

Abstract
Interactions between cholinergic and glutamatergic neurotransmitter systems influence synaptic transmission and plasticity. While previous studies have examined cross-talk between acetylcholine (ACh) and NMDA or AMPA receptors, little is known about the effect of ACh on kainate receptors (KARs). We show that stimulation of m1 or m3 muscarinic ACh receptors (mAChRs) for 2 min potentiates recombinant KAR currents in a long lasting fashion. Muscarinic AChR activation potentiates heteromeric GluK2/GluK4 and GluK2/GluK5 receptors, but not homomeric GluK2 receptors. In hippocampal slices kainate potentiates mossy fiber axon excitability. Transient mAChR activation enhances this action of kainate, suggesting a novel mechanism through which acetylcholine could modulate synaptic transmission in the hippocampus. KAR over-activation has been implicated in excitotoxic cell death. To establish the functional significance of the interaction between mAChRs and KARs we examined the effect of mAChR activation on KAR-mediated excitotoxicity. We find that during pharmacological blockade of NMDA and AMPA receptors, KAR activation with AMPA produces significant cell death in primary cortical culture. Concanavalin A (Con A), which selectively blocks KAR desensitization, markedly increases this KAR-mediated neurotoxicity. Brief activation of mAChRs with pilocarpine significantly enhances KAR-mediated excitotoxicity both in the presence and absence of Con A. We conclude that KARs are modulated in a subunit dependent manner by mAChRs. We suggest that ACh may induce long lasting alterations in neuronal excitability and enhance excitotoxicity in part by potentiating KAR function.
Keywords: Excitotoxicity, GluK2, GluK5, Hippocampus, Glutamate, Acetylcholine
1. Introduction
Interactions between cholinergic and glutamatergic neurotransmitter systems modulate synaptic transmission and neuronal excitability. These interactions also regulate forms of synaptic plasticity thought to be important in learning and memory (Blokland, 1995). Dysfunctional interactions between these transmitter systems have been proposed to contribute to a number of neurodegenerative and psychiatric disorders. Brain acetylcholine levels fluctuate greatly with changes in behavioral state (i.e. stress, arousal, fear) and during the sleep-waking cycle (Marrosu et al., 1995; Acquas et al., 1996). These behaviorally-induced changes in acetylcholine levels have the potential to regulate glutamatergic function in a state dependent manner. Because of this, a great deal of effort has been focused on understanding the interaction between cholinergic and glutamatergic neurotransmission.
It is well established that acetylcholine, acting on muscarinic receptors (mAChRs) potentiates currents through the N-methyl-D-aspartate (NMDA) subtype of glutamate receptor (NMDAR) (Markram and Segal, 1992; Marino et al., 1998). This potentiation of NMDAR-mediated currents is present only during mAChR stimulation. In contrast, mAChRs can produce lasting changes in synaptic efficacy at glutamatergic synapses by influencing AMPA receptor currents. Both mAChR-induced long term potentiation (Auerbach and Segal, 1994; Fernandez de Sevilla et al., 2008) and long term depression (Kirkwood et al., 1999) have been reported. Muscarinic receptors appear to influence AMPA receptor currents through indirect mechanisms, including alterations in AMPA receptor trafficking (Fernandez de Sevilla et al., 2008). An interaction between mAChRs and KARs has not been reported. Therefore, one goal of this study was to determine whether mAChRs influence KAR currents.
Kainate receptors (KARs) are widely expressed throughout the CNS where they contribute to synaptic transmission and plasticity (Lerma, 2003). KARs are tetramers composed of combinations of GluK1-3 subunits that have a low affinity for glutamate and GluK4-5 subunits that have a high affinity for glutamate. Only the GluK1-3 subunits can produce functional homomeric receptors. KARs are located at both presynaptic and postsynaptic sites (Lerma, 2003), where they modulate neurotransmitter release or mediate excitatory neurotransmission, respectively.
Over-activation or dysfunction of KARs has been implicated in a number of neurological disorders, including schizophrenia (Bah et al., 2004) and epilepsy (Vincent and Mulle, 2009). In the hippocampus, activation of axonal KARs with a low concentration of kainate can enhance the excitability of mossy fibers (Schmitz et al., 2000; Kamiya and Ozawa, 2000). KARs have also been implicated in excitotoxic cell death. In vitro, KAR over-activation is neurotoxic in primary neuronal culture (Jensen et al., 1999; Giardina and Beart, 2001). In vivo, KAR antagonists are neuroprotective in both global and focal ischemia models (O'Neill et al., 2000). Muscarinic modulation of KAR function could therefore regulate neuronal excitability and alter excitotoxicity in pathological states.
