with increases in cortisol (Gruenewald et al., 2004).
Rejection-related stressors and their emotional sequelae are also powerful activators of the SAM axis. For example, individuals exposed to social rejection exhibit strong cardiovascular responses (Mendes et al., 2008), as do those exposed to social-evaluative threat (Gruenewald et al., 2004). In addition, social-evaluative threat elicits increases in epinephrine and norepinephrine (Wirtz et al., 2006). Finally, individuals with high levels of trait shame exhibit elevated production of alpha amylase, a biomarker of SAM axis activation (Rohleder et al., 2008).
3.2. Social rejection and inflammation
Exposure to social rejection may also activate the immune system, initiating processes that promote inflammation. Pro-inflammatory cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α), are an important component of this immune response. These cytokines are notable because they signal the central nervous system to elicit sickness behaviors. Sickness behaviors are thought to facilitate an organism’s recuperation and recovery from illness or injury, and include depressotypic symptoms such as anhedonia, fatigue, psychomotor slowing, and social-behavioral withdrawal (Dantzer et al., 2008; Yirmiya et al., 2000).
Studies investigating the association between rejection-related emotions and inflammation have found that writing about personal situations involving self-blame increases feelings of shame; increases in shame, in turn, are associated with greater inflammatory activity (Dickerson et al., 2004). Trait shame, or the tendency to feel shame in everyday life, also has been associated with elevated in vitro lipopolysaccharide-stimulated levels of IL-6 (Rohleder et al., 2008). Finally, there is evidence that aspects of social rejection, specifically social isolation (Cole et al., 2007) and interpersonal stress (Miller et al., 2009b), upregulate the expression of genes that promote inflammation.
Experimental studies of social threat paint a similar picture. For example, laboratory stressors involving social-evaluative threat reliably elicit increases in IL-1β, IL-6, and TNF-α (Kemeny, 2009). In one such study, individuals who performed a speech and math task in the presence of an evaluative audience exhibited greater in vitro lipopolysaccharide-stimulated production of TNF-α than those who performed the same task without being evaluated (Dickerson et al., 2009). These two conditions were rated as being equally challenging, controllable, and difficult. However, feeling more evaluated during the task was associated with greater increases in TNF-α. Social-evaluative threat, therefore, appears to be a key characteristic of stressors that promote inflammation.
3.3. Inflammation and depression
These findings have direct implications for the relation of social rejection to depression because inflammatory mediators have been implicated in the pathophysiology of depression. In this context, it has been observed that: (1) depression is commonly comorbid with inflammatory disorders, such as arthritis, diabetes, and cardiovascular disease; (2) depression confers increased risk for the development of inflammatory disorders in physically healthy individuals; and (3) depressed individuals with and without comorbid physical illness exhibit elevated levels of inflammatory molecules (Kiecolt-Glaser and Glaser, 2002).
Pro-inflammatory cytokines, in fact, may directly evoke depression (Miller et al., 2009). Three lines of research support this formulation. First, administration of the pro-inflammatory cytokines IL-1β and TNF-α elicits a depressotypic behavioral state in rodents (Dantzer et al., 2008). Second, administration of cytokines that promote inflammatory activity, specifically interferon-α (IFN-α) and interleukin-2 (IL-2), evokes clinically-significant levels of depression in up to 50% patients who receive them as treatments for infectious diseases or cancer (Capuron and Miller, 2004; Raison et al., 2006). Finally, vaccination and administration of endotoxin, both of which increase pro-inflammatory cytokine production, induce depressed mood in physically healthy individuals (Eisenberger et al., 2009; Glaser et al., 2003).
The only multilevel study of social rejection, inflammation, and depression that we know of manipulated both social rejection and inflammation while assessing neural responses using fMRI. Administration of endotoxin had the predicted effect of increasing both inflammatory activity (as indexed by IL-6) and depressive symptoms. Increases in IL-6, in turn, were associated with activation in the anterior insula and dACC in response to social exclusion. Moreover, activity in these brain regions mediated the relation between increases in IL-6 and depressive mood in females (Eisenberger et al., 2009). In related work, administration of IFN-α was found to be associated with greater dACC activation in participants completing a visuospatial attention task (Harrison et al., 2009). Also, endotoxin-induced mood deterioration has been associated with activation in the subgenual anterior cingulate cortex, a brain region implicated in mood regulation and in the etiology of depression (Capuron et al., 2005; see also O’Connor et al., 2009). Inflammatory cytokines, in fact, may act on the brain in multiple ways leading to depression (Dantzer et al., 2008; Miller et al., 2009).
