

Abstract
Biological processes are regulated with a high level of spatial and temporal resolution. In order to understand and manipulate these processes, scientists need to be able to regulate them with Nature’s level of precision. In this context, light is a unique regulatory element because it can be precisely controlled in location, timing and amplitude. Moreover, most biological laboratories have a wide range of light sources as standard equipment. This review article summarizes the most recent advances in light-mediated regulation of protein function and the application in a cellular context. Specifically, the photocaging of small molecule modulators of protein function and of select amino acid residues in proteins will be discussed. In addition, examples of the photochemical control of protein function through the application of natural light-receptors are presented.
Introduction
Proteins are indispensable biological macromolecules within cells. They have a wide range of biological function, from structural support and regulatory mechanisms to the catalysis of synthetic transformations. In nature, protein function is precisely regulated by a variety of complex mechanisms. Myriad effector molecules interact to control the activity of proteins in a precise spatial and temporal fashion. To better understand the function, mechanism of action, and regulation of a specific protein, it is desirable to be able to externally modulate the protein’s activity and observe the effects produced by its intentional activation and deactivation. In this context, light represents a unique external control element since its intensity can be easily controlled with high spatiotemporal resolution1–7. The examples discussed in this review illustrate some of the most successful and most recent strategies implemented to date for controlling protein activity with light.
Light-removable protecting groups, typically composed of aromatic rings, so-called “caging groups”, installed on amino acid residues or small organic effector molecules allow for control over the function of a specific protein in a precise spatiotemporal manner using light irradiation. Caging groups are covalently bound to small organic effector molecules or specific amino acid residues in such a way that their natural activities are inhibited. When irradiated (most commonly with UV light), these molecules undergo photolytic cleavage to expose the previously caged small molecule effector or amino acid in its natural form (commonly referred to as “decaging”), thereby either inactivating (Figure 1a) or activating (Figure 1b) protein activity. The most commonly employed caging groups for these applications are based on ortho-nitrobenzyl moieties8, but other chromophores with enhanced photochemical properties have been developed as well9–12. Furthermore, researchers have developed systems where naturally occurring photoreactive proteins, such as LOV (light-, oxygen- and voltage-sensitive) domains and plant phytochromes, are conjugated to the protein-of-interest, enabling modulation of protein activity by light irradiation, often in a reversible fashion (Figure 1c and 1d). Advantages of such systems are that they do not require any chemical modifications and can often be genetically encoded. A disadvantage is that the protein activity before and after light-switching is difficult to predict, thus often requiring laborious optimization of the system under study through trial and error. Examples of the four different approaches illustrated in Figure 1 are discussed below.
Figure 1.
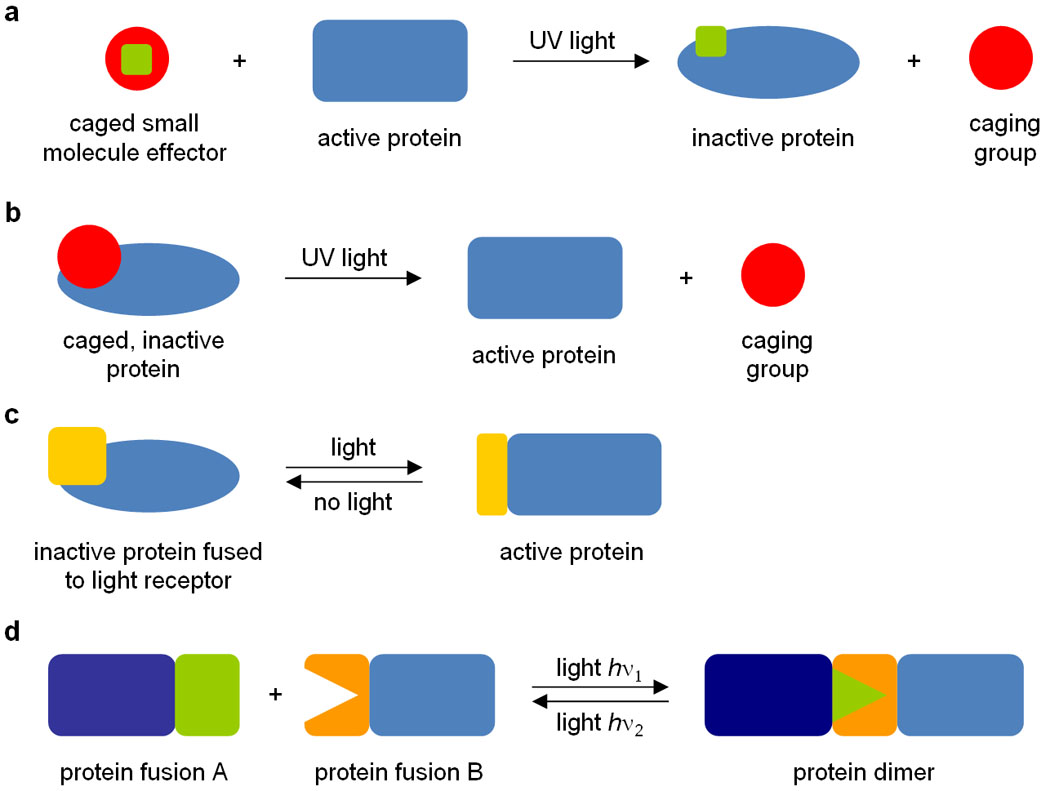
Four different approaches to control protein function with light. (a) Caged small effector molecules can inhibit protein function after caging group removal via UV irradiation. (b) Caged proteins, expressed using genetically encoded caged amino acids, can be activated via UV irradiation through caging group removal. (c) The activity of proteins can be reversibly regulated by light irradiation when fused to a light receptor (e.g. a natural light oxygen voltage (LOV) domain or a synthetic photoswitchable affinity label (PAL)). (d) Two proteins can be dimerized by light irradiation when fused to a natural plant phytochrome (e.g. PhyB) and a phytochrome interaction factor (PIF).