In this study, we investigated the functional interaction between mAChRs and KARs. Our goal was to determine whether mAChR activation regulates KAR function and whether this regulation influences excitotoxicity mediated by KARs. Using recombinant KARs expressed in Xenopus oocytes, we found that stimulation of m1 or m3 mAChRs for 2 min produces a long lasting and subunit-dependent potentiation of KAR currents. In hippocampal slices, we found that kainate-induced potentiation of mossy fiber axon excitability was enhanced by prior activation of mAChRs. The ability of mAChR activation to potentiate KAR currents suggested that mAChR activation would also potentiate KAR-mediated excitotoxicity. Indeed, we found that KAR- mediated excitotoxicity in primary cortical culture was markedly increased by prior mAChR stimulation. These results indicate a significant interaction between mAChR and KAR transmitter systems and suggest that ACh may induce long lasting alterations in neuronal excitability and enhance excitotoxicity in part by potentiating KAR function. This neurotransmitter interaction may have important implications in a variety of neurological disorders, including epilepsy.
2. Results
2.1 Muscarinic receptor activation potentiates kainate receptor-mediated current
Because of the widespread expression of GluK2 in brain and its reported physiological importance (Lerma, 2003), this study focused on muscarinic regulation of GluK2-containing KARs. Kainate currents were evoked in Xenopus oocytes expressing GluK2, GluK2/GluK4 or GluK2/GluK5 receptors alone or in combination with m1 or m3 mAChRs. Steady state kainate currents at GluK2 receptors were evoked with domoate (10 μM), while steady state currents at GluK2/GluK4 and GluK2/GluK5 receptors were evoked with AMPA (30 μM). At 30 μM, AMPA selectively activates GluK4 and GluK5-containing KARs. Muscarinic receptors were stimulated with a 2 min application of pilocarpine (100 μM), a selective mAChR agonist. In oocytes in which m1 or m3 mAChRs had not been injected, pilocarpine produced no current, indicating that oocytes in this study did not express native mAChRs. However, in oocytes expressing m1 or m3 mAChRs, pilocarpine produced a large inward current attributable to Cl- efflux through Ca2+-activated Cl- channels endogenous to the oocytes. The presence of this pilocarpine-induced current indicated effective surface expression of mAChRs.
Potentiation of KAR currents immediately followed pilocarpine application (Figures 1A & B), but was dependent on the KAR subunits expressed (Figure 1C). Stimulation of m1 or m3 mAChRs with pilocarpine significantly potentiated current at GluK2/GluK4 (m1, 133 ± 3%, n = 8, P < 0.01; m3, 119 ± 4%, n = 6, P < 0.01) and GluK2/GluK5 (m1, 172 ± 10%, n = 8, P < 0.01; m3, 168 ± 16%, n = 8, P < 0.01) receptors 30 minutes after pilocarpine washout (Figure 1C). In contrast, activation of m1 mAChRs did not potentiate current at GluK2 homomeric receptors (106 ± 5%, n = 6), suggesting that muscarinic potentiation of KARs requires the GluK4 or GluK5 subunit. Current elicited by GluK2/GluK5 receptors was significantly more potentiated than that at GluK2/GluK4 receptors (P < 0.05). However, potentiation of KARs produced by m1 and m3 mAChRs was similar. To determine whether pilocarpine potentiated KAR currents through activation of mAChRs, we tested its effects on KAR current in oocytes lacking mAChRs. Omission of the m1 or m3 mAChR prevented potentiation of KARs (Figures 1B & C), indicating that the enhancement was a direct result of mAChR activation (GluK2/GluK4, 101 ± 3%, n = 8; GluK2/GluK5, 95 ± 6%, n = 6). Muscarinic potentiation of KAR currents persisted for the duration of each experiment. Three of eight experiments with oocytes expressing GluK2/GluK5 receptors and m1 mAChRs were continued for more than 2 hours before termination. In these oocytes we observed potentiation of KARs that lasted for the duration of the experiment (157 ± 9% at 2 hours).
Figure 1.

Subunit dependent potentiation of KARs by mAChRs. A. Sample trace showing potentiation of the GluK2/GluK5 current by a 2 min exposure to pilocarpine (100 μM). GluK2/GluK5 currents were evoked by application of AMPA (30 μM). B. Time course of the effect of pilocarpine (100 μM, gray bar) on currents at GluK2/GluK5 receptors in oocytes expressing (n = 8) or not expressing (n = 6) m1 mAChRs. Steady state current amplitudes were binned in 5 min intervals throughout the experiment. C. Average potentiation of AMPA or NMDA evoked current at the indicated receptor by 100 μM pilocarpine measured 30 min after pilocarpine exposure (n = 4-11, **P < 0.01). Stimulation of m1 mAChRs potentiated GluK2/GluK5 receptors to a significantly greater degree than GluK2/GluK4 receptors (+P < 0.05).