3.4. Biological pathways of sustained inflammation
Although the majority of studies reviewed here investigated acute responses to rejection-related stress, several pathways may be involved in sustaining inflammation, leading to increased risk for depression. Glucocorticoid resistance is one such pathway. As previously described, exposure to social threat promotes the production of cortisol and catecholamines. Binding of circulating cortisol and catecholamines to receptors on immune cells can modulate the release of pro-inflammatory cytokines (Padgett and Glaser, 2003). Typically, cortisol acts on the glucocorticoid receptor with anti-inflammatory effects (Sapolsky et al., 2000). Prolonged exposure to cortisol, however, may down-regulate the glucocorticoid receptor on immune cells, leading to glucocorticoid resistance and to sustained inflammation (Stark et al., 2001). Pro-inflammatory cytokines themselves also may induce glucocorticoid resistance (Miller et al., 2009a). Importantly, this process occurs in response to social threat. For example, disrupting the social status of rodents leads to dysregulation of the inflammatory response via glucocorticoid resistance (Avitsur et al., 2001). Exposure to social-evaluative threat in humans also promotes glucocorticoid resistance (Dickerson et al., 2009). In combination with these effects, rejection-related SAM activation could contribute to sustained inflammation through binding of catecholamines on immune cells and through sympathetic innervation of immune organs (Bierhaus et al., 2003; Elenkov et al., 2000b; however, see Elenkov et al., 2000a).
Sustained inflammation may also occur via immune cell aging, as indexed by telomere length. Telomeres are DNA-protein complexes that cap the ends of chromosomes and protect against DNA damage (Blackburn, 1991). Immune cell telomeres shorten with each cycle of cell division, and the biological age of an immune cell may thus be indexed by telomere length. Although the association between social rejection and telomere length has not yet been examined, plausible pathways exist by which rejection may accelerate telomere shortening. In particular, correlates of social-evaluative threat, specifically cortisol and pro-inflammatory cytokines, are known to accelerate telomere shortening (Choi et al., 2008; O’Donovan et al., 2009). Cells with short telomere length, in turn, produce larger quantities of pro-inflammatory cytokines (Dagarag et al., 2004), thereby increasing risk for depression. Elevated levels of cortisol and inflammation in depressed individuals could further accelerate telomere shortening. Consistent with this formulation, there is evidence that depressed individuals have shorter telomeres (Simon et al., 2006). Psychosocial factors relating to rejection, such as interpersonal loss (Parks et al., 2009) and chronic social stress (Damjanovic et al., 2007; Epel et al., 2004), also have been associated with short telomeres (see also Cherkas et al., 2006).
4. A psychobiological model of social rejection and depression
Drawing on the research reviewed here, we propose a psychobiological model of social rejection and depression (see Fig. 1). In this model, social rejection events activate brain regions involved in processing negative affect and rejection-related distress (e.g., anterior insula and dACC). They also elicit negative self-referential cognitions (e.g., “I’m undesirable,” “Other people don’t like me”) and related self-conscious emotions (e.g., shame, humiliation). Downstream biological consequences include upregulation of the HPA axis, SAM axis, and inflammatory response. The resulting release of pro-inflammatory cytokines induces sickness behaviors that increase risk for depression, especially when sustained via glucocorticoid resistance, catecholamines, sympathetic innervation of immune organs, and immune cell aging.
Fig. 1.