Caged small organic effector molecules
Many small effector molecules are known that can bind to proteins, prompting a protein to assume a biologically active or inactive state. Installing caging groups on the small molecule effectors (organic protein ligands with molecular weights typically <1000 Da) of proteins gives researchers the ability to control protein activity without attaching caging groups to the actual proteins. These caged small molecules can be utilized in vivo in a variety of prokaryotic and eukaryotic systems, simply by adding the caged effector molecule to the growth medium. Examples of caged small-molecule effectors that specifically affect gene expression include caged doxycycline13, ecdysone14, estradiol15 and tamoxifen16.
Caged isopropyl-β-D-thiogalactoside (IPTG) 1 (Figure 2a; the caging group is shown in red) was successfully used to control the expression of genes under control of a lac operator in bacterial cells17. The caged molecule is unable to bind to the lac repressor; however, UV-mediated decaging produced active IPTG, which subsequently removed the lac repressor from the promoter sequence on the DNA and induced gene expression. Using β-galactosidase as a reporter gene, a tenfold activation of gene expression was observed after a brief 5 min UV irradiation of cells exposed to 1. Irradiation – and thus decaging - of 1 led to a similar level of gene expression as direct addition of non-caged IPTG to the growth medium. Spatial control of gene expression was also demonstrated and thus enabled bacterial lithography (Figure 2b). Cultures grown on solid media were irradiated on half of the area of the plate, with the other half masked from irradiation. The irradiated side showed light-induced gene expression, with the masked side remaining unaffected. Two different constructs were used for the bacterial lithography experiments, one being the same β-galactosidase expression system used before, and the other having a GFP gene under control of the lac operator. Expression from both constructs was controlled equally well spatially using the developed caged IPTG molecule. Another excellent example of bacterial lithography took advantage of a photoreactive cyanobacterial protein (an approach similar to the PhyB/PIF system discussed herein) to produce images on a lawn of bacteria with high resolution18.
Figure 2.
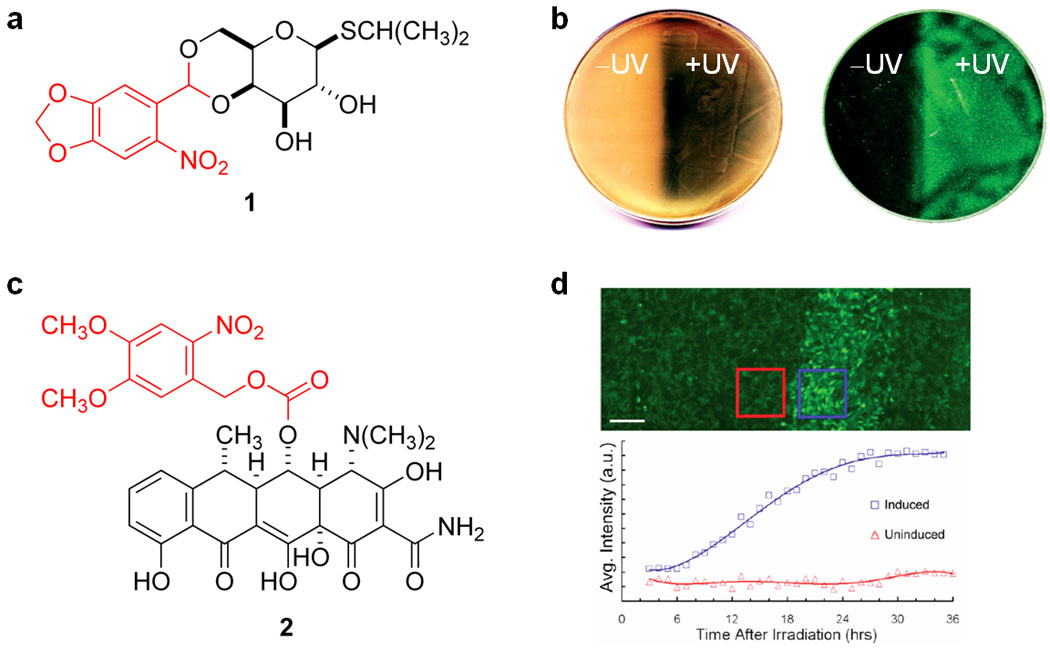
Light-activatable small molecule inhibitors of gene expression repressor proteins. (a) Caged IPTG 1. (b) Bacterial lithography experiment with 1, showing light-controlled induction and spatial control of β-galactosidase (left) and GFP (right) in a bacterial lawn (∅ 10 cm). (c) Caged doxycycline 2. (d) Spatial control of GFP expression using 2, with irradiation through a 344-µm photomask (scale bar = 250 µm); irradiated (blue) and non-irradiated (red) cells were quantified and fluorescence intensity is shown as a function of time. The light-removable caging groups are shown in red.
More defined spatial control over protein expression was recently demonstrated in mammalian cells using a caged doxycycline molecule 2 (Figure 2c; the caging group is shown in red)19. The doxycycline-inducible gene expression system RetroTET-Art (containing a doxycycline-reactive repressor/activator pair)20 was employed in conjunction with the caged molecule 2 19. Patterned GFP expression was achieved in a monolayer of NIH 3T3 mouse fibroblast cells pre-treated with 2 by placing photomasks on the bottoms of culture dishes and irradiating with UVA light of 330–380 nm. Within the irradiated region, 70–85% of cells showed induced expression of GFP, as evidenced by a 5-fold higher level of GFP expression than non-irradiated areas (Figure 2d). The ability to spatially pattern cells with light was also demonstrated using this system, potentially providing an opportunity for very precise tissue engineering. 3T3 cells expressing a membrane-bound ligand, ephrin A5, under control of RetroTET-Art were grown to a monolayer on tissue culture plates. Spatially restricted irradiation allowed the patterning of HEK293T cells expressing either the membrane receptor EphA7-T1 (which interacts attractively with ephrin A5) or EphA7 (which interacts repulsively with ephrin A5). Upon irradiation, HEK293T cells expressing EphA7-T1 attached to the 3T3 cells, while the HEK293T cells expressing EphA7 preferentially avoided the irradiated 3T3 cells when settling on top of them.
