

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and is the most common form of dementia in elderly people. The accumulation of amyloid β (Aβ) is one of the histopathological hallmarks of AD. Aβ is aggregated to form oligomers which are toxic to neurons and are critical to the onset and progression of AD. In a Caenorhabditis elegans (C. elegans) model of AD, human Aβ is expressed intracellularly in the body wall muscle. The expression and subsequent aggregation of Aβ in the muscle lead to progressive paralysis. Although the mechanism of action is unknown, antidepressants have been used with FDA approved drugs for dementia in AD and have been shown to enhance cognitive function in human and in animal models of AD. We found that the antidepressant fluoxetine, a selective serotonin reuptake inhibitor, significantly delayed Aβ-induced paralysis in the C. elegans model of Aβ toxicity by reducing Aβ oligomers. Our result showed that insulin signaling and DAF-16/FOXO transcription factor were required for fluoxetine-mediated delayed paralysis. We also found that fluoxetine increased thermal stress resistance and extended life span. This finding suggests that fluoxetine may have benefit for the treatment of AD by reduction of proteotoxicity.
Keywords: fluoxetine, Alzheimer’s disease, amyloid β, selective serotonin reuptake inhibitor, C. elegans
1 INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and is the most common form of dementia in elderly people. The treatment of patients with AD is only symptomatic treatment with the available FDA approved drugs which are acetylcholinesterase inhibitors (donepezil, rivastigmine, galantamine) and N-methyl-D-aspartate receptor antagonist memantine. These drugs have a limited efficacy and are considered to be effective only in a short period of time ranging from 6 to 12 months (Shah et al., 2006; Raina et al., 2008). The formation of β-amyloid plaques is one of the key causes of the disease (Hardy et al., 2002). Aβ aggregation is toxic to neurons and eventually leads to clinical symptoms characterized by progressive and irreversible dementia.
AD affects not only cognitive function, but also other behaviors. Increasing evidence suggests that depression is a common psychiatric manifestation in AD patients There is a high prevalence rate (30-50%) of Alzheimer’s disease (AD) and depression comorbidity (Lee et al., 2003). Since depressive symptoms are commonly detected before AD patients manifest cognitive deterioration or are clinically diagnosed (Danion, 1993; Geerlings et al., 2000; Visser et al., 2000), antidepressants may serve as a rational complementary therapy for the treatment of Alzheimer’s disease (Aboukhatwa et al., 2010). In the recent years, clinical studies showed positive role of chronic administration of selective serotonin reuptake inhibitor (SSRI) antidepressants in hindering the progression of the AD and improving patient cognitive performance (Altman et al., 1987) either alone or in combination with other FDA approved drugs for treatment of AD (Lai et al., 2002; Finkel et al., 2004; Vythilingam et al., 2004; Schmitt et al., 2006; Mowla et al., 2007; Thompson et al., 2007).
Citalopram, an SSRI, has shown significantly improvement in the score of depressed patients in the Clinical Global Impression Scale (CGIS) and Montgomery Asberg Depression Scale (MADRS), as well as improvement of the emotional and cognitive functions in subgroup of patients who suffer from dementia (Nyth et al., 1992). Drug combination trials reported that patients treated with rivastigmine, a FDA proved drug for AD plus fluoxetine, a SSRI, had better performance in daily life activities and overall function which highlightens some of the benefits that may be obtained by adding a serotonin regimen to FDA approved drugs for AD patients (Mowla et al., 2007). Another study also reports the beneficial outcome of combining sertaline (SSRI) with donepzil treatment especially for AD patients with moderate to severe dementia (Finkel et al., 2004).Among currently available antidepressants, selective serotonin reuptake inhibitors (SSRIs) are commonly prescribed to patients with AD to relieve depression, anxiety and agitation (Nyth et al., 1990; Pollock et al., 1999). SSRIs are also frequently used concomitantly with acetylcholinesterase inhibitors (AChEIs) in patients with AD (Pollock et al., 1999). In addition, SSRIs may exert some degree of protection against the negative effects of depression on cognition in AD patients treated with AChEIs (Rozzini et al., 2010). SSRIs increase extracellular serotonin (5-HT) concentration by inhibiting serotonin reuptake transporter and is thought to improve depressive symptom through compensation for a 5-HT deficit in AD. Currently available SSRIs include fluoxetine, sertraline, paroxetine, fluvoxamine, citalopram and escitalopram. Pre-clinic studies also demonstrated restore of cognitive impairments and retards the development of amyloid and tau pathologies in 3xTgAD mice (Duman, 1998). However, the mechanism of action is unknown.
Fluoxetine has emerged as the treatment of choice for depression due to a better safety profile, and fewer side effects compared to tricyclic antidepressants. Moreover, fluoxetine stands out among other SSRIs for its adverse event profile and requirement for dosage titration (Wilde et al., 1998). Fluoxetine may also have therapeutic potential as neuroprotective and disease-modifying agent. There are several studies demonstrating that fluoxetine increased neurogenesis in the hippocampus (Jacobs et al., 2000; Malberg et al., 2000; David et al., 2009). Furthermore, fluoxetine enhanced memory and cognition in the patients with mild cognitive impairment (Mowla et al., 2007). However, the underlying mechanisms of its efficacy remain unclear.
The nematode model organism Caenorhabditis elegans (C. elegans) is an important model to study the molecular mechanisms of drug action and disease pathogenesis. For example, some behaviors of C. elegans such as egg laying have been used as a simple genetic system for identification of the action of fluoxetine and imipramine (Dempsey et al., 2005). C. elegans has been used as a model for various age-associated neurodegenerative diseases, including Alzheimer’s (Link, 1995; Link, 2001), Parkinson’s (Nass et al., 2002; Lakso et al., 2003) and Huntington’s diseases (Faber et al., 1999). There is strong genetic conservation between C. elegans and mammals as demonstrated by the fact that 60-80% of human gene have been identified in C. elegans (Kaletta et al., 2006). The transgenic C. elegans model of AD has been developed by expressing human Aβ in the muscle. The expression and subsequent aggregation of Aβ in the muscle lead to progressive paralysis (Link, 2006).