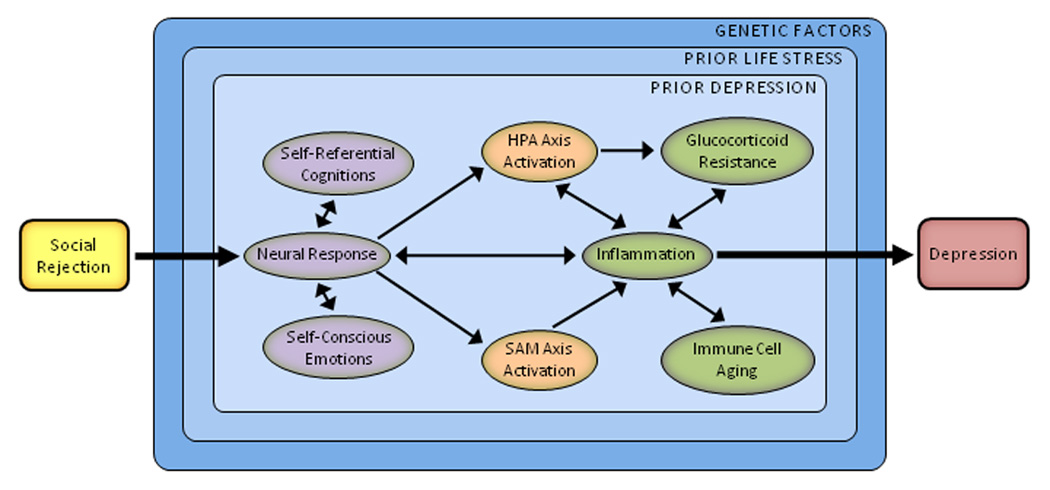
A psychobiological model of social rejection and depression. Stressors involving social rejection include elements of social-evaluative threat, social demotion, and social exclusion. Because of these attributes, rejection-related stressors elicit a distinct and integrated set of cognitive, emotional, and biological changes that promote behavioral disengagement and withdrawal. The process begins with the perception of threat. Social rejection events activate brain regions involved in processing negative affect and rejection-related distress (e.g., anterior insula, dorsal anterior cingulate cortex). They also elicit negative self-referential cognitions (e.g., “I’m undesirable,” “I’m unlovable,” and “Other people don’t like me”) and related self-conscious emotions (e.g., shame, humiliation). Downstream biological consequences include upregulation of the hypothalamic-pituitary-adrenal (HPA) axis, sympathetic-adrenal-medullary (SAM) axis, and inflammatory response. Resulting increases in inflammation may be indexed by the pro-inflammatory cytokines interleukin-1β, interleukin-6, and tumor necrosis factor-α. These cytokines are important because they induce a constellation of depressotypic behaviors called sickness behaviors. Although these changes can be short-lived, sustained inflammation may occur via several pathways, including glucocorticoid resistance, catecholamines, sympathetic innervation of immune organs, and immune cell aging. This response also may be moderated by several factors, including prior life stress, prior depression, and genes implicated in stress reactivity.
5. Moderators of the psychobiological stress response
The preceding sections describe a psychobiological model of social rejection and depression. However, not everyone mounts a significant cytokine response to social or biological provocation. Similarly, many individuals who experience social rejection never get depressed. Differential risk for depression following rejection is likely influenced by several factors that moderate responsivity to stress in general. We review three such factors here, focusing on prior exposure to stress, previous experiences with depression, and genes implicated in stress reactivity.
5.1. Prior life stress
Prior major life stress and previous depressive episodes are two of the strongest risk factors for depression. Although biological reactivity to stress may be altered by exposure to prior stressors occurring at any time, early life stress may be particularly hazardous. Indeed, humans may experience the effects of environmental insults as early as gestational day 14, when in utero blood flow is established (Goodman and Brand, 2009). Fetuses of stressed mothers can be exposed to high levels of corticotrophin-releasing factor (CRF), potentiating reactivity to stress over the life course (Lupien et al., 2009). Childhood maltreatment may also lead to elevated risk for depression. For instance, individuals with histories of childhood physical or sexual abuse exhibit greater HPA axis responses to stress in adulthood (Heim et al., 2000). Early maltreatment also predicts elevations in inflammation in adulthood (Danese et al., 2008).
These findings suggest that environmental adversity may increase risk for depression in part by sensitizing individuals biologically to stress. Post (1992) proposed a framework for this effect, focusing on the neurobiological changes that follow exposure to prior stress or depression. The model posits that neurobiological kindling sensitizes individuals to stress such that they consequently succumb to less severe forms of adversity. Consistent with this prediction, individuals exposed to early parental loss, abuse, or neglect exhibit lower levels of pre-onset stress than those without such early adversity (Harkness et al., 2006). Slavich and colleagues extended these findings by showing that individuals with early parental loss have lower levels of interpersonal loss occurring prior to onset of depression. However, this association was not observed for other types of stress (Slavich et al., 2010). Exposure to early interpersonal loss, therefore, may sensitize people uniquely to subsequent situations involving loss or rejection.