Reversible photochemical regulation of ion channel activity
An interesting approach to the light-regulation of protein activity is the application of photoswitchable affinity labels (PALs) to introduce photo-activatable ion channels in neurons. Caged molecules have traditionally been used in a wide range of studies of neuronal function21. Some of these approaches involved photo-activatable caged glutamate22, 23, genetically-modified nicotinic acetylcholine receptors24, potassium ion (K+) channels25 and glutamate receptors26, and the incorporation of retinal27, a naturally-occurring photoactive molecule, into neurons. Light-activated nanopores that penetrate a membrane and form a functional channel after light irradiation have also been engineered28.
A light-regulated K+ channel was successfully created using the PAL method29. The PALs used had three distinct features: a quaternary ammonium group, a photo-isomerizable azobenzene group, and an electrophilic, cysteine-reactive group (e.g. acrylamide, chloroacetamide, epoxide or maleimide). The ammonium group is comparable in size to a potassium cation and thus blocks the pore of the K+ channel; the electrophilic group reacts with the nucleophilic amino acid residues, typically cysteines, creating a covalent attachment to the ion channel. The azobenzene moiety enables reversible photo-isomerization of the molecule. When the PAL is in its trans form, the pore is blocked; however, when converted to the cis geometry by irradiaton with 360–400 nm UV light, the blockage is removed, enabling the passage of potassium ions (Figure 3a). Conversion from cis back to trans can be accomplished either by slow thermal reversion in the dark or rapidly by irradiation with 450–560 nm light. Through the reversible blocking of a K+ channel, it was possible to produce changes in membrane potential a cultured cell via irradiation with 380 nm and 500 nm light (Figure 3b). This approach was also applied to the photochemical control over neuronal firing in cultured cells and live tissues (rat cerebral slices and ganglia taken from medicinal leeches) by light-triggering of action potentials.
Figure 3.
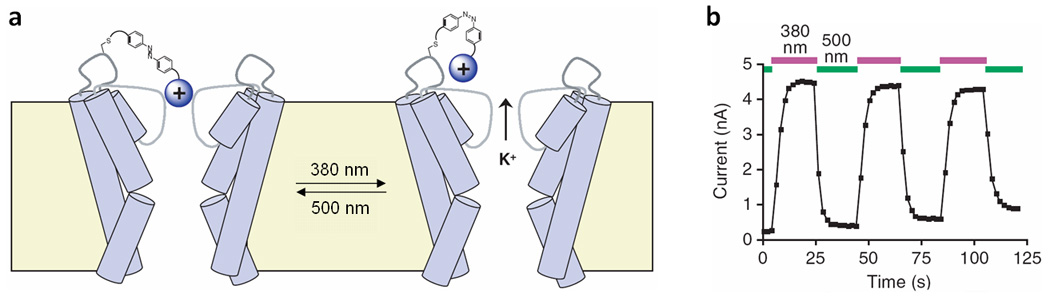
Reversible photochemical activation of a potassium ion (K+) channel. (a) Schematic of PAL-gated K+ channel. The sphere labeled with a plus sign represents a quaternary ammonium group, which blocks the channel when the diazobenzene is in the trans conformation. Irradiation at 380 nm switches the diazobenzene from trans to cis, thus enabling K+ flow. Irradiation with 500 nm light reverses the switching event, thus blocking the ion channel. (b) Light-controlled membrane potential of a PAL-gated K+ channel measured in dependence of light irradiation.
Genetically encoded caged amino acids
Instead of photochemically regulating protein function through the caging of small molecule effectors of protein function, it is also possible to directly install caging groups on the protein itself. Traditionally, caged proteins were produced by reacting surface lysine residues of the isolated protein with caging groups in a non-specific fashion (similar to the PAL installation discussed above)30, 31. This approach is problematic because it requires purification of the protein-of-interest, attachment of caging groups, and re-introduction of the caged protein into cellular systems (for in vivo applications). Perhaps most importantly, the caging of residues crucial to activity cannot be guaranteed using this method, and the location and number of caged residues cannot be controlled. In vitro transcription/translation systems can be used instead to successfully engineer caged proteins to increase residue specificity32. Rather than targeting the side chains of amino acids, caging groups can be installed on α-amino groups, as demonstrated recently in the creation of a photo-activatable intein splicing system33. In contrast, unnatural amino acid incorporation through site-directed mutagenesis allows incorporation of caged amino acids at specific sites, and caged proteins can be expressed in bacterial, yeast, and mammalian cells using orthogonal synthetase/tRNA pairs34–36. Caged tyrosine37–40, cysteine41, serine42 and lysine43,44 have been incorporated successfully into proteins using site-directed unnatural amino acid mutagenesis.