Previous studies have reported m1 mAChR-mediated potentiation of NMDAR-mediated currents (Marino et al., 1998). To determine if a similar potentiation was apparent in our recombinant system, we compared muscarinic potentiation of KARs with that of NMDA receptors. We found that m1 mAChR activation with pilocarpine significantly potentiated current at GluN1a/GluN2B receptors (Figure 1C). The extent of this potentiation (162 ± 8%, n = 5) was similar to that of GluK2/GluK5 receptors (172 ± 10%, n = 8).
2.2 Muscarinic receptors potentiate the kainate receptor-mediated increase in MF axon excitability
Activation of KARs with a low concentration of kainate potentiates fiber volleys and antidromic spikes in the MF pathway (Kamiya and Ozawa, 2000). To determine whether neuronal KARs were potentiated by mAChR activation, we evaluated the effect of mAChR activation on the KAR-mediated increase in MF excitability.
Antidromic spikes were evoked in the MFs by stimulation in stratum lucidum of CA3b and recorded from the dentate granule cell layer. KARs were selectively activated with kainate (500 nM) in the presence of GYKI 52466 (100 μM), d-APV (100 μM) and bicuculline methochloride (100 μM) to block AMPA, NMDA and GABAA receptors, respectively. KAR activation increased the antidromic spike amplitude (140 ± 6%, n = 5, P < 0.01; Figure 2) in agreement with previous reports (Kamiya and Ozawa, 2000). This increase was completely blocked by CNQX (100 μM; 103 ± 2%, n = 5).
Figure 2.

Muscarinic potentiation of KARs on mossy fibers in the hippocampal slice preparation. A. Sample antidromic spikes recorded in the dentate granule cell layer at the time points indicated by the numbers in panel B. B. Averaged antidromic spike amplitudes during the course of an experiment designed to test the effect of mAChR activation on kainate-induced potentiation of the antidromic spike (n = 5). Note that KAR activation with kainate (500 nM) potentiates antidromic spikes in the dentate granule cells. Activation of muscarinic receptors with CCh (30 μM) has no effect on the antidromic spike amplitude but significantly increases subsequent potentiation of the antidromic spike by kainate. C. Bar graph showing the effect of CCh (30 μM), kainate (500 nM) and kainate 30 min after CCh treatment on antidromic spike amplitude (n = 5). Kainate causes a significant potentiation of the antidromic spike amplitude (**P < 0.01) while CCh causes a significant potentiation of this kainate receptor-mediated increase (#P < 0.05, n=5).
Carbachol (CCh) potentiates NMDA currents by acting on m1 mAChRs (Marino et al., 1998). We investigated whether native KARs interacted with mAChRs by testing whether CCh (30 μM) could influence kainate induced changes in MF excitability. Kainate-induced potentiation of the antidromic spike was measured before and after a 10 min CCh application. By itself, CCh had no effect on MF excitability (97 ± 2%, n = 5). However, following CCh washout, KAR activation produced a significantly greater increase in the antidromic spike (139 ± 6% before CCh vs 183 ± 10% after CCh, n = 5, P < 0.01; Figure 2). CCh-induced potentiation was completely blocked by atropine (10 μM), indicating that the effects of CCh were caused by mAChR activation, rather than nicotinic receptor activation or a non-specific run-up of the effect of kainate. As with recombinant receptors, muscarinic potentiation of neuronal KARs was long lasting in that a significant increase in kainate-induced potentiation was still evident 40 min after CCh washout.
2.3 Muscarinic receptors potentiate kainate receptor-mediated excitotoxicity
KAR agonists are neurotoxic in primary neuronal cultures (Jensen et al., 1999; Giardina and Beart, 2001). We hypothesized that muscarinic receptor activation would potentiate KAR-mediated excitotoxicity in primary cultures of rat cerebral cortex. To test this hypothesis we first developed an excitotoxicity assay in which cell death resulted from the selective activation of KARs.
We began by examining AMPA/KA receptor-mediated excitotoxicity. To activate AMPA/KA receptors we chose not to use kainate because it strongly desensitizes KARs and previous work has demonstrated that it produces excitotoxicity primarily by stimulating AMPA receptors (Jensen et al., 1999). Instead, AMPA was used as the agonist because it is strongly desensitizing at AMPA receptors, but only weakly desensitizing at KARs. In addition, AMPA activates heteromeric GluK2/GluK5 receptors, but not homomeric GluK2 receptors (Herb et al., 1992). Neurons in primary cortical culture contain both GluK2 and GluK5 KAR subunits (Jensen et al., 1999). Thus, any KAR-mediated excitotoxicity observed in these experiments should reflect activation of GluK5-containing receptors, the receptor type that shows maximal muscarinic potentiation.