5.2. Prior depression
Prior depressive episodes may also moderate individuals’ responses to stress. For example, individuals who experience interpersonal loss in childhood may develop knowledge structures, or schemas, that include themes of inferiority, worthlessness, and rejection. These schemas may become activated later in life when the individual experiences subsequent loss or rejection. Once activated, the schemas direct attention to—and enhance memory for—schema-congruent information, such as loss- or rejection-related experiences, and negatively skew the interpretation of neutral or ambiguous social cues. Activated schemas also give rise to specific negative thoughts (e.g., “I’m unlovable,” “Other people don’t like me”) and emotions (e.g., shame, humiliation, sadness). With successive episodes of depression, these schemas become more interconnected and come to include more related memories; as a result, they may be activated more easily—that is, by more minor and more diverse forms of stress (Beck, 2008). Consistent with this formulation, risk for depression increases, and levels of pre-onset stress decrease, over successive depressive episodes (Monroe et al., 2007b). Again, this increase in sensitization may be specific to particular types of stress, such as interpersonal loss and rejection (see Slavich et al., 2010).
5.3. Genetic factors
Finally, research has begun to identify genetic factors that moderate reactivity to stress. The most widely studied gene in this context is 5-HTT, which encodes the serotonin transporter and affects regional up- and down-regulation of specific serotonin receptors. Particular attention has been paid to the promoter region of this gene, 5-HTTLPR, because of its involvement in serotonin signaling, and, consequently, mood, cognition, and behavior. In a landmark study, Caspi et al. (2003) found that individuals with one or two copies of the short variant of 5-HTTLPR were more likely than long/long carriers to develop depression following early maltreatment or recent life stress. Subsequent studies have yielded mixed results (Risch et al., 2009). These inconsistencies, however, are likely due at least in part to methodological issues, one of which concerns the problematic assessment of life stress (Monroe and Reid, 2008; Rutter et al., 2009; Uher and McGuffin, 2010).
Depression is a complex, etiologically-heterogeneous disorder. Robust associations between specific genotypes and risk for depression may therefore be elusive. As a result, some researchers have begun investigating intermediate phenotypes for depression. In this context, individuals with risk variants of the 5-HTTLPR polymorphism have been shown to exhibit greater amygdala responses to emotional stimuli (Munafò et al., 2008); greater amygdala reactivity, in turn, has been associated with greater chronicity of depression (Dannlowski et al., 2008). Short/short carriers also appear to exhibit potentiated biological responses to stress, as indexed by greater and more prolonged elevations in cortisol (Gotlib et al., 2008). Research in this area is advancing rapidly, with new, potentially-relevant genotypes being identified regularly.
6. Final considerations and future directions
We began by suggesting that not all stressful life events are equivalent with respect to their fundamental attributes or associated risk for depression. Rather, social rejection events are more likely to precipitate depression, and also appear to bring about depression more quickly, than non-rejection events. The model we propose elucidates pathways that may underlie this effect.
Although this model provides a blueprint for future research on social rejection and depression, interpersonal factors and depression are related in complex ways. For example, social rejection may cause depression, but depressive behaviors and personality styles may also engender rejection (e.g., via excessive reassurance seeking or stress generation). Yet other factors may increase the likelihood of experiencing both rejection and depression (e.g., trait rejection sensitivity, neuroticism, and the genetic factors underlying neuroticism). Additional research is clearly needed to clarify how these factors interact in relation to depression. Studies investigating the neural, cognitive, emotional, and biological correlates of different stressors are also needed to more precisely identify the specific characteristics that make stressors potentially depressogenic. This research can inform the development of better taxonomies of life stress. It also has the ability to improve our understanding of how stress causes depression, one of our most common and costly psychiatry disorders.