Site-directed mutagenesis and an evolved orthogonal cellular machinery have recently been used to incorporate the caged lysine 3 (Figure 4a) into proteins in human embryonic kidney (HEK293) cells44. The proteins contained the nuclear localization sequences (NLS) of nucleoplasmin and the tumor suppressor p53, which allowed photo-regulation of the cellular localization of those proteins. The orthogonal cellular machinery was first evolved by using the pyrrolysyl-tRNA synthetase of the methanogenic bacterium Methanosarcina barkeri and its cognate amber codon tRNA, both of which are functional in bacterial and mammalian cells and are orthogonal to all endogenous tRNAs and tRNA synthetases45. The active site of the synthetase was mutagenized and the obtained library of synthetase mutants was subjected to a double-sieve selection yielding a synthetase/tRNA that selectively incorporated 3, but no endogenous amino acid, in response to the amber stop codon (UAG)44. This pair was then used to incorporate 3 into a fusion protein featuring the NLS of tumor suppressor p53 and EGFP (p53-EGFP; gene diagram shown in Figure 4b), where the crucial lysine residue K305 was replaced by the amber stop codon, thus encoding the caged lysine analog (K305→3). Without irradiation, fluorescence microscopy showed the EGFP localized to the cytoplasm, thus demonstrating that the caging group on the lysine had blocked nuclear import. After irradiation at 365 nm for 5 s, an increase in nuclear import was observed, indicating that the function of the NLS could be restored by photolysis of the caging group (Figure 4c). The generality of this process was demonstrated by obtaining similar results for the caging of a lysine residue necessary for nuclear import in the NLS of nucleoplasmin.
Figure 4.
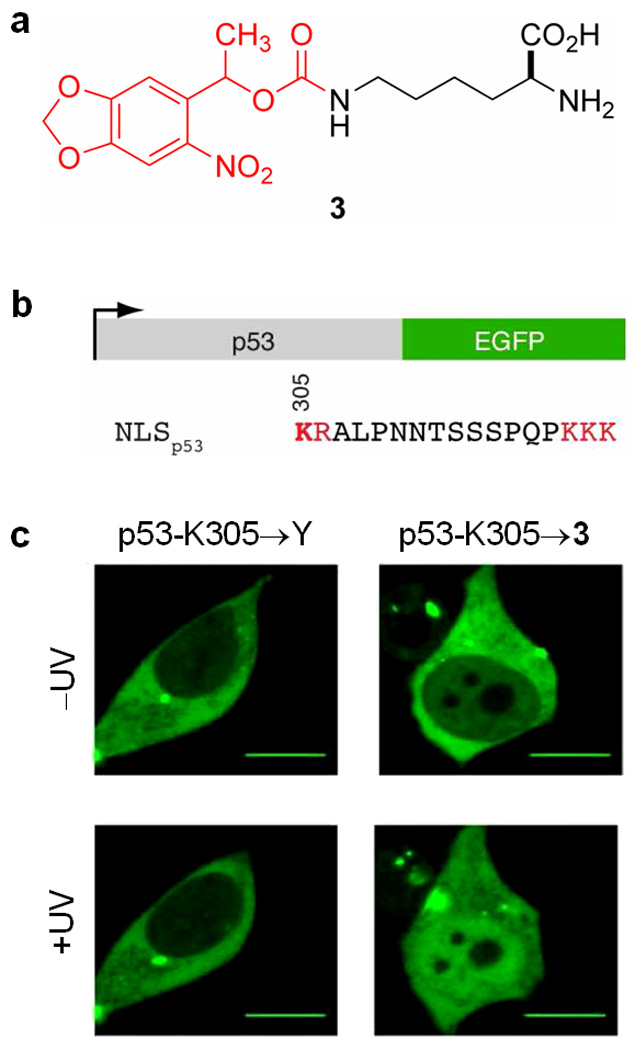
Photochemical control of protein localization in mammalian cells. (a) Genetically encoded caged lysine 3; light-removable caging group shown in red. (b) Gene diagram of p53-egfp, with the crucial lysine K305 bolded. (c) Nuclear import of EGFP in HEK293 cells after introduction and photolysis of 3 introduced into position 305 in NLSp53. A mutation of K305 to tyrosine (K305→Y) blocks transport into the nucleus, regardless of light irradiation (left). Introduction of the caged lysine 3 at position 305 (K305→3) blocks transport of p53-EGFP-HA, but enables translocation into the nucleus after a brief light irradiation (365 nm, 5 s) (right). Scale bar = 10 µm.
Genetically encoded photoresponsive protein domains
The incorporation of caged amino acids into proteins in live cells enables precise, light-mediated regulation of protein function in an in vivo environment. However, once decaged, no photochemical deactivation of the protein is possible, thus preventing the reversible regulation of protein activity. The reversible photochemical switching of protein function was recently achieved by fusing LOV domains, which are naturally found in plant phototropin proteins, to the protein-of-interest46. The LOV domain is bound by a flavin mononucleotide (FMN) cofactor, which triggers a conformational change in the protein when exposed to blue light (approximately 450–470 nm). The conformation change occurs as an unwinding of a helical domain (Jα) adjacent to the LOV domain, serving as a signaling message to an attached effector protein. Once blue-light irradiation ceases, the protein assumes its original, inactive conformation. Fusing the LOV domain to effector proteins has enabled the light-regulation of a histidine-kinase47, a GTPase48, 49 and dihydrofolate reductase50.
A system that illustrates a successful light-activated LOV domain fusion is LovTAP (Lov- and tryptophan-activated protein) – a fusion of a LOV domain to an E. coli trp repressor protein that requires L-tryptophan and blue light irradiation for activity51. In this protein, the conformation of the allosteric Jα region separating the LOV domain from the trp repressor region determines whether or not the repressor region can bind DNA. In the dark, the fusion protein is inactive because Jα is associated with the LOV domain, and the repressor region becomes distorted and cannot bind DNA. When exposed to blue light, the FMN chromophore cofactor of the LOV domain induces a conformational change in the protein. As a result, Jα dissociates from the LOV domain, restoring activity to the Trp repressor protein and enabling DNA binding (Figure 5a). A DNA protection assay has been conducted where an RsaI cleavage site was inserted into the trp operator region of a plasmid (Figure 5b). When irradiated with blue light, the fusion protein would bind to the trp operator and prevent cleavage by the RsaI restriction enzyme, with the proportion of digested DNA decreasing as the concentration of LovTAP (µM) increased (dashed lines). In the dark, however, the cleavage site was left unprotected and the DNA was cleaved to a greater extent (solid lines). The preference of the fusion protein for the trp operator for successful protection indicated that the Trp repressor region of the fusion protein retained its native activity, and the photoactive properties of the LOV domain enabled light-regulation of the system. The next logical step will be the application of LOV-regulated trp repressor in the photocontrol of gene function in live cells, as has been demonstrated for other LOV-protein fusions, including the histidine kinases FixL47 and Rac48, 49.