In C. elegans, the insulin/IGF-1 signaling pathway controls many biological processes such as lifespan, metabolism and stress response. This pathway is comprised of many proteins including insulin/IGF-1 receptor (DAF-2), PI 3-kinase (AGE-1) and FOXO transcription factor (DAF-16). In mammals, DAF-16 homologues are forkhead (FOXO) transcription factors, of which specific functions have been identified for multiple FOXO proteins such as FOXO1a and FOXO3a (Kuningas et al., 2007). Some FOXO proteins initiate stress-induced apoptosis, which eliminate damaged dysfunctional cells. FOXO proteins also up-regulate antioxidant defense and DNA repair-facilitating gene (Vijg et al., 2008). Recently it was reported that modulation of the insulin/IGF-1 signaling pathway increases thermotolerance and delays the onset of Aβ toxicity in C. elegans expressing human Aβ (Cohen et al., 2006). In this study, we aim to determine if fluoxetine protects against Aβ toxicity in transgenic C. elegans expressing human Aβ. We found that fluoxetine significantly delayed Aβ-induced paralysis in the C. elegans model of Aβ toxicity by reducing Aβ oligomers. In addition, DAF-16/FOXO transcription factor was required for fluoxetine protection against Aβ toxicity in C. elegans. These findings suggested that some antidepressants used in AD patients might have additional therapeutic benefits beyond their efficacy to ameliorate depressive symptoms.
2. MATERIALS AND METHODS
2.1 Strain
The wild type C. elegans strain N2, the transgenic strain CL2006 , TJ356 and daf-16 were obtained from the Caenorhabditis Genetic Center (University of Minnesota).
Maintenance of all strains was routinely performed at 20°C on Nematode Growth Medium (NGM) plates with Escherichia coli strain OP50 as a food source as previously described (Brenner, 1974).
2.2 Drug treatment
Fluoxetine (fluoxetine hydrochloride; Sigma-Aldrich, USA), citalopram (citalopram hydrobromide; Sigma-Aldrich, USA) and paroxetine (paroxetine hydrochloride hemihydrates; Sigma-Aldrich, USA) were added to the OP50 bacteria to a desired final concentration. The treatment was given to the transgenic worm from egg stage onward.
2.3 Paralysis assay
Transgenic C. elegans strain CL2006 maintained at 20°C was egg-synchronized onto the 35×10 mm culture plates containing either a vehicle or drug. The worms were tested everyday for paralysis. To identify the paralysis, each worm was gently touched with a platinum loop. The worm was considered paralyzed if it did not move or moved head only after touching.
2.4 Thermotolerance assay
Wild-type (N2) and daf-16 C. elegans at 3 days of age was incubated at 35 °C. At 7 hr plates were removed and worms were scored as alive or dead. Worms failing to respond to the gentle touch with platinum loop were scored as dead.
2.5 Life span assay
Synchronized eggs of N2 C. elegans were maintained at 20 °C on the 35×10 mm culture plates (~35 eggs/plate) containing vehicle or fluoxetine. Once the worms reached to young adult stage, they were shifted everyday to fresh plates until they stop laying egg. Then they were shifted to fresh plates every 2 days. The life span was counted from adult day 1 onward. The worm was considered death if it did not move at all after gently touch with the platinum loop.
2.6 Subcellular DAF-16 localization
Subsequent to the treatment, living worms were placed on microscope slides, capped with cover slips and the subcellular DAF-16 distribution was analyzed by fluorescence microscopy. TJ356 worms were classified into three categories (cytosolic, intermediate and nuclear) with respect to the major localization of the DAF-16::GFP fusion protein.
2.7 RNA Interference (RNAi)
RNAi was performed in C. elegans by feeding the worms with dsRNA-containing bacteria. C. elegans was fed with E.coli HT115 strains expressing dsRNA specific to daf-16 or hsf-1 or tph-1gene. After 3-4 h, worms were removed and eggs were permitted to mature to L4 young larvae. The L4 larvae were transferred to another plate containing dsRNA and allowed to lay eggs. The resultant adult worms were used for the paralysis assay.
2.8 Dot blot of Aβ species
After the experimental treatments, the worms were collected by washing with M9 and distrilled water, quickly frozen in liquid nitrogen, homogenized in the cell lysis buffer (25 mM Tris 7.5, 5 mM NaCl, 1 mM DTT, 5 mM EDTA) with protease inhibitor cocktail (sigma, Saint Louis, MO). Equal protein samples (2 μg) were applied to nitrocellulose membrane (Bio-rad, Hercules, CA). The membranes were probed with a primary anti-Aβ oligomer antibody NU-4 (1:1,000) (Lambert et al., 2007) or anti-Aβ42 antibody 6E10 (1:1,000) followed by corresponding secondary antibodies.
2.9 Western blotting of Aβ species
The Aβ species in the transgenic C. elegans strains was identified by immunoblotting using a Tris-Tricine gel and the standard Western blotting protocol. After the experimental treatments, the worms were collected by washing with distilled water, quickly frozen in liquid nitrogen, sonicated in cell lysis buffer (50 mM HEPES, pH 7.5, 6 mM MgCl2, 1 mM EDTA, 75 mM sucrose, 25 mM benzamide, 1 mM DTT, 1% Triton X-100) with protease inhibitor cocktail (sigma, Saint Louis, MO). The samples were then heated with loading buffer containing 5% β-mercaptoethanol (2:1; Bio-Rad, Hercules, CA). After mixing with the loading buffer, proteins were unheated and loaded on the gel. Equal amounts of the total protein (50 μg) were loaded in each lane. The membranes were probed with a primary anti-Aβ oligomer antibody NU-4 (1:1,000) (Lambert et al., 2007) or anti-Aβ42 antibody 6E10 (1:1,000) followed by corresponding secondary antibodies.
2.10 Cell culture
2.10.1 N2a cells (Aβ expressing mutated cell line)
This cell line expresses a Swedish mutation in APP 695 and another mutation in PS1 where exon-9 is deleted. These double mutations are similar to the one seen in early stage familial Alzheimer’s disease. N2a cells produce high levels of amyloid peptide. This cell line is a gift from Dr H Xu at University of California in San Diego, CA, USA. This cell line is used to test the effect of fluoxetine on the levels of amyloid species intracellulary and extracellulary (in the medium).
2.10.1 Western blotting
N2a cells were maintained in non stimulating medium (47% Dulbecco’s modified eagle medium, 5% fetal bovine serum, 47% opti-MEM, 0.2 mg/ml G418 and 1% antibiotics) at 37°C and 5% CO2/95% O2. When the cells reach 80% confluency, they were shifted to stimulating medium (50% Dulbecco’s modified eagle medium, 0.5% fetal bovine serum, 50% opti-MEM, 0.2 mg/ml G418, 1% antibiotics and 10 mM sodium butyric acid). N2a cells were treated with increasing concentration of fluoxetine for 24 hr. After 24 hr, the cells were washed twice with cold phosphate buffer saline (PBS) and then collected from the plates. The cells were lysed by sonication in lysis buffer (75 mM Tris-HCl, 1 mM DTT, 50 mM NaCl, 1 mM EGTA and 5 mM EDTA) with added protease inhibitor cocktail. The undisrupted cells were removed by centrifugation (12,000 g, 15 min, at 4 °C). Equal amount of protein (50 μg) from each sample were loaded in each lane. The membranes were probed with a primary anti-Aβ42 antibody 6E10 (1:1,000) followed by a secondary anti-mouse IgG antibody (1:2,000).