Acknowledgements
Preparation of this review was supported by a Society in Science: Branco Weiss Fellowship and by Ruth L. Kirschstein National Research Service Award MH019391-17 to George Slavich, and by a Fulbright Scholarship and Rotary International Ambassadorial Scholarship to Aoife O’Donovan. We thank Naomi Eisenberger and Keely Muscatell, and two anonymous reviewers, for their helpful comments on a previous version of this report.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Horm. Behav. 2001;39:247–257. doi: 10.1006/hbeh.2001.1653. [DOI] [PubMed] [Google Scholar]
- Baumeister RF, Leary MR. The need to belong: desire for interpersonal attachments as a fundamental human motivation. Psychol. Bull. 1995;117:497–529. [PubMed] [Google Scholar]
- Beck AT. Depression: clinical, experimental, and theoretical aspects. New York: Harper and Row; 1967. [Google Scholar]
- Beck AT. The evolution of the cognitive model of depression and its neurobiological correlates. Am. J. Psychiatry. 2008;165:969–977. doi: 10.1176/appi.ajp.2008.08050721. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, Ferstl R, von Eynatten M, Wendt T, Rudofsky G, Joswig M, Morcos M, Schwaninger M, McEwen B, Kirschbaum C, Nawroth PP. A mechanism converting psychosocial stress into mononuclear cell activation. PNAS. 2003;100:1920–1925. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH. Structure and function of telomeres. Nature. 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- Blatt SJ. Experiences of depression: theoretical, clinical, and research perspectives. Washington, DC: American Psychological Association; 2004. [Google Scholar]
- Bowlby J. Loss: Sadness and depression. Vol. 3. New York: Basic Books; 1980. Attachment and loss. [Google Scholar]
- Brown GW, Harris TO. Social origins of depression: a study of psychiatric disorder in women. New York: Free Press; 1978. [Google Scholar]
- Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-α. Biol. Psychiatry. 2004;56:819–824. doi: 10.1016/j.biopsych.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Capuron L, Pagnoni G, Demetrashvili M, Woolwine BJ, Nemeroff CB, Berns GS, Miller AH. Anterior cingulate activation and error processing during interferon-alpha treatment. Biol. Psychiatry. 2005;58:190–196. doi: 10.1016/j.biopsych.2005.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Cherkas LF, Aviv A, Valdes AM, Hunkin JL, Gardner JP, Surdulescu GL, Kimura M, Spector TD. The effects of social status on biological aging as measured by white-blood-cell telomere length. Aging Cell. 2006;5:361–365. doi: 10.1111/j.1474-9726.2006.00222.x. [DOI] [PubMed] [Google Scholar]
- Choi J, Fauce SR, Effros RB. Reduced telomerase activity in human T lymphocytes exposed to cortisol. Brain Behav. Immun. 2008;22:600–605. doi: 10.1016/j.bbi.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SW, Hawkley LC, Arevalo JM, Sung CY, Rose RM, Cacioppo JT. Social regulation of gene expression in human leukocytes. Genome Biol. 2007;8:R189. doi: 10.1186/gb-2007-8-9-r189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagarag M, Evazyan T, Rao N, Effros RB. Genetic manipulation of telomerase in HIV-specific CD8+ T cells: enhanced antiviral functions accompany the increased proliferative potential and telomere length stabilization. J. Immunol. 2004;173:6303–6311. doi: 10.4049/jimmunol.173.10.6303. [DOI] [PubMed] [Google Scholar]
- Damjanovic AK, Yang Y, Glaser R, Kiecolt-Glaser JK, Nguyen H, Laskowski B, Zou Y, Beversdorf DQ, Weng N. Accelerated telomere erosion is associated with a declining immune function of caregivers of Alzheimer’s disease patients. J. Immunol. 2007;179:4249–4254. doi: 10.4049/jimmunol.179.6.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch. Gen. Psychiatry. 2008;65:409–416. doi: 10.1001/archpsyc.65.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannlowski U, Ohrmann P, Bauer J, Deckert J, Hohoff C, Kugel H, Arolt V, Heindel W, Kersting A, Baune BT, Suslow T. 5-HTTLPR biases amygdala activity in response to masked facial expressions in major depression. Neuropsychopharmacology. 2008;33:418–424. doi: 10.1038/sj.npp.1301411. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nature Rev. Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denson TF, Spanovic M, Miller N. Cognitive appraisals and emotions predict cortisol and immune responses: a meta-analysis of acute laboratory social stressors and emotion inductions. Psychol. Bull. 2009;135:823–853. doi: 10.1037/a0016909. [DOI] [PubMed] [Google Scholar]