Figure 5.
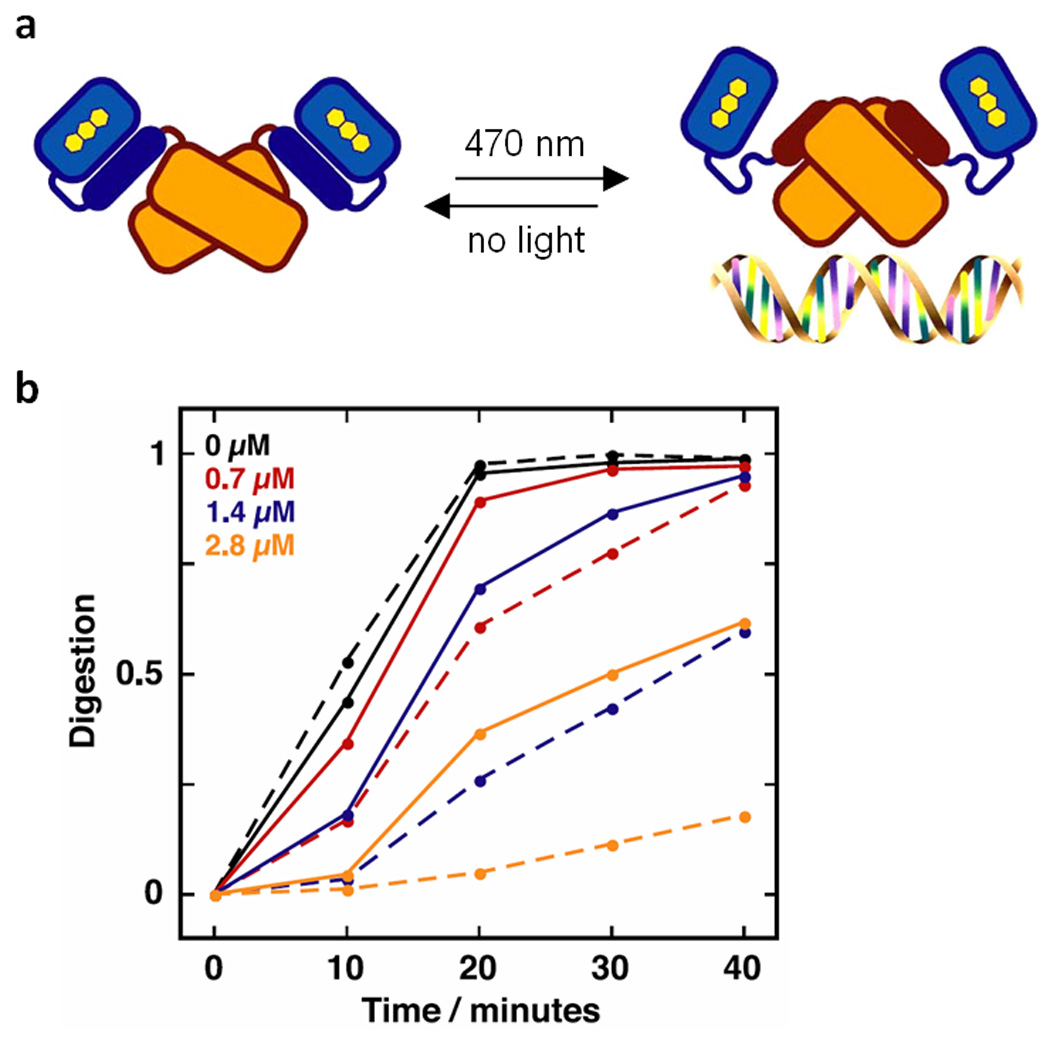
Mechanism of LovTAP, a reversibly switchable DNA binding protein. (a) Before exposure to blue light, Jα (dark blue) is associated with the LOV domain (light blue), which renders the Trp repressor region (light orange) inactive. After irradiation with blue light (470 nm) a conformational change occurs and Jα (now dark orange) dissociates from the LOV domain, in turn activating the Trp repressor. The active protein binds to DNA at a lac operator region. The original conformation of the protein is resumed after incubation in the dark, and the Trp repressor region dissociates from the DNA, thus making the activity of LovTAP reversible. (b) LovTAP protects DNA against RsaI digestion at the lac operator site. Increasing the concentration of LovTAP (0–2.8 µM) decreases digestion, as does irradiation with blue light (dashed lines: irradiated; solid lines: non-irradiated).
Genetically encoded protein dimerization
Plant phytochromes are photosensory molecules that alternate between two conformations in response to red and infrared light52. They are two-domain proteins bound to a chromophore, phycocyanobilin (PCB), which absorbs light of the appropriate wavelengths. One protein conformation is “active”, meaning that it can interact with other proteins in signaling pathways related to light absorption in plants, whereas the other conformation is “inactive” in this context53. The inactive form, referred to as Pr, absorbs red light of approximately 660 nm, which converts it to the active form, Pfr. When Pfr absorbs IR light (~750 nm), it is converted back into Pr, resulting in a loss of biological activity. These conversions are extremely fast, and can be conducted repetitively without degradation of the phytochrome.