2.11 APP and PS1 double transgenic mouse model of AD
These mice are heterozygous double transgenic mice (APPswe/PS1 Δ9) that have been purchased from Jackson laboratory. The mice are conserved by cross breeding the transgenic mice with the wild type (B6C3) and then the offspring is genotyped. This mouse model is used in western blotting.
2.12 Statistic analysis
Data are represented as mean ± SEM. Significant differences between groups were assessed by unpaired t test. A p value < 0.05 was considered statistically significant. GraphPad Prism 5 software was used for the paralysis analysis. P value calculations were made between treated and untreated animals using the log-rank (Mantel-Cox) test.
3. RESULTS
3.1 Fluoxetine and paroxetine but not citalopram, delays Aβ-induced paralysis in C. elegans strain CL2006
According to the amyloid hypothesis, AD is thought to be caused by the production and deposition of neurotoxic Aβ-peptide in the brain (Blennow et al., 2006). The deposition of Aβ in the brain leads to many consequences such as the formation of neurofibrillary tangles, oxidative stress, glutamatergic excitotoxicity, inflammation, neuronal cell death and eventually the clinical symptoms of AD (Cummings, 2004; van Marum, 2008). In transgenic C. elegans model of AD, human Aβ42 protein has been expressed intracellularly in the body wall muscle and the expression and subsequent aggregation of Aβ in the muscle lead to progressive paralysis (Link, 1995; Link, 2006). To investigate the protective effect of fluoxetine, the worm strain CL2006 which produces Aβ constitutively in the muscle was used. The worms were treated with fluoxetine at concentration of 1, 10, 100 and 1,000 μM (fig 1A). Fluoxetine at 10 and 1,000 μM significantly delayed Aβ-induced paralysis in this transgenic worm (p = 0.02 and 0.0001 respectively). As a comparison, paroxetine and citalopram, other selective serotonin reuptake inhibitors, were also tested. Paroxetine at dose 100 nM significantly delayed Aβ-induced paralysis (p = 0.05) (fig 1C). However, citalopram at dose up to 2,000 μM did not delay Aβ-induced paralysis (fig 1B) (p = 0.29).
Fig. 1.
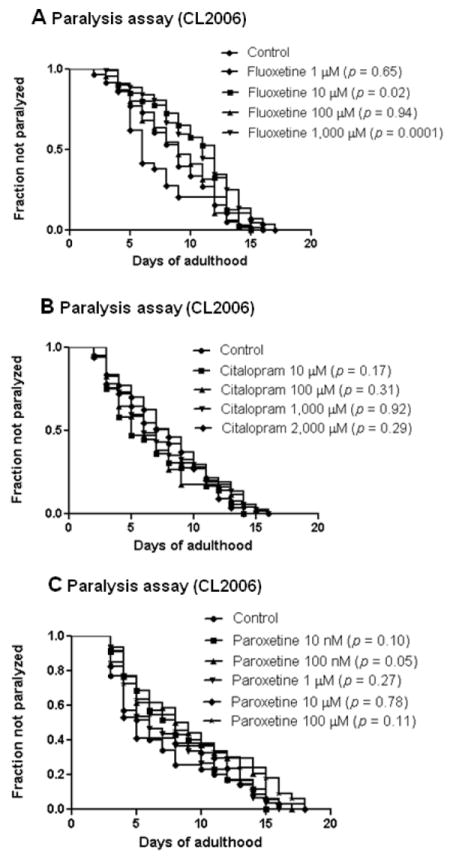
Aβ-induced paralysis in C. elegans strain CL2006 fed with antidepressants. (A) Effect of fluoxetine at concentration of 1, 10, 100, 1000 μM on Aβ-induced paralysis. (B) Effect of citalopram at concentration of 10, 100, 1,000 and 2,000 μM on Aβ-induced paralysis. (C) Effect of paroxetine at concentration of 10 nM, 100 nM, 1 μM, 10 μM and 100 μM. Synchronized eggs of CL2006 C. elegans were maintained at 20 °C, on the 35×10 mm culture plates (~35 eggs/plate) containing vehicle (control) or drugs. The treatment was given to the worm from egg stage onward until the worm completely paralyzed.(35 worms in each experiment, 3 independent experiments).
3.2 Fluoxetine reduces Aβ oligomer in transgenic C. elegans and N2a neuroblastoma cells
Growing evidence suggests that amyloid plaques do not always correlate with neurodegeneration and cognitive decline (Resende et al., 2008). In stead, the concentration of soluble Aβ (oligomers) is well correlated with cognitive deficits (McLean et al., 1999). To determine whether fluoxetine treatment affected the toxic species Aβ oligomer in the worm we measured Aβ oligomer using an oligomer specific antibody NU4. Fluoxetine 1 mM showed a trend of decreased levels of Aβ oligomer in comparison with the untreated control, suggesting that fluoxetine may alleviated Aβ toxicity by reducing Aβ oligomers (fig 2A; p = 0.09, n = 3). However, fluoxetine 1 mM did not decrease total Aβ as shown in figure 2B (p = 1.2, n = 3), supporting a role of fluoxetine in specifically inhibition of toxic species of Aβ. We also analyzed Aβ oligomer in C.elegans fed with 1 mM fluoxetine by Western blotting using antibody against Aβ oligomers (NU-4). Fig 2C shows Aβ-immunoreactive band (MW ~6-53 kDa) detected in the tissues from transgenic worms (CL2006) fed with 1mM fluoxetine or 100nM paroxetine, another SSRI antidepressant drug as a control. The treatment of CL2006 C.elegans strain with two selective serotonin reuptake inhibitors (fluoxetine and paroxetine) results in reduction in Aβ oligomers species with molecular weight ~ 12 and ~53 kDa (Fig 2C).
Fig 2.