- Dickerson SS, Gable SL, Irwin MR, Aziz N, Kemeny ME. Social-evaluative threat and proinflammatory cytokine regulation: an experimental laboratory investigation. Psychol. Sci. 2009;20:1237–1244. doi: 10.1111/j.1467-9280.2009.02437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson SS, Kemeny ME. Acute stressors and cortisol responses: a theoretical integration and synthesis of laboratory research. Psychol. Bull. 2004;130:355–391. doi: 10.1037/0033-2909.130.3.355. [DOI] [PubMed] [Google Scholar]
- Dickerson SS, Kemeny ME, Aziz N, Kim KH, Fahey JL. Immunological effects of induced shame and guilt. Psychosom. Med. 2004;66:124–131. doi: 10.1097/01.psy.0000097338.75454.29. [DOI] [PubMed] [Google Scholar]
- Eisenberger NI, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. An fMRI study of cytokine-induced depressed mood and social pain: the role of sex differences. Neuroimage. 2009;47:881–890. doi: 10.1016/j.neuroimage.2009.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenkov IJ, Papanicolaou DA, Wilder RL, Chrousos GP. Modulatory effects of glucocorticoids and catecholamines on human interleukin-12 and interleukin-10 production: clinical implications. Proc. Assoc. Am. Phys. 2000a;108:374–381. [PubMed] [Google Scholar]
- Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol. Rev. 2000b;52:595–638. [PubMed] [Google Scholar]
- Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. Accelerated telomere shortening in response to life stress. PNAS. 2004;101:17312–17315. doi: 10.1073/pnas.0407162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freud S. Mourning and melancholia. In: Strachey J, translator. The Standard Edition of the Complete Psychological Works of Sigmund Freud. Vol. 14. London: Hogarth Press; 1957. pp. 243–258. (Original work published 1917) [Google Scholar]
- Gilbert P. Depression: The Evolution of Powerlessness. New York: Guilford Press; 1992. [Google Scholar]
- Glaser R, Robles TF, Sheridan J, Malarkey WB, Kiecolt-Glaser JK. Mild depressive symptoms are associated with amplified and prolonged inflammatory responses after influenza virus vaccination in older adults. Arch. Gen. Psychiatry. 2003;60:1009–1014. doi: 10.1001/archpsyc.60.10.1009. [DOI] [PubMed] [Google Scholar]
- Goodman SH, Brand SR. Depression and early adverse experiences. In: Gotlib IH, Hammen CL, editors. Handbook of Depression. second ed. New York: Guilford Press; 2009. pp. 249–274. [Google Scholar]
- Gotlib IH, Joormann J, Minor KL, Hallmayer J. HPA axis reactivity: a mechanism underlying the associations among 5-HTTLPR, stress, and depression. Biol. Psychiatry. 2008;63:847–851. doi: 10.1016/j.biopsych.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenewald TL, Kemeny ME, Aziz N, Fahey JL. Acute threat to the social self: shame, social self-esteem, and cortisol activity. Psychosom. Med. 2004;66:915–924. doi: 10.1097/01.psy.0000143639.61693.ef. [DOI] [PubMed] [Google Scholar]
- Hammen C. Stress and depression. Annu. Rev. Clin. Psychol. 2005;1:293–319. doi: 10.1146/annurev.clinpsy.1.102803.143938. [DOI] [PubMed] [Google Scholar]
- Harkness KL, Bruce AE, Lumley MN. The role of childhood abuse and neglect in the sensitization to stressful life events in adolescent depression. J. Abnorm. Psychol. 2006;115:730–741. doi: 10.1037/0021-843X.115.4.730. [DOI] [PubMed] [Google Scholar]
- Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol. Psychiatry. 2009;66:407–414. doi: 10.1016/j.biopsych.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, Miller AH, Nemeroff CB. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA. 2000;284:592–597. doi: 10.1001/jama.284.5.592. [DOI] [PubMed] [Google Scholar]