PhyA and PhyB are two plant phytochromes which have been used to create photoactivatable fusion proteins. In their active Pfr forms, they undergo a binding interaction with the protein PIF (phytochrome interaction factor)54. This interaction has been exploited to modulate the dimerization of other proteins, where one protein of interest is fused to PhyA or PhyB, and the other protein is fused to PIF. A system similar to the one described here, utilized a cyanobacterial phytochrome in the afore-mentioned bacterial lithography experiment to display high-resolution images on a bacterial lawn18. The coupling of a cyanobacterial phytochrome to a bacterial kinase has recently been engineered as a light-sensing component of a genetically-encoded “edge detection program”, where bacteria incubated in the dark secreted the chemical messenger homoserine lactone to nearby cells. If nearby cells detected both the chemical messenger and light, they catalyzed production of a dark pigment, allowing the bacteria to mark the edge of a photomask55. A necessary component of these systems is the addition of the chromophore co-factor, which imparts photoactivity to the phytochrome molecule. While other chromophores can be used effectively, the most common is PCB56.
An elegant application of the light-regulated Phy-PIF interaction is the creation of a light-controlled transcriptional activation system57. Here, the transcriptional activation domain of GAL4 was fused to PIF, and the DNA-binding domain of GAL4 was fused to the photoactive N-terminal domain of PhyB (Figure 6a). Yeast expressing these constructs were used to demonstrate transcriptional control over β-galactosidase expression with constant red light. Upon further investigation, it was found that maximal activation of gene expression could be achieved by using shorter pulses of red light rather than constant exposure. By using increasing red light pulses of 5, 10 and 30 min, they observed 2-, 4- and 30-fold increased LacZ expression and thus β-galactosidase activity. The reversibility of this light-controlled gene expression system was demonstrated with 5-min pulses of IR light (Figure 6b). β-Galactosidase accumulation slowed within 10 min after IR light exposure, and completely ceased within 15 min. Even though the expression levels of the light-inducible system were three orders of magnitude above background expression after light activation, they only reached one-sixth of wild-type expression levels observed with the native GAL4 promoter.
Figure 6.
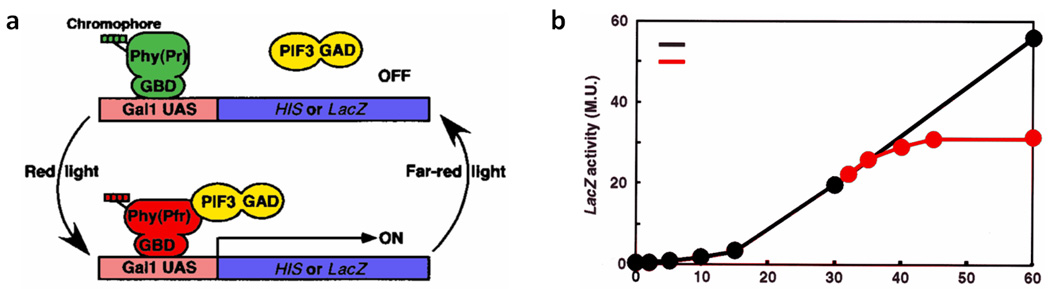
Light-controlled expression of LacZ by PhyB-PIF protein dimerization. (a) Schematic of the light-controlled transcriptional activator. Phytochrome (Phy), which is fused to the DNA-binding region of GAL4 (GBD), reversibly binds to PIF. PIF is fused to the transcriptional activating domain of GAL4 (GAD), thus activating gene expression upon exposure to red light and ceasing activity by dissociation upon exposure to IR light. (b) LacZ (β-galactosidase) activity induced with pulses of red light (Rp, black line), and arrested with pulses of IR light (FRp, red line).
Recently, the Phy-PIF dimerization was utilized in the construction of a light-controlled system for the regulation of protein localization to the cell membrane in mammalian cells54, 58. A membrane anchored PhyB was used in the red light-activated recruitment of yellow fluorescent protein. Spatial control over this system was demonstrated by irradiation of sub-cellular areas with red or IR light, thus projecting patterns onto the cell surface. In addition, light-induced localization of the guanine nucleotide exchange factor Tiam to the cell membrane using the same system, resulted in photo-controlled lamellipodia formation in mammalian cells. Phy-PIF dimerization has also been used recently in yeast to engineer a light-activated conditional protein splicing system59.
Conclusion
Photochemical control over protein function has been achieved through a variety of distinct approaches. Small organic effector molecules that regulate the activity of proteins have been caged, allowing the activities of the proteins to be regulated by light. Proteins themselves have been rendered inactive through the installation of caging groups or photoswitchable groups, and have then been activated through UV irradiation. Such light-responsive groups have been attached through chemical reactions (i.e. photochemical regulation of K+ channels) and through the site-specific incorporation of genetically encoded unnatural amino acids using orthogonal tRNA/tRNA synthetase pairs in bacterial and mammalian cells (i.e. photochemical control of intracellular protein localization). Further optimization of these existing technologies and development of new caging groups for amino acids and the corresponding engineered cellular machinery will enable a higher degree of precision and versatility in controlling protein activity with light.
Naturally occurring photoresponsive proteins have also been adopted for the purpose of controlling the activities of other proteins-of-interest with light. These systems involve fusing the protein that will be the object of light-regulation to a photoactive domain that changes its conformation through irradiation (i.e. LOV domain-fusion proteins), or take advantage of light-induced protein dimerization to reconstitute a dissociated protein-of-interest, thereby restoring its activity (i.e. the PhyB-PIF system). An advantage of those light-regulatory elements is their reversibility while being fully genetically encoded. Each type of system discussed here illustrates how light can be used as a tool to control protein activity both in vitro and in vivo, often with a high degree of spatiotemporal precision. These technologies will prove useful in further investigations of important biological questions regarding the regulation and function of proteins and genes in cells and multi-cellular model organisms. However, moving forward, open sharing of developed genetic constructs between laboratories and the commercial availability of photocaged monomeric building blocks (e.g. amino acids) and small molecule modifiers will be necessary.