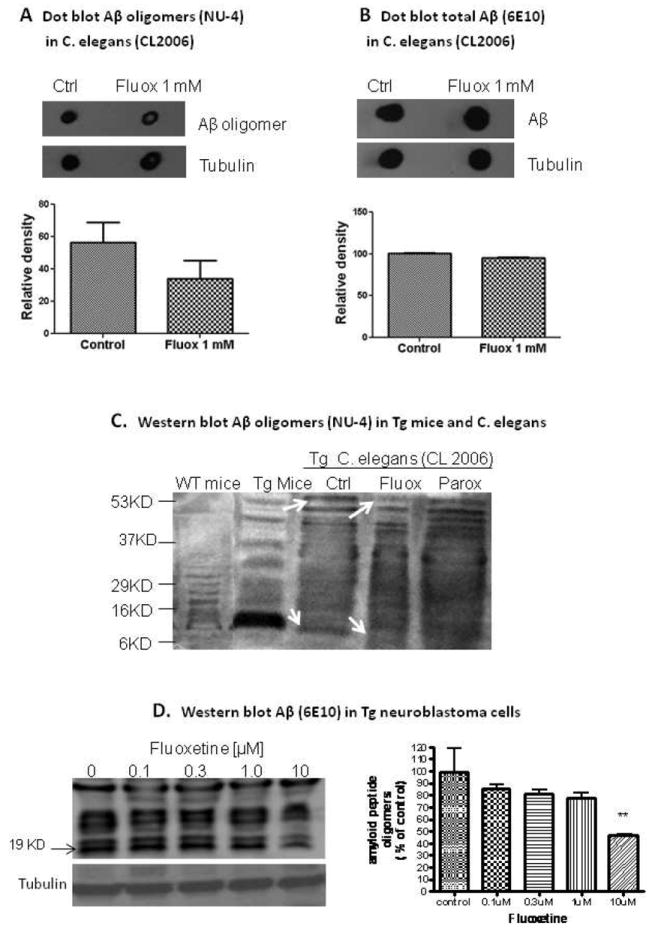
Dot blot assay of Aβ oligomers in transgenic C. elegans treated with or without fluoxetine (Fluox at 1 mM). (A) Relative density of Aβ oligomer immunoreactivity (NU-4) in CL2006 worms untreated (control) or treated with fluoxetine (n = 3). Inset, representative dot blot of Aβ oligomers in untreated worms and treated worm. (B) Relative density of total Aβ immunoreactivity (6E10) in worms untreated (control) or treated with fluoxetine (n = 3). Inset, representative dot blot of total Aβ in untreated worms and treated worm. Equal amount of protein (2μg) were loaded on the membrane. Tubulin was used to normalize protein loading. Error bars indicate SEM. C: The treatment of C. elegans with 1mM fluoxetine and 100nM paroxetine result in reduction in Aβ oligomers in comparison to the control. D: Effect of fluoxetine on amyloid peptide species is determined by western blotting. Fluoxetine significantly decreased the levels of intracellular Aβ oligomers (ANOVA, P=0.0230). posthoc Dunnett test shows that 10μM fluoxetine treatment causes significant reduction in Aβ oligomers in comparison to the control (P < 0.01). Data is expressed as mean± SEM (N=3). ** represents P < 0.01.
The brain extract from wild and double transgenic APP/PS1 mice was used as a positive control for the endogenous Aβ oligomers. C.elegans was treated with 1mM fluoxetine or 100nM paroxetine from the egg stage of the worm to day one of the adulthood (3-4 days). The doses of drugs used in this experiment are determined based on the effective concentration of the drugs in paralysis assay (Fig 1). In this experiment, Nu4 antibody which is specific for Aβ oligomers was used.
The effect of Fluoxetine on Aβ oligomers was further confirmed by using N2a cells that express Aβ transgene. The effect of increasing concentrations of fluoxetine (0.1-10μM) on intracelullar Aβ oligomers in N2a cells was tested using Western blotting with 6E10 antibody to determine the effect on different Aβ species (Fig 2D). The results showed that 10μM fluoxetine treatment resulted in significant reduction of Aβ oligomers in N2a cells. One-way ANOVA showed that there was a significant difference for the fluoxetine treatment (p=0.0230). posthoc Dunnett test shows that 10μM fluoxetine treatment caused a significant reduction in Aβ oligomers in comparison to the control (p < 0.01).
3.3 DAF-16 and TPH-1 are required for the protective effect of fluoxetine on delaying paralysis
It has been revealed recently that DAF-16, HSF-1 and insulin signaling pathway play a role in the protection against Aβ toxicity (Cohen et al., 2006; Cohen et al., 2008). It is also well known that C. elegans FOXO transcription factor DAF-16 is a key mediator for regulating longevity and stress resistance (Mukhopadhyay et al., 2006; Murphy, 2006; Schaffitzel et al., 2006). To test whether DAF-16 and HSF-1 are required for the protective effect of fluoxetine against Aβ toxicity, we performed the experiment by using RNAi knockdown of DAF-16 or HSF-1 expression. We found that fluoxetine at 1 mM significantly delayed Aβ-induced paralysis in worms grown on bacteria containing empty vector (fig 3A, p = 0.01) but not on daf-16 RNAi bacteria (fig 3B, p = 0.1). This result indicated that reducing the activity of DAF-16 abolished the protective effect of fluoxetine, suggesting requirement of DAF-16 for protective effect of fluoxetine. On the other hand, fluoxetine at1 mM still significantly delayed Aβ-induced paralysis in worms fed with hsf-1 RNAi bacteria (fig 3C, p = 0.001), indicating that HSF-1 is not required for the protective effect of fluoxetine. In addition, we knocked down tph-1 gene which is responsible for transcription of tryptophan hydroxylase. This enzyme is a rate-limiting step enzyme for the synthesis of serotonin. Because fluoxetine blocks the reuptake of serotonin into the cell and increases serotonin in synaptic cleft, so we test if serotonin is required for the protective effect of fluoxetine. We found that fluxetine did not delay paralysis in worms fed with tph-1 RNAi bacteria (fig 3D, p = 0.17) when compared to control, indicating that serotonin is required for the protective effect of fluoxetine.
Fig. 3.