Exposure of cortical cultures to AMPA (1-30 μM) produced excitotoxicity, as indicated by an increase in lactate dehydrogenase release in the culture well. AMPA-induced excitotoxicity was dose-dependent with an EC50 = 3.9 ± 2 μM (n = 6; Figure 3A) in good agreement with previous findings (Larm et al., 1997). NMDA (30 μM) was also neurotoxic (n = 4, P < 0.05; Figure 3B) and this neurotoxicity was completely blocked by D-APV (100 μM). In sister cultures the maximal extent of AMPA-mediated LDH release in the presence of D-APV (207 ± 30%; n = 4; Figure 3B) was substantially less than that produced by NMDA.
Figure 3.

AMPA produces excitotoxic cell death in primary cortical culture. A. AMPA induces cell death in a dose dependent manner (EC50 = 3.9 ± 2 μM). Experiments were performed in the presence of d-APV. B. NMDA produces significant cell death (**P < 0.01, n = 4). d-APV has no effect alone, but blocks NMDA-induced LDH release. In sister cultures AMPA induced significant cell death (P < 0.01, n = 4) that was about half of that produced by NMDA. All experiments were performed in the presence of 1 μM TTX.
To determine the contribution of KARs to AMPA-evoked excitotoxicity, we compared cell death produced by AMPA (30 μM) in the absence and presence of CP-465,022. CP-465,022 is a potent, selective and non-competitive AMPA-receptor antagonist (IC50 at AMPA receptors = 15 nM) that produces minimal inhibition of KAR-mediated currents at concentrations below 1 μM (Lazzaro et al., 2002). At a concentration that has previously been reported to selectively block AMPA receptor-mediated currents (Lazzaro et al., 2002), CP-465,022 (300 nM) suppressed AMPA-induced cell death by 56 ± 11%, suggesting that ∼44% of cell death in these experiments was produced by KAR activation (Figure 4A, n = 6). To further confirm a neurotoxic role for KARs we used Con A, a plant lectin that selectively reduces KAR desensitization to potentiate KAR activity. In the presence of CP-465,022 (300 nM), AMPA (30 μM) produced significant LDH release (145 ± 9%, n = 21, P < 0.01). Application of Con A (0.3 mg/ml) to sister cultures significantly enhanced this AMPA-induced excitotoxicity (n = 21, P < 0.01; Figure 4B). These results are consistent with the interpretation that KAR activation is neurotoxic and that this neurotoxicity is limited by KAR desensitization. Excitotoxicity produced by the agonist AMPA in the presence of CP-465,022 and Con A was blocked by the AMPA and KA receptor antagonist CNQX (100 μM, n = 5; Figure 4B), further supporting a role for KARs in producing cell death in these experiments.
Figure 4.
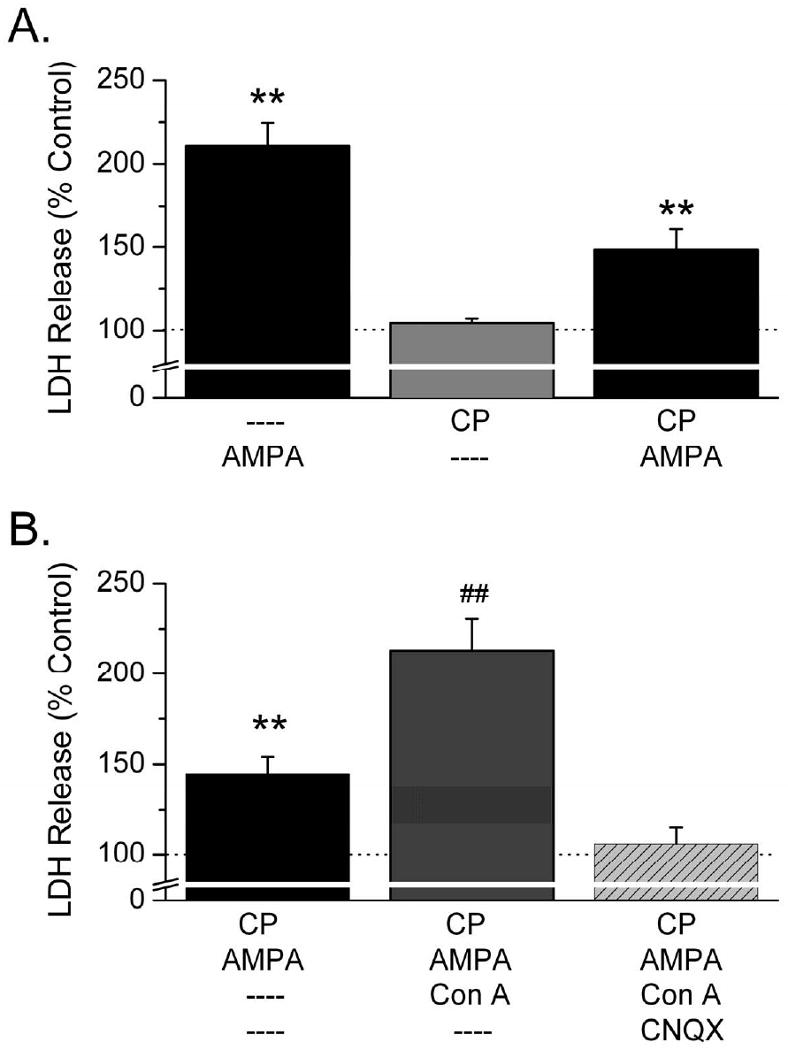
AMPA produces excitotoxicity by activating KARs. A. The selective AMPA-receptor antagonist CP-465,022 (300 nM) has no effect on LDH release when applied in the absence of agonist. However, cell death produced by AMPA in the presence of CP-465,022 is reduced by 56%, indicating that ∼44% of AMPA-mediated excitotoxicity is produced by KARs (n = 6). B. In the presence of CP-465,022 AMPA produces significant excitotoxicity (**P < 0.01) which is increased by 46 ± 8% (n = 21) in the presence of Con A and blocked by CNQX.