Acknowledgements
Financial support from the National Institutes of Health (R01GM079114) is acknowledged. AD is a Beckman Young Investigator and a Cottrell Scholar. The authors apologize to those researchers whose work could not be discussed as a result of space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Young DD, Deiters A. Photochemical control of biological processes. Org Biomol Chem. 2007;5(7):999–1005. doi: 10.1039/b616410m. [DOI] [PubMed] [Google Scholar]
- 2.Lee HM, et al. Illuminating the chemistry of life: design, synthesis, and applications of "caged" and related photoresponsive compounds. ACS Chem Biol. 2009;4(6):409–427. doi: 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deiters A. Principles and applications of the photochemical control of cellular processes. Chembiochem. 2010;11(1):47–53. doi: 10.1002/cbic.200900529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deiters A. Light activation as a method of regulating and studying gene expression. Curr Opin Chem Biol. 2009;13(5–6):678–686. doi: 10.1016/j.cbpa.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casey JP, et al. Caged siRNAs for spatiotemporal control of gene silencing. Mol Pharm. 2009;6(3):669–685. doi: 10.1021/mp900082q. [DOI] [PubMed] [Google Scholar]
- 6.Mayer G, Heckel A. Biologically active molecules with a "light switch". Angew Chem Int Ed. 2006;45(30):4900–4921. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- 7.Tang X, Dmochowski IJ. Regulating gene expression with light-activated oligonucleotides. Mol Biosyst. 2007;3(2):100–110. doi: 10.1039/b614349k. [DOI] [PubMed] [Google Scholar]
- 8.Adams SR, Tsien RY. Controlling cell chemistry with caged compounds. Annu Rev Physiol. 1993;55:755–784. doi: 10.1146/annurev.ph.55.030193.003543. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y, et al. 8-Bromo-7-hydroxyquinoline as a photoremovable protecting group for physiological use: mechanism and scope. J Am Chem Soc. 2006;128(13):4267–4276. doi: 10.1021/ja0555320. [DOI] [PubMed] [Google Scholar]
- 10.Momotake A, et al. The nitrodibenzofuran chromophore: a new caging group for ultra-efficient photolysis in living cells. Nat Methods. 2006;3(1):35–40. doi: 10.1038/nmeth821. [DOI] [PubMed] [Google Scholar]
- 11.Furuta T, Noguchi K. Controlling cellular systems using Bhc-caged compounds. Trends Anal Chem. 2004;23:501–509. [Google Scholar]
- 12.Furuta T, et al. Brominated 7-hydroxycoumarin-4-ylmethyls: photolabile protecting groups with biologically useful cross-sections for two photon photolysis. Proc Natl Acad Sci U S A. 1999;96(4):1193–1200. doi: 10.1073/pnas.96.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cambridge SB, et al. Doxycycline-dependent photoactivated gene expression in eukaryotic systems. Nat Methods. 2009;6(7):527–531. doi: 10.1038/nmeth.1340. [DOI] [PubMed] [Google Scholar]
- 14.Lin W, et al. Spatially discrete, light-driven protein expression. Chem Biol. 2002;9(12):1347–1353. doi: 10.1016/s1074-5521(02)00288-0. [DOI] [PubMed] [Google Scholar]
- 15.Cruz FG, et al. Light-activated gene expression. J Am Chem Soc. 2000;122:8777–8778. [Google Scholar]
- 16.Link KH, et al. Light activated recombination. J Am Chem Soc. 2005;127(38):13088–13089. doi: 10.1021/ja0531226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young DD, Deiters A. Photochemical activation of protein expression in bacterial cells. Angew Chem Int Ed. 2007;46(23):4290–4292. doi: 10.1002/anie.200700057. [DOI] [PubMed] [Google Scholar]
- 18.Levskaya A, et al. Synthetic biology: engineering Escherichia coli to see light. Nature. 2005;438(7067):441–442. doi: 10.1038/nature04405. [DOI] [PubMed] [Google Scholar]
- 19.Sauers DJ, et al. Light-Activated Gene Expression Directs Segregation of Co-cultured Cells in Vitro. ACS Chem Biol. 2010 doi: 10.1021/cb9002305. [DOI] [PubMed] [Google Scholar]
- 20.Rossi FM, et al. Tetracycline-regulatable factors with distinct dimerization domains allow reversible growth inhibition by p16. Nat Genet. 1998;20(4):389–393. doi: 10.1038/3871. [DOI] [PubMed] [Google Scholar]
- 21.Callaway EM, Yuste R. Stimulating neurons with light. Curr Opin Neurobiol. 2002;12(5):587–592. doi: 10.1016/s0959-4388(02)00364-1. [DOI] [PubMed] [Google Scholar]
- 22.Fedoryak OD, et al. Synthesis of a caged glutamate for efficient one- and two-photon photorelease on living cells. Chem Commun (Camb) 2005;(29):3664–3666. doi: 10.1039/b504922a. [DOI] [PubMed] [Google Scholar]
- 23.Callaway EM, Katz LC. Photostimulation using caged glutamate reveals functional circuitry in living brain slices. Proc Natl Acad Sci U S A. 1993;90(16):7661–7665. doi: 10.1073/pnas.90.16.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Philipson KD, et al. Incorporation of caged cysteine and caged tyrosine into a transmembrane segment of the nicotinic ACh receptor. Am J Physiol Cell Physiol. 2001;281(1):C195–C206. doi: 10.1152/ajpcell.2001.281.1.C195. [DOI] [PubMed] [Google Scholar]