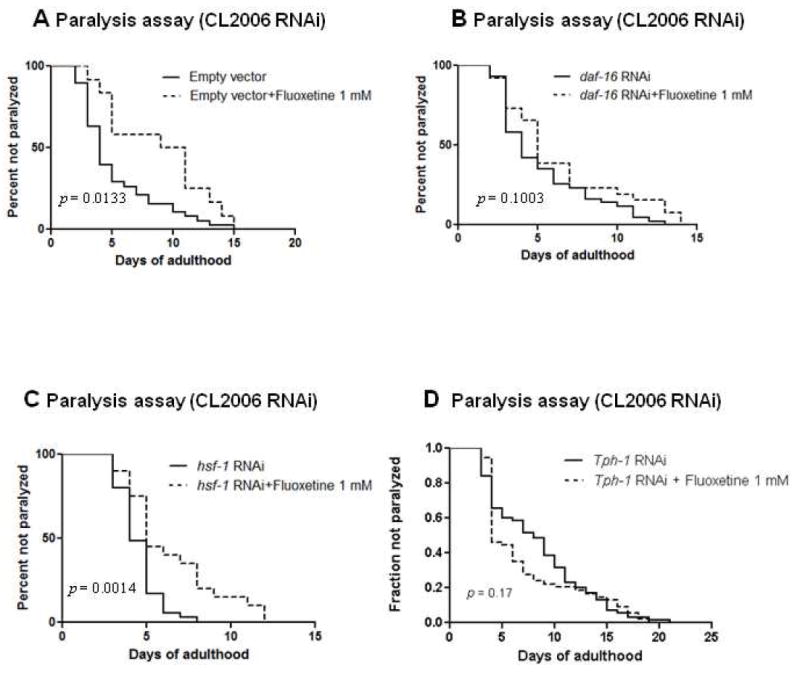
Paralysis in CL2006 with or without DAF-16 ,HSF-1 and TPH-1 knock down by RNAi. (A) Paralysis in CL2006 treated with or without fluoxetine in C. elegans fed with vector control RNAi. Solid line, worms grown on bacteria containing empty vector alone; dotted line, worms grown on bacteria containing empty vector and treated with fluoxetine. (B) Paralysis in CL2006 treated with or without fluoxetine in C. elegans fed with RNAi to down regulate DAF-16. Solid line, worms grown on daf-16 RNAi bacteria alone; dotted line, worms grown on daf-16 RNAi bacteria and treated with fluoxetine. (C) Paralysis in CL2006 treated with or without fluoxetine in C. elegans fed with RNAi to down regulate HSF-1. Solid line, worms grown on hsf-1 RNAi bacteria alone; dotted line, worms grown on hsf-1 RNAi bacteria and treated with fluoxetine. (D) Paralysis in CL2006 treated with or without fluoxetine in C. elegans fed with RNAi to down regulate TPH-1. Solid line, worms grown on tph-1 RNAi bacteria alone; dotted line, worms grown on tph-1 RNAi bacteria and treated with fluoxetine. (70 worms in each experiment, 2 independent experiments)
3.4 Fluoxetine, but not citalopram, induces the translocation of DAF-16 from cytosol to nucleus
The localization of DAF-16 in the nucleus is an essential prerequisite for its ability to initiate the transcription of target genes. To further confirm the involvement of DAF-16 in fluoxetine-reduced Aβ toxicity, we examined the effect of fluoxetine on subcellular distribution of DAF-16 using the transgenic C. elegans strain TJ356. This strain of worm carries a fusion construct of DAF-16 coupled to GFP as a reporter gene allowing the determination of the subcellular localization of this transcription factor. The worms can be scored in three stages; 1) for cytosolic localization, fluorescences are evenly distributed throughout C. elegans body (Fig. 4A), 2) for nuclear localization, the distribution of the fluorescence is enclosed in the nucleus as arrow shown in fig 4B, 3) for intermediate localization, the fluorescences are distributed in both nuclear and cytosole. Eighty percent of the control group showed a cytosolic localization of DAF-16 and only small fraction showed an intermediate localization (fig 4B). Fluoxetine 1 mM induced the translocation of DAF-16 from cytosol to the nucleus by increasing the percentage of worms with intermediate DAF-16 localization by 92.38%. This result indicated that fluoxetine has partial effect on the translocation of DAF-16 from cytosol to nucleus. On the other hand, citalopram 2 mM did not alter the subcellular distribution of DAF-16 (fig 4C). The observation that fluoxetine caused a translocation of DAF-16 suggested that fluoxetine might affect insulin signaling by regulating the activity of DAF-16.
Fig. 4.
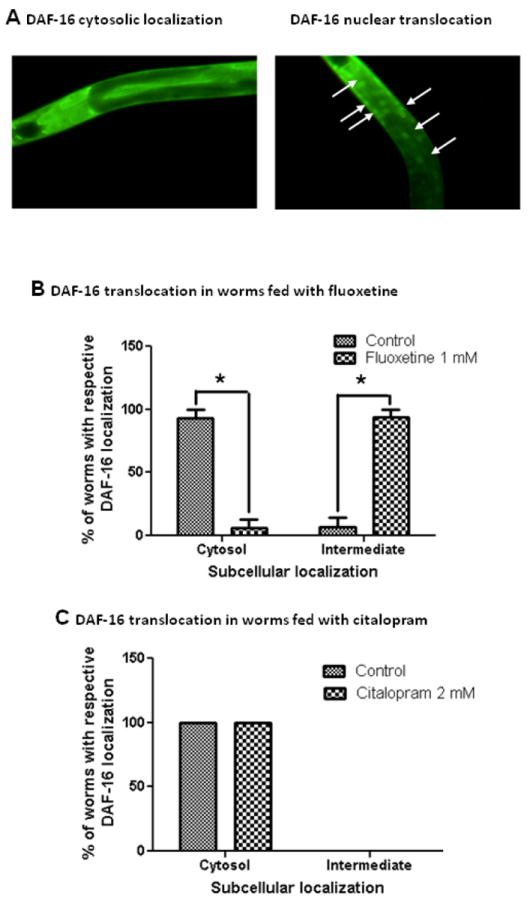
The effect of fluoxetine and citalopram on subcellular DAF-16 localization. (A) Representative fluorescence images of transgenic strain TJ356 showing cytosolic (left) and nucleus (right, arrows) localization of DAF-16. Subcellular distribution of DAF-16 in worms treated with fluoxetine (B) or citalopram (C) was analyzed by fluorescence microscopy. The worms were classified into 2 categories: cytosol, translocate according to their localization phenotype. (*p < 0.05 vs control, n = 24)
3.5 Fluoxetine increases thermal stress resistance and extends life span in wild type C. elegans
Insulin signaling pathway regulates aging, stress resistance and neurodegeneration (Cohen et al., 2008). To determine if fluoxetine specifically delay Aβ-induced paralysis, but not physiological wellness, we further evaluated whether fluoxetine could protect against heat stress and prolong survival. We tested the effect of fluoxetine on thermal stress resistance by exposing the worm to a higher temperature (35°C). Worm normally live at 20-25°C, when exposed to 35°C, they will die gradually within 7-8 hr. We found that worms fed with fluoxetine (1 mM) showed 38% increase in thermal stress resistance and the effect was lost in daf-16 mutation worms (Fig 5A). These results suggest that thermal protection by fluoxetine required DAF-16. Survival assays was also conducted in worms fed with or without increasing concentrations of fluoxetine. In worms fed with 100 μM fluoxetine, there was a significant prolongation of mean life span of the C. elegans (p=0.04, n=70).
Fig. 5.