We then tested whether mAChR activation with pilocarpine would potentiate KAR-mediated cell death. KARs were isolated by the presence of CP-465,022 (300 nM), d-APV (100 μM) and TTX (1 μM) throughout the experiment. Cultures were incubated either with or without 100 μM pilocarpine for 30 min. In the absence of pilocarpine, 30 μM AMPA induced significant LDH release (137 ± 7%, n = 12, P < 0.01), indicating that KAR activation induced cell death. Following pilocarpine treatment, AMPA-induced excitotoxicity was significantly enhanced (200 ± 17%, n = 12, P < 0.01; Figure 5A). Cultures from which AMPA was omitted showed no more cell death than untreated controls, indicating that pilocarpine by itself was not toxic. These results strongly suggest that mAChR activation potentiates KAR-mediated neurotoxicity. Furthermore, the ability of pilocarpine treatment to enhance AMPA-induced excitotoxicity after pilocarpine washout indicates that muscarinic potentiation of KARs is long lasting.
Figure 5.

KAR-mediated excitotoxicity is potentiated by mAChR stimulation. A. LDH release produced by AMPA was measured after a 30 min treatment with either saline or pilocarpine. In saline pre-treated cultures AMPA induced a significant increase in LDH release in comparison to cultures not subjected to AMPA (**P < 0.01). However, the increase in LDH release was significantly greater when cultures were treated with pilocarpine prior to AMPA (n = 12, ##P < 0.01). Pilocarpine alone had no effect on LDH release. In a subset of these experiments we used sister cultures to test the effect of Con A on pilocarpine potentiation of AMPA-mediated excitotoxicity. Con A further potentiated AMPA-induced LDH release after both saline (#P < 0.05) and pilocarpine (+P < 0.05) pretreatment (n = 5). B. Pilocarpine produced a similar percentage increase in AMPA-induced LDH release in both the presence and absence of Con A. All experiments were performed in the presence of 1 μM TTX, 100 μM D-APV and 300 nM CP-465,022.
KAR desensitization limits excitotoxicity (Figure 4B). To determine if pilocarpine enhanced AMPA-induced cell death by reducing KAR desensitization, we compared pilocarpine potentiation of AMPA-induced excitotoxicity in the absence and presence of Con A in a subset of sister cultures. As previously observed (Figure 4B), Con A significantly enhanced AMPA-mediated neurotoxicity in this subset of sister cultures (n = 5, P < 0.05; Figure 5A). Pilocarpine potentiated AMPA-induced excitotoxicity to a similar degree both in the presence and absence of Con A (Figure 5B). This indicates that Con A did not occlude the actions of pilocarpine. These results suggest that mAChR activation does not potentiate KAR activity by reducing KAR desensitization. Furthermore, the ability of pilocarpine to potentiate excitotoxicity in the presence of Con A supports our conclusion that KARs contribute to AMPA-induced cell death and that pilocarpine treatment potentiates this KAR-mediated excitotoxicity.