- Kemeny ME. The psychobiology of stress. Curr. Dir. Psychol. Sci. 2003;12:124–129. [Google Scholar]
- Kemeny ME. Psychobiological responses to social threat: evolution of a psychological model in psychoneuroimmunology. Brain Behav. Immun. 2009;23:1–9. doi: 10.1016/j.bbi.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Hettema JM, Butera F, Gardner CO, Prescott CA. Life event dimensions of loss, humiliation, entrapment, and danger in the prediction of onsets of major depression and generalized anxiety. Arch. Gen. Psychiatry. 2003;60:789–796. doi: 10.1001/archpsyc.60.8.789. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Kessler RC, Walters EE, MacLean CJ, Sham PC, Neale MC, Heath AC, Eaves LJ. Stressful life events, genetic liability, and onset of an episode of major depression in women. Am. J. Psychiatry. 1995;152:833–842. doi: 10.1176/ajp.152.6.833. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Birnbaum H, Bromet E, Hwang I, Sampson N, Shahly V. Age differences in major depression: results from the National Comorbidity Survey Replication (NCS-R) Psychol. Med. 2010;40:225–237. doi: 10.1017/S0033291709990213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, Glaser R. Depression and immune function: central pathways to morbidity and mortality. J. Psychosom. Res. 2002;53:873–876. doi: 10.1016/s0022-3999(02)00309-4. [DOI] [PubMed] [Google Scholar]
- Lieberman MD, Eisenberger NI. Neuroscience. Pains and pleasures of social life. Science. 2009;323:890–891. doi: 10.1126/science.1170008. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Mazure CM. Life stressors as risk factors in depression. Clin. Psychol. Sci. Pract. 1998;5:291–313. [Google Scholar]
- Mendes WB, Major B, McCoy S, Blascovich J. How attributional ambiguity shapes physiological and emotional responses to social rejection and acceptance. J. Pers. Soc. Psychol. 2008;94:278–291. doi: 10.1037/0022-3514.94.2.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry. 2009a;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Rohleder N, Cole SW. Chronic interpersonal stress predicts activation of pro- and anti-inflammatory signaling pathways 6 months later. Psychosom Med. 2009b;71:57–62. doi: 10.1097/PSY.0b013e318190d7de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe SM, Reid MW. Gene-environment interactions in depression research: genetic polymorphisms and life-stress polyprocedures. Psychol. Sci. 2008;19:947–956. doi: 10.1111/j.1467-9280.2008.02181.x. [DOI] [PubMed] [Google Scholar]
- Monroe SM, Rohde P, Seeley JR, Lewinsohn PM. Life events and depression in adolescence: relationship loss as a prospective risk factor for first onset of major depressive disorder. J. Abnorm. Psychol. 1999;108:606–614. doi: 10.1037//0021-843x.108.4.606. [DOI] [PubMed] [Google Scholar]
- Monroe SM, Slavich GM, Georgiades K. The social environment and life stress in depression. In: Gotlib IH, Hammen CL, editors. Handbook of Depression. second ed. New York: Guilford Press; 2009. pp. 340–360. [Google Scholar]
- Monroe SM, Slavich GM, Torres LD, Gotlib IH. Severe life events predict specific patterns of change in cognitive biases in major depression. Psychol. Med. 2007a;37:863–871. doi: 10.1017/S0033291707000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe SM, Slavich GM, Torres LD, Gotlib IH. Major life events and major chronic difficulties are differentially associated with history of major depressive episodes. J. Abnorm. Psychol. 2007b;116:116–124. doi: 10.1037/0021-843X.116.1.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafò MR, Brown SM, Hariri AR. Serotonin transporter (5-HTTLPR) genotype and amygdala activation: a meta-analysis. Biol. Psychiatry. 2008;63:852–857. doi: 10.1016/j.biopsych.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor MF, Irwin MR, Wellisch DK. When grief heats up: pro-inflammatory cytokines predict regional brain activation. Neuroimage. 2009;47:891–896. doi: 10.1016/j.neuroimage.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan A, Hughes BM. Access to social support in life and in the laboratory: combined impact on cardiovascular reactivity to stress and state anxiety. J. Health Psychol. 2008;13:1147–1156. doi: 10.1177/1359105308095968. [DOI] [PubMed] [Google Scholar]