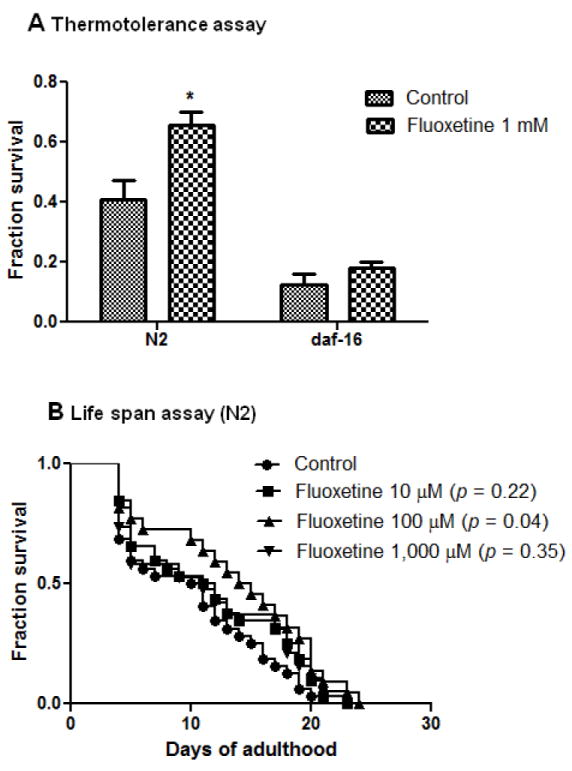
Effect of fluoxetine on thermal stress resistance and life span in C. elegans. (A) Thermotolerance in wild type (N2) and daf-16 C.elegans treated with or without Fluoxetine 1 mM (*p = 0.01vs control) and (B) Life span assay in wild type (N2) C. elegans treated with vehicle (control) or with increasing dose of fluoxetine (10, 100 and 1,000 μM). Synchronized eggs of N2 and daf-16 C. elegans were maintained at 20 °C , on the 35×10 mm culture plates (~35 eggs/plate) containing vehicle or drug. The treatment was given to the worm from egg stage onward.
4. DISCUSSION
In the present study, we found that fluoxetine delayed Aβ-induced paralysis in transgenic C. elegans model of Aβ toxicity. We also found that paroxetine, another SSRIs, which has been reported to improve cognitive performance in 3xTgAD mouse model of AD and reduce the concentrations of Aβ in the hippocampus (Nelson et al., 2007), delayed Aβ-induced paralysis. However, this benefit does not apply to all SSRIs because citalopram did not delay Aβ-induced paralysis. Furthermore, Aβ oligomers were reduced in the transgenic worm treated with fluoxetine. It is well recognized that Aβ oligomers are toxic to neurons and are critical to the onset and progression of AD and that Aβ oligomers are particularly damaging to the synapse (Lambert et al., 1998; Lacor et al., 2004; Lacor et al., 2007). Therefore, our result suggested that fluoxetine ameliorated Aβ toxicity in transgenic C. elegans by reducing the toxic form of Aβ. Fluoxetine only affected on Aβ oligomers but not on total concentration of Aβ. The potential reason is that Aβ species are at a dynamic balance between monomer, oligomers and fibrils. Our previous results shown that when Aβ oligomers were reduced by EGb 761 , the levels of Aβ monomers were enhanced (Wu et al., 2006). This could be one of the explanations why the total concentrations were unchanged.
We further investigated the possible mechanisms underlying the reduction of Aβ oligomers in C. elegans by fluoxetine. There is evidence from human and different model organisms indicating a role of insulin receptor/IGF-1 receptor signaling in Aβ metabolism and clearance (Townsend et al., 2007; Zhao et al., 2008; Zhao et al., 2009). It has been recently shown that knocking down DAF-2 in C. elegans, the homolog of the mammalian insulin receptor/IGF-1 receptor, reduces Aβ1-42 toxicity (Cohen et al., 2008). The authors hypothesized that DAF-16 regulates the process that convert the toxic form (oligomers) to less-toxic form (high-molecular-mass aggregates). Our results indicated that DAF-16 is required for the protective effect of fluoxetine, which is further supported by our observation that fluoxetine induced, at least partially, the translocation of DAF-16 from cytosol to the nucleus. In contrast, citalopram did not affect translocation of DAF-16 which might explain why citalopram could not delay Aβ-induced paralysis. However, how DAF-16 mediates the protection by fluoxetine against Aβ toxicity remains to be elucidated.
Translation of results from extending life span in C.elegans to mammals is not straight forward. However, mammalian homologues of C.elegans daf-16, FOXO1 and FOXO3 genes variants were segregated with survival to age 85 and older, and in female, gene variants that reduce insulin/IGF-1 signaling are associated with longevity. Recently, heterozygous mutations in the IGF-IR were found overrepresented in centenarians (Vijg and Campisi, 2008), further validity that indeed C.elegans would be a model of importance for modulating this signaling and potentially predictive for other drugs.
Fluoxetine is a potent antidepressant that increases serotonin at the synapse via inhibition of the serotonin reuptake transporter. However, fluoxetine can act on additional targets other than the serotonin transporter (Kaletta et al., 2006). Because citalopram did not show the effect on delaying paralysis, this support the idea that fluoxetine might act on alternative target which is the insulin signaling pathway. In addition, although the SSRIs share a common primary pharmacology by inhibition of the serotonin reuptake transporter, their secondary pharmacology is remarkably heterogeneous (Carrasco et al., 2005), which may account for the different outcomes between fluoxetine and citalopram found in this study. For example, fluoxetine is a more potent inhibitor of 5-HT2C receptor while citalopram has the highest affinity for histamine H1 receptors compared to other SSRIs (Palvimaki et al., 1999; Owens et al., 2001). This might be an explanation why fluoxetine showed the effect on delaying paralysis whereas citalopram did not.
Fluoxetine might act directly or indirectly through the insulin signaling pathway, since Murakami et al. (2007) revealed an interaction between the serotonin pathway and the insulin/IGF-1 pathway (Murakami et al., 2007). Their study showed that a mutation of certain serotonin receptor genes increased longevity and was likely mediated through the insulin/IGF-1 pathway. Our results are in agreement with this finding because we found that serotonin was also required for the protective effect of fluoxetine. Nevertheless, additional investigation is needed to explore the mechanisms involved in the relationship between serotonin pathway and insulin/IGF-1 signaling pathway.
There are controversies about the interaction of insulin signaling and pathophysiology of Alzheimer’s disease. It has been shown that insulin signaling has a protective role in prevention or slowing pathogenesis of Alzheimer’s disease (Cole et al., 2007; De Felice et al., 2009) while insulin resistance increases the risk of Alzheimer’s disease (Craft, 2007). In addition, insulin and insulin sensitizers improve cognitive and memory functions in animal models of Alzheimer’s disease (Craft et al., 2003; Watson et al., 2005). In contrast, metformin, an insulin-sensitizer, significantly increases the generation of both intracellular and extracellular Aβ species (Chen et al., 2009). Impaired insulin signaling reduces Aβ accumulation and protects against premature death in a model of Alzheimer’s disease (Freude et al., 2009). Our results are in agreement with this finding since fluoxetine reduced Aβ oligomer in part by working through DAF-16. The increase of the activity of DAF-16 was revealed by the impairment of insulin signaling by knocking down the insulin receptor (Cohen et al., 2006).
Other antidepressants have also been reported for their efficacy in reduction of Aβ toxicity. Most recently, imipramine, a tricyclic antidepressant, has been reported to prevent cognitive decline and Aβ accumulation in a mouse model of Alzheimer’s disease (Chavant et al., 2009).Taken together, these findings and our results provide insight that some SSRIs and tricyclic antidepressant might have disease-modifying properties in addition to providing depressive symptoms relief.