3. Discussion
This study is the first to demonstrate mAChR potentiation of KAR currents. mAChR potentiation of GluK2-containing KARs requires the presence of GluK4 or GluK5 subunits; with GluK2/GluK5-containing receptors exhibiting the greatest potentiation (Figure 1C). mAChR potentiation of KARs is apparent in hippocampal mossy fibers (Figure 2), suggesting that these KARs contain GluK4 or GluK5 subunits. Finally, mAChR activation potentiates KAR-mediated excitotoxicity (Figures 3 – 5).
The ability of mAChR activation to potentiate KARs suggests a novel mechanism of KAR plasticity. Compared to AMPA and NMDA receptor-mediated neurotransmission, little is known about the plasticity of KAR EPSCs. Several studies have reported long term depression of KARs. For example, early in postnatal development at thalamocortical synapses, stimulation protocols that induce long term potentiation (LTP) of AMPA receptor-mediated responses, induce long term depression (LTD) of the KAR-mediated response (Kidd and Isaac, 1999). LTD of KAR EPSCs has also been reported in perirhinal cortical neurons (Park et al., 2006). At mossy fiber synapses on hippocampal CA3 neurons stimulation of NMDA receptors produces short term depression of KAR EPSCs (Rebola et al., 2007). In contrast to these studies showing depression of KAR EPSCs, Cho and colleagues found that mGluR5 activation produces a short term potentiation of KAR currents in perirhinal cortical neurons (Cho et al., 2003). These prior studies all report plasticity of KAR EPSCs through mechanisms involving activation of glutamate receptors. In contrast, we have found that activation of muscarinic receptors can also potentiate KAR currents. This interaction between mAChRs and KARs indicates that mechanisms regulating synaptic efficacy of KARs may be more diverse than previously considered.
We found that activation of mAChRs with pilocarpine enhances both KAR currents and KAR excitotoxicity. These findings are consistent with the idea that pilocarpine enhances excitotoxicity by potentiating KAR-mediated responses. The long-lasting nature of this potentiation enables pilocarpine to facilitate excitotoxicity even after it has been washed out. Although unlikely, an alternate mechanism could exist in which pilocarpine produces sub-lethal neuronal injury that persists after pilocarpine washout and sums with the AMPA-induced neurotoxicity.
KAR excitotoxicity was induced with AMPA. Excitotoxicity produced by AMPA in primary cortical culture has been previously reported (Koh et al., 1990; Jensen et al., 1998) and has been attributed to activation of AMPA receptors or indirect activation of NMDA receptors (Jensen et al., 1998). This study is the first to report a KAR-mediated component of neurotoxicity induced by the agonist AMPA; although KAR toxicity in response to other KAR agonists in primary cultured neurons is well established (Jensen et al., 1999; Giardina and Beart, 2001). Furthermore, other studies of AMPA-induced excitotoxicity do not rule out a KAR-mediated component. For example, Jensen et al. (Jensen et al., 1998) found that the AMPA-selective antagonist GYKI 53655 did not completely block AMPA-induced excitotoxicity in mature cortical cultures, leaving open the possibility of a KAR contribution.
The molecular mechanism by which mAChR activation potentiates KARs has not been elucidated. It is possible that muscarinic potentiation of KARs may be mediated by m1 or m3 receptors. Indeed, m1 receptors have been reported to potentiate both NMDA (Marino et al., 1998) and AMPA currents (Fernandez de Sevilla et al., 2008). Muscarinic m1 and m3 receptors act through Gq proteins to increase intracellular calcium and activate several kinases, including PKC (Felder, 1995), PKA (Beguin et al., 1996) and CaMKII (Muller et al., 1992). Each of these kinases has been reported to phosphorylate GluK1 or GluK2 subunits (Traynelis and Wahl, 1997; Hirbec et al., 2003; Hao et al., 2005). Phosphorylation of GluK4 or GluK5 subunits has not been reported.
PKC activation potentiates KAR EPSCs in perirhinal cortex (Park et al., 2006) and at mossy fiber synapses [(Hirbec et al., 2003) but see (Selak et al., 2009)]. mGluRs potentiate KARs through activation of PKC in perirhinal cortical neurons. mAChRs activate similar signaling cascades as mGluRs and may be expected to potentiate KARs through a similar mechanism. However, mGluR potentiation was different from that reported here because it was short-lived; occurring only in the presence of the mGluR agonist (Cho et al., 2003; Park et al., 2006). Alternately, mAChR potentiation of kainate receptor function may follow a different cascade. Muscarinic receptors induce long term potentiation of excitatory postsynaptic currents in hippocampal neurons by increasing AMPA receptor expression in dendritic spines (Fernandez de Sevilla et al., 2008). This effect requires activation of m1 mAChRs, production of IP3, and release of Ca2+ from IP3-sensitive stores by the activation of IP3 receptors. A similar mechanism may contribute to muscarinic potentiation of KARs. Further studies are needed to address these possibilities.