- 25.Banghart M, et al. Light-activated ion channels for remote control of neuronal firing. Nat Neurosci. 2004:1381–1386. doi: 10.1038/nn1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volgraf M, et al. Allosteric control of an ionotropic glutamate receptor with an optical switch. Nat Chem Biol. 2006;2(1):47–52. doi: 10.1038/nchembio756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446(7136):633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 28.Chang CY, et al. A photogenerated pore-forming protein. Chem Biol. 1995;2(6):391–400. doi: 10.1016/1074-5521(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 29.Fortin DL, et al. Photochemical control of endogenous ion channels and cellular excitability. Nature Methods. 2008;5:331–338. doi: 10.1038/nmeth.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curley K, Lawrence DS. Light-Activated Proteins. Curr Opin Chem Biol. 1999;3:84–88. doi: 10.1016/s1367-5931(99)80015-5. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence DS. The preparation and in vivo applications of caged peptides and proteins. Curr Opin Chem Biol. 2005;9(6):570–575. doi: 10.1016/j.cbpa.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Petersson EJ, et al. Caging proteins through unnatural amino acid mutagenesis. Methods Enzymol. 2003;360:258–273. doi: 10.1016/s0076-6879(03)60114-x. [DOI] [PubMed] [Google Scholar]
- 33.Vila-Perello M, et al. Activation of protein splicing by protease- or light-triggered O to N acyl migration. Angew Chem Int Ed. 2008;47(40):7764–7767. doi: 10.1002/anie.200802502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie J, Schultz PG. Adding amino acids to the genetic repertoire. Curr Opin Chem Biol. 2005;9(6):548–554. doi: 10.1016/j.cbpa.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Schultz PG. Expanding the genetic code. Angew Chem Int Ed. 2004;44(1):34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 36.Cropp TA, Schultz PG. An expanding genetic code. Trends Genet. 2004;20(12):625–630. doi: 10.1016/j.tig.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 37.Chou C, et al. Photocaged T7 RNA Polymerase for the Light-Activation of Transcription and Gene Function in Pro- and Eukaryotic Cells. ChemBioChem. 2010;11 doi: 10.1002/cbic.201000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou C, et al. A light-activated DNA polymerase. Angew Chem Int Ed. 2009;48(32):5950–5953. doi: 10.1002/anie.200901115. [DOI] [PubMed] [Google Scholar]
- 39.Edwards WF, et al. Light-activated Cre recombinase as a tool for the spatial and temporal control of gene function in mammalian cells. ACS Chem Biol. 2009;4(6):441–445. doi: 10.1021/cb900041s. [DOI] [PubMed] [Google Scholar]
- 40.Deiters A, et al. A genetically encoded photocaged tyrosine. Angew Chem Int Ed. 2006;45(17):2728–2731. doi: 10.1002/anie.200600264. [DOI] [PubMed] [Google Scholar]
- 41.Wu N, et al. A genetically encoded photocaged amino acid. J Am Chem Soc. 2004;126(44):14306–14307. doi: 10.1021/ja040175z. [DOI] [PubMed] [Google Scholar]
- 42.Lemke EA, et al. Control of protein phosphorylation with a genetically encoded photocaged amino acid. Nat Chem Biol. 2007;3(12):769–772. doi: 10.1038/nchembio.2007.44. [DOI] [PubMed] [Google Scholar]
- 43.Chen PR, et al. A facile system for encoding unnatural amino acids in mammalian cells. Angew Chem Int Ed. 2009;48(22):4052–4055. doi: 10.1002/anie.200900683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gautier A, et al. Genetically Encoded Photo-control of Protein Localization in Mammalian Cells. J Am Chem Soc. 2010;132 doi: 10.1021/ja910688s. [DOI] [PubMed] [Google Scholar]
- 45.Neumann H, et al. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat Chem Biol. 2008;4(4):232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 46.Crosson S, Moffat K. Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch. Plant Cell. 2002;14(5):1067–1075. doi: 10.1105/tpc.010475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moglich A, et al. Design and signaling mechanism of light-regulated histidine kinases. J Mol Biol. 2009;385(5):1433–1444. doi: 10.1016/j.jmb.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu YI, et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;7260(461):104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoo SK, et al. Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Dev Cell. 2010;18(2):226–236. doi: 10.1016/j.devcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee J, et al. Surface sites for engineering allosteric control in proteins. Science. 2008;322(5900):438–442. doi: 10.1126/science.1159052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strickland D, et al. Light-activated DNA binding in a designed allosteric protein. Proc Natl Acad Sci U S A. 2008;105(31):10709–10714. doi: 10.1073/pnas.0709610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quail PH. Phytochrome photosensory signalling networks. Nat Rev Mol Cell Biol. 2002;3(2):85–93. doi: 10.1038/nrm728. [DOI] [PubMed] [Google Scholar]
- 53.Ni M, et al. Binding of phytochrome B to its nuclear signalling partner PIF3 is reversibly induced by light. Nature. 1999;400(6746):781–784. doi: 10.1038/23500. [DOI] [PubMed] [Google Scholar]
- 54.Georgianna WE, Deiters A. Reversible light switching of cell signalling by genetically encoded protein dimerization. Chembiochem. 2010;11(3):301–303. doi: 10.1002/cbic.200900754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tabor JJ, et al. A synthetic edge detection program. Cell. 2009;137(7):1272–1281. doi: 10.1016/j.cell.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Losi A, Gartner W. Bacterial bilin- and flavin-binding photoreceptors. Photochem Photobiol Sci. 2008;7(10):1168–1178. doi: 10.1039/b802472c. [DOI] [PubMed] [Google Scholar]
- 57.Shimizu-Sato S, et al. A light-switchable gene promoter system. Nature Biotechnology. 2002;20:1041–1044. doi: 10.1038/nbt734. [DOI] [PubMed] [Google Scholar]
- 58.Levskaya A, et al. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461(7266):997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tyszkiewicz AB, Muir TW. Activation of protein splicing with light in yeast. Nat Methods. 2008;5(4):303–305. doi: 10.1038/nmeth.1189. [DOI] [PubMed] [Google Scholar]