In conclusion, this study is the first to demonstrate that fluoxetine can delay Aβ-induced paralysis in a model organism. The mechanism underlying its protective efficacy was shown in part through DAF-16 and insulin signaling pathway. However, an additional investigation is needed to better understand the mechanisms underlying the protective effect of fluoxetine and the possibility of the interaction between serotonin and insulin signaling pathway.
Acknowledgments
This study was supported by NIH grant R01 AT001928-03A1 (YL) from the National Center for Complementary and Alternative Medicine. We thank Dr. Edward Moreton of University of Maryland for reading, editing and commenting on the manuscript and Drs. Pascale N. Lacor and William Klein of Northwestern University for providing Aβ oligomers specific antibody NU4.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboukhatwa M, Dosanjh L, Luo Y. Antidepressants are a rational complementary therapy for the treatment of Alzheimer’s disease. Mol Neurodegener. 2010;5:10. doi: 10.1186/1750-1326-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman HJ, Stone WS, Ogren SO. Evidence for a possible functional interaction between serotonergic and cholinergic mechanisms in memory retrieval. Behav Neural Biol. 1987;48(1):49–62. doi: 10.1016/s0163-1047(87)90574-7. [DOI] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco JL, Sandner C. Clinical effects of pharmacological variations in selective serotonin reuptake inhibitors: an overview. Int J Clin Pract. 2005;59(12):1428–34. doi: 10.1111/j.1368-5031.2005.00681.x. [DOI] [PubMed] [Google Scholar]
- Chavant F, Deguil J, Pain S, Ingrand I, Milin S, Fauconneau B, Perault MC, Lafay-Chebassier C. Imipramine, in part through TNF-{alpha} inhibition, prevents cognitive decline and A{beta} accumulation in a mouse model of Alzheimer’s disease. J Pharmacol Exp Ther. 2009 doi: 10.1124/jpet.109.162164. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao FF. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci U S A. 2009;106(10):3907–12. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313(5793):1604–10. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci. 2008;9(10):759–67. doi: 10.1038/nrn2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer’s Disease. Exp Gerontol. 2007;42(1-2):10–21. doi: 10.1016/j.exger.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4(2):147–52. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- Craft S, Asthana S, Cook DG, Baker LD, Cherrier M, Purganan K, Wait C, Petrova A, Latendresse S, Watson GS, Newcomer JW, Schellenberg GD, Krohn AJ. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28(6):809–22. doi: 10.1016/s0306-4530(02)00087-2. [DOI] [PubMed] [Google Scholar]
- Cummings JL. Alzheimer’s disease. N Engl J Med. 2004;351(1):56–67. doi: 10.1056/NEJMra040223. [DOI] [PubMed] [Google Scholar]
- Danion JM. Antidepressive agents and memory. Encephale. 1993;19(Spec No 2):417–22. [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62(4):479–93. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009;106(6):1971–6. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey CM, Mackenzie SM, Gargus A, Blanco G, Sze JY. Serotonin (5HT), fluoxetine, imipramine and dopamine target distinct 5HT receptor signaling to modulate Caenorhabditis elegans egg-laying behavior. Genetics. 2005;169(3):1425–36. doi: 10.1534/genetics.104.032540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS. Novel therapeutic approaches beyond the serotonin receptor. Biol Psychiatry. 1998;44(5):324–35. doi: 10.1016/s0006-3223(98)00031-6. [DOI] [PubMed] [Google Scholar]
- Faber PW, Alter JR, MacDonald ME, Hart AC. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci U S A. 1999;96(1):179–84. doi: 10.1073/pnas.96.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel SI, Mintzer JE, Dysken M, Krishnan KR, Burt T, McRae T. A randomized, placebo-controlled study of the efficacy and safety of sertraline in the treatment of the behavioral manifestations of Alzheimer’s disease in outpatients treated with donepezil. Int J Geriatr Psychiatry. 2004;19(1):9–18. doi: 10.1002/gps.998. [DOI] [PubMed] [Google Scholar]
- Freude S, Hettich MM, Schumann C, Stohr O, Koch L, Kohler C, Udelhoven M, Leeser U, Muller M, Kubota N, Kadowaki T, Krone W, Schroder H, Bruning JC, Schubert M. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. Faseb J. 2009;23(10):3315–24. doi: 10.1096/fj.09-132043. [DOI] [PubMed] [Google Scholar]
- Geerlings MI, Schoevers RA, Beekman AT, Jonker C, Deeg DJ, Schmand B, Ader HJ, Bouter LM, Van Tilburg W. Depression and risk of cognitive decline and Alzheimer’s disease. Results of two prospective community-based studies in The Netherlands. Br J Psychiatry. 2000;176:568–75. doi: 10.1192/bjp.176.6.568. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, Praag H, Gage FH. Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol Psychiatry. 2000;5(3):262–9. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- Kaletta T, Hengartner MO. Finding function in novel targets: C. elegans as a model organism. Nat Rev Drug Discov. 2006;5(5):387–98. doi: 10.1038/nrd2031. [DOI] [PubMed] [Google Scholar]
- Kuningas M, Magi R, Westendorp RG, Slagboom PE, Remm M, van Heemst D. Haplotypes in the human Foxo1a and Foxo3a genes; impact on disease and mortality at old age. Eur J Hum Genet. 2007;15(3):294–301. doi: 10.1038/sj.ejhg.5201766. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24(45):10191–200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27(4):796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MK, Tsang SW, Francis PT, Keene J, Hope T, Esiri MM, Spence I, Chen CP. Postmortem serotoninergic correlates of cognitive decline in Alzheimer’s disease. Neuroreport. 2002;13(9):1175–8. doi: 10.1097/00001756-200207020-00021. [DOI] [PubMed] [Google Scholar]
- Lakso M, Vartiainen S, Moilanen AM, Sirvio J, Thomas JH, Nass R, Blakely RD, Wong G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem. 2003;86(1):165–72. doi: 10.1046/j.1471-4159.2003.01809.x. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HB, Lyketsos CG. Depression in Alzheimer’s disease: heterogeneity and related issues. Biol Psychiatry. 2003;54(3):353–62. doi: 10.1016/s0006-3223(03)00543-2. [DOI] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92(20):9368–72. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD. Transgenic invertebrate models of age-associated neurodegenerative diseases. Mech Ageing Dev. 2001;122(14):1639–49. doi: 10.1016/s0047-6374(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Link CD. C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer’s disease. Exp Gerontol. 2006;41(10):1007–13. doi: 10.1016/j.exger.2006.06.059. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20(24):9104–10. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46(6):860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Mowla A, Mosavinasab M, Haghshenas H, Haghighi AB. Does serotonin augmentation have any effect on cognition and activities of daily living in Alzheimer’s dementia? A double-blind, placebo-controlled clinical trial. J Clin Psychopharmacol. 2007;27(5):484–7. doi: 10.1097/jcp.0b013e31814b98c1. [DOI] [PubMed] [Google Scholar]