Although it is not known whether GluK1 or GluK3-containing KARs exhibit a similar interaction with muscarinic receptors, our results indicate that muscarinic potentiation of GluK2-containing KARs requires the presence of the GluK4 or GluK5 subunit. Addition of the GluK4 or GluK5 subunit to the GluK2 homomer has previously been shown to influence both its pharmacological and functional properties. For example, addition of GluK4 or GluK5 subunits increases agonist sensitivity (Barberis et al., 2008), increases channel conductance (Swanson et al., 1996), alters deactivation and desensitization rates in an agonist-dependent manner (Barberis et al., 2008; Mott et al., 2010) and increases sensitivity to a variety of modulators (Mott et al., 2003; Mott et al., 2008). The present study demonstrates that GluK4 and GluK5 subunits also enable regulation of KARs by mAChRs. Thus, GluK4 and GluK5 subunits serve important roles in regulating KAR function. Alterations in the expression of GluK4 or GluK5 subunits could influence the plasticity of KARs as well as the excitatory effect of mAChR activation.
GluK5 subunit expression is increased in the hippocampus in epilepsy (Mathern et al., 1998). An increase in the percentage of GluK5-containing KARs in this brain region could increase potentiation of the KAR current by mAChRs in epilepsy. The hippocampus is extensively innervated by cholinergic fibers arising from the medial septal nucleus (Dutar et al., 1995). Brain acetylcholine levels are dynamic and fluctuate greatly with changes in behavioral state and during the sleep-waking cycle (Marrosu et al., 1995; Acquas et al., 1996). In addition, acetylcholine levels increase right before seizures and remain elevated during seizures (Gardner and Webster, 1977; Jope and Gu, 1991). Results of this study suggest that increased acetylcholine before or during seizures may enhance neuronal excitability in part by potentiating KAR function. Overactivation of KARs could contribute to excitotoxicity and seizure development (Vincent and Mulle, 2009). Thus, endogenous elevations in ACh levels may enhance KAR function thereby contributing to hyperexcitability and excitotoxicity in epilepsy.
4. Experimental Procedures
4.1 Preparation and Recording from Hippocampal Slices
All procedures involving animals have been approved and monitored by the Institutional Animal Care and Use Committees of Emory University and the University of South Carolina. Transverse brain slices (500 μm thick) were prepared from isoflurane-anesthetized male Sprague-Dawley rats using a vibratome (Leica VT1000S). Brain slices were incubated in ACSF (artificial cerebrospinal fluid) containing (in mM): 120 NaCl, 3.3 KCl, 1.2 NaH2PO4, 25 NaHCO3, 10 glucose, 0.5 CaCl2, 5 MgCl2, bubbled with a 95% O2/5% CO2 gas mixture to pH 7.4 and stored at room temperature until use. Osmolarity was 300-305 mOsm.
For recording, slices were placed into a small submersion chamber maintained at 32 - 34° C and held in place by a bent piece of platinum wire resting on the surface of the slice. Slices were perfused with recording ACSF containing (in mM): 120 NaCl, 3.3 KCl, 1.2 NaH2PO4, 25 NaHCO3, 10 glucose, 1.5 CaCl2, 1.5 MgCl2, bubbled with a 95% O2/5% CO2 gas mixture at pH 7.4 (32-34°C). Osmolarity was 300-305 mOsm.
Stimuli were 0.1 ms, monophasic, cathodal, rectangular, constant current pulses (10 - 1000 μA) delivered through a monopolar platinum-irridium electrode referenced to a bath ground. Mossy fibers were stimulated either in the granule cell layer or in stratum lucidum of CA3c. For extracellular recording a glass pipette (resistance 5 MΩ) filled with recording ACSF was placed in the granule cell layer to record mossy fiber antidromic spikes. Responses were recorded using an Axopatch 1D amplifier and filtered at 1 kHz. Responses were digitized by a Digidata 1200 A-D board (Molecular Devices, Union City, CA) in a PC-based computer using pClamp 9 software.