- O’Donovan A, Lin J, Dhabhar FS, Wolkowitz O, Tillie JM, Blackburn E, Epel E. Pessimism correlates with leukocyte telomere shortness and elevated interleukin-6 in post-menopausal women. Brain Behav. Immun. 2009;23:446–449. doi: 10.1016/j.bbi.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett DA, Glaser R. How stress influences the immune response. Trends Immunol. 2003;24:444–448. doi: 10.1016/s1471-4906(03)00173-x. [DOI] [PubMed] [Google Scholar]
- Parks CG, Miller DB, McCanlies EC, Cawthon RM, Andrew ME, DeRoo LA, Sandler DP. Telomere length, current perceived stress, and urinary stress hormones in women. Cancer Epidemiol. Biomarkers Prev. 2009;18:551–560. doi: 10.1158/1055-9965.EPI-08-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post RM. Transduction of psychosocial stress into the neurobiology of recurrent affective disorder. Am. J. Psychiatry. 1992;149:999–1010. doi: 10.1176/ajp.149.8.999. [DOI] [PubMed] [Google Scholar]
- Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, Griem A, Kovacs M, Ott J, Merikangas KR. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009;301:2462–2471. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohleder N, Chen E, Wolf JM, Miller GE. The psychobiology of trait shame in young women: extending the social self preservation theory. Health Psychol. 2008;27:523–532. doi: 10.1037/0278-6133.27.5.523. [DOI] [PubMed] [Google Scholar]
- Rutter M, Thapar A, Pickles A. Gene-environment interactions: biologically valid pathway or artifact? Arch. Gen. Psychiatry. 2009;66:1287–1289. doi: 10.1001/archgenpsychiatry.2009.167. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocrine Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Selye H. The Stress of Life. New York: McGraw-Hill; 1956. [Google Scholar]
- Shrout PE, Link BG, Dohrenwend BP, Skodol AE, Stueve A, Mirotznik J. Characterizing life events as risk factors for depression: the role of fateful loss events. J. Abnorm. Psychol. 1989;98:460–467. doi: 10.1037//0021-843x.98.4.460. [DOI] [PubMed] [Google Scholar]
- Simon NM, Smoller JW, McNamara KL, Maser RS, Zalta AK, Pollack MH, Nierenberg AA, Fava M, Wong KK. Telomere shortening and mood disorders: preliminary support for a chronic stress model of accelerated aging. Biol. Psychiatry. 2006;60:432–435. doi: 10.1016/j.biopsych.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Slavich GM, Monroe SM, Gotlib IH. Early adversity and depression history: associations with recent life stress in major depressive disorder. Under review. 2010 doi: 10.1016/j.jpsychires.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavich GM, Thornton T, Torres LD, Monroe SM, Gotlib IH. Targeted rejection predicts hastened onset of major depression. J. Soc. Clin. Psychol. 2009b;28:223–243. doi: 10.1521/jscp.2009.28.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001;280:1799–1805. doi: 10.1152/ajpregu.2001.280.6.R1799. [DOI] [PubMed] [Google Scholar]
- Uher R, McGuffin P. Mol. Psychiatry. Vol. 15. 2010. The moderation by the serotonin transporter gene of environmental adversity in the etiology of depression: 2009 update; pp. 18–22. [DOI] [PubMed] [Google Scholar]
- Weiner H. Perturbing the Organism: The Biology of Stressful Experience. Chicago: University of Chicago Press; 1992. [Google Scholar]
- Wirtz PH, von Kanel R, Mohiyeddini C, Emini L, Ruedisueli K, Groessbauer S, Ehlert U. Low social support and poor emotional regulation are associated with increased stress hormone reactivity to mental stress in systemic hypertension. J. Clin. Endocrinol. Metab. 2006;91:3857–3865. doi: 10.1210/jc.2005-2586. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Pollak Y, Morag M, Reichenberg A, Barak O, Avitsur R, Shavit Y, Ovadia H, Weidenfeld J, Morag A, Newman ME, Pollmacher T. Illness, cytokines, and depression. Ann. N.Y. Acad. Sci. 2000;917:478–487. doi: 10.1111/j.1749-6632.2000.tb05412.x. [DOI] [PubMed] [Google Scholar]