- Mowla A, Mosavinasab M, Pani A. Does fluoxetine have any effect on the cognition of patients with mild cognitive impairment? A double-blind, placebo-controlled, clinical trial. J Clin Psychopharmacol. 2007;27(1):67–70. doi: 10.1097/JCP.0b013e31802e0002. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A, Oh SW, Tissenbaum HA. Worming pathways to and from DAF-16/FOXO. Exp Gerontol. 2006;41(10):928–34. doi: 10.1016/j.exger.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Murakami H, Murakami S. Serotonin receptors antagonistically modulate Caenorhabditis elegans longevity. Aging Cell. 2007;6(4):483–8. doi: 10.1111/j.1474-9726.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- Murphy CT. The search for DAF-16/FOXO transcriptional targets: approaches and discoveries. Exp Gerontol. 2006;41(10):910–21. doi: 10.1016/j.exger.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99(5):3264–9. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson RL, Guo Z, Halagappa VM, Pearson M, Gray AJ, Matsuoka Y, Brown M, Martin B, Iyun T, Maudsley S, Clark RF, Mattson MP. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol. 2007;205(1):166–76. doi: 10.1016/j.expneurol.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyth AL, Gottfries CG. The clinical efficacy of citalopram in treatment of emotional disturbances in dementia disorders. A Nordic multicentre study. Br J Psychiatry. 1990;157:894–901. doi: 10.1192/bjp.157.6.894. [DOI] [PubMed] [Google Scholar]
- Nyth AL, Gottfries CG, Lyby K, Smedegaard-Andersen L, Gylding-Sabroe J, Kristensen M, Refsum HE, Ofsti E, Eriksson S, Syversen S. A controlled multicenter clinical study of citalopram and placebo in elderly depressed patients with and without concomitant dementia. Acta Psychiatr Scand. 1992;86(2):138–45. doi: 10.1111/j.1600-0447.1992.tb03242.x. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Knight DL, Nemeroff CB. Second-generation SSRIs: human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol Psychiatry. 2001;50(5):345–50. doi: 10.1016/s0006-3223(01)01145-3. [DOI] [PubMed] [Google Scholar]
- Palvimaki EP, Kuoppamaki M, Syvalahti E, Hietala J. Differential effects of fluoxetine and citalopram treatments on serotonin 5-HT(2C) receptor occupancy in rat brain. Int J Neuropsychopharmacol. 1999;2(2):95–99. doi: 10.1017/S1461145799001406. [DOI] [PubMed] [Google Scholar]
- Pollock BG, Rosen J, Mulsant BH. Antipsychotics and selective serotonin reuptake inhibitors for the treatment of behavioral disturbances in dementia of the Alzheimer type. Consultant Pharmacist. 1999;11:1251–58. [Google Scholar]
- Raina P, Santaguida P, Ismaila A, Patterson C, Cowan D, Levine M, Booker L, Oremus M. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008;148(5):379–97. doi: 10.7326/0003-4819-148-5-200803040-00009. [DOI] [PubMed] [Google Scholar]
- Resende R, Ferreiro E, Pereira C, Resende de Oliveira C. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience. 2008;155(3):725–37. doi: 10.1016/j.neuroscience.2008.06.036. [DOI] [PubMed] [Google Scholar]
- Rozzini L, Chilovi BV, Conti M, Bertoletti E, Zanetti M, Trabucchi M, Padovani A. Efficacy of SSRIs on cognition of Alzheimer’s disease patients treated with cholinesterase inhibitors. Int Psychogeriatr. 2010;22(1):114–9. doi: 10.1017/S1041610209990184. [DOI] [PubMed] [Google Scholar]
- Schaffitzel E, Hertweck M. Recent aging research in Caenorhabditis elegans. Exp Gerontol. 2006;41(6):557–63. doi: 10.1016/j.exger.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Schmitt JA, Wingen M, Ramaekers JG, Evers EA, Riedel WJ. Serotonin and human cognitive performance. Curr Pharm Des. 2006;12(20):2473–86. doi: 10.2174/138161206777698909. [DOI] [PubMed] [Google Scholar]
- Shah S, Reichman WE. Treatment of Alzheimer’s disease across the spectrum of severity. Clin Interv Aging. 2006;1(2):131–42. doi: 10.2147/ciia.2006.1.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S, Herrmann N, Rapoport MJ, Lanctot KL. Efficacy and safety of antidepressants for treatment of depression in Alzheimer’s disease: a metaanalysis. Can J Psychiatry. 2007;52(4):248–55. doi: 10.1177/070674370705200407. [DOI] [PubMed] [Google Scholar]
- Townsend M, Mehta T, Selkoe DJ. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282(46):33305–12. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- van Marum RJ. Current and future therapy in Alzheimer’s disease. Fundam Clin Pharmacol. 2008;22(3):265–74. doi: 10.1111/j.1472-8206.2008.00578.x. [DOI] [PubMed] [Google Scholar]
- Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008;454(7208):1065–71. doi: 10.1038/nature07216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser PJ, Verhey FR, Ponds RW, Kester A, Jolles J. Distinction between preclinical Alzheimer’s disease and depression. J Am Geriatr Soc. 2000;48(5):479–84. doi: 10.1111/j.1532-5415.2000.tb04992.x. [DOI] [PubMed] [Google Scholar]
- Vythilingam M, Vermetten E, Anderson GM, Luckenbaugh D, Anderson ER, Snow J, Staib LH, Charney DS, Bremner JD. Hippocampal volume, memory, and cortisol status in major depressive disorder: effects of treatment. Biol Psychiatry. 2004;56(2):101–12. doi: 10.1016/j.biopsych.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13(11):950–8. doi: 10.1176/appi.ajgp.13.11.950. [DOI] [PubMed] [Google Scholar]
- Wilde MI, Benfield P. Fluoxetine. A pharmacoeconomic review of its use in depression. Pharmacoeconomics. 1998;13(5 Pt 1):543–61. doi: 10.2165/00019053-199813050-00007. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wu Z, Butko P, Christen Y, Lambert MP, Klein WL, Link CD, Luo Y. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci. 2006;26(50):13102–13. doi: 10.1523/JNEUROSCI.3448-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Amyloid beta oligomers induce impairment of neuronal insulin receptors. Faseb J. 2008;22(1):246–60. doi: 10.1096/fj.06-7703com. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Lacor PN, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric a{beta} J Biol Chem. 2009;284(28):18742–53. doi: 10.1074/jbc.M109.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]