4.2 Oocyte Preparation and Injection
Xenopus oocytes were prepared and injected as previously described (Mott et al., 2003). Briefly, stage V-VI oocytes were isolated from anesthetized frogs, enzymatically treated by gentle shaking with collagenase (Worthington, Type IV, 1.7 mg/ml for 45 - 120 min) in a calcium-free Barth's solution and then (in some cases) manually defolliculated. Cells were injected with up to 50 ng of mRNA transcribed from linearized constructs in either a pGEM-HE or pSGEM vector. For heteromeric receptors, mRNA was injected at a 1:3 ratio (GluK2:GluK4, GluK2:GluK5 or GluN1a:GluN2B). In experiments examining the effects of muscarinic receptors, m1 or m3 mRNA was injected with glutamate receptor mRNA at a 1:1 ratio (GluK2:m1) or 1:3:1 ratio (GluK2:GluK5:m1, GluK2:GluK5:m3, GluK2:GluK4:m1, GluK2:GluK4:m3 or GluN1a:GluN2B:m1). Injected oocytes were maintained at 17°C in Barth's solution containing gentamycin (100 μg/ml), penicillin (10 U/ml) and streptomycin (10 μg/ml) for 3 - 10 days, after which two electrode voltage clamp recordings were made at room temperature (23 - 25°C) from oocytes continually perfused in a standard frog Ringer's solution. This solution contained (in mM): 90 NaCl, 1 KCl, 15 HEPES, 0.4 BaCl2 and 0.1 MgCl2. Recording pipettes were filled with 3 M CsCl plus 0.4 M EGTA, pH 7.5 to chelate Ca2+ and thereby minimize the activation of calcium-dependent chloride currents. GluK2/GluK4 and GluK2/GluK5 were activated with S-AMPA (30 μM). AMPA selectively activates heteromeric kainate receptors containing GluK2 and GluK4 or GluK5 subunits, but not GluK2 homomeric receptors (Herb et al., 1992). Homomeric GluK2 receptors were activated by domoate (10 μM). NMDA receptors were activated using NMDA (100 μM) and glycine (10 μM). Currents were elicited from a holding potential of -70 mV. Agonist was delivered for 40-60 seconds per application. The interval between agonist pulses was set to be the time necessary for the current from the prior application to decay to baseline, but no shorter than 90 seconds (range: 90-120 sec). Steady state current amplitudes were measured. After at least 10 min of stable baseline currents, pilocarpine (100μM) was applied for two minutes and then washed off. Kainate or NMDA receptor steady state currents were measured for up to 2 hours after pilocarpine washout, but specifically at the 30 min time point. Current signals were digitized at 1 kHz using a Digidata 1200 analogue to digital converter (Axon Instruments). Application of each of the agonists produced a stable, rapidly-rising and non-desensitizing or weakly desensitizing current in the majority of oocytes. Oocytes in which the current was not stable or in which the baseline holding current drifted by more than 10% were discarded.
4.3 In Vitro Excitotoxicity
Primary cultures of rat cerebral cortex were prepared from Sprague-Dawley rat embryos (E16–E19) essentially as described (Mott et al., 1998). Cells were plated into 24-well plates at a density of 3 × 105 per well. Plating medium consisted of Neurobasal medium supplemented with L-glutamine (2 mM), penicillin (5 U/ml), streptomycin (10 g/ml) and B-27 supplement. Cells were maintained in a humidified incubator at 37°C and 5% CO2 and fed every four days with plating medium. After 14–21 days in culture, excitotoxicity was induced by treating cultures with s-AMPA (1 - 30 μM) in the presence of d-APV (100 μM) and tetrodotoxin (1 μM) for 24 hours at 37°C. Concanavalin A (Con A; 0.3 mg/ml), CNQX (100 μM) or CP-465,022 (300 nM) was added with AMPA as indicated. NMDA-evoked excitotoxicity was induced by 24 hour exposure of the cultures to NMDA (30 μM) and glycine (10 μM) in the presence of TTX (1 μM). After 24 hours, excitotoxic damage produced by AMPA or NMDA was assessed by measuring the amount of lactate dehydrogenase (LDH) released into the culture medium (Cytotoxicity Detection Kit; Roche Applied Science, Indianapolis, IN). Released LDH was expressed as the fraction of total LDH present in each well. AMPA-evoked excitotoxicity was defined as the amount of LDH released by AMPA as a percentage of LDH released in the absence of AMPA. All experiments were performed in quadruplicate from sister cultures and comparisons were made between wells on the same 24 well culture plate. Cultures in which the AMPA-evoked excitotoxic cell death was less than 10% were discarded.
The effect of muscarinic receptor activation on AMPA-induced excitotoxicity was assessed by treating cultures with pilocarpine (100 μM) in the presence of d-APV (100 μM) and tetrodotoxin (1 μM) for 30 min. Cultures were then rinsed with Neurobasal medium and AMPA-evoked excitotoxicity induced and measured as described above.
4.4 Data Analysis
Analysis was performed using pClamp (Axon Instruments), Origin (MicroCal, Northampton, MA) and Prism (GraphPad, San Diego, CA) software packages. Statistical comparisons were performed using the appropriate Student's t test or ANOVA with post hoc tests. Values are given as mean ± SE.
4.5 Materials
GluK2 mRNA in pGEM-HE and the pSGEM vector were a generous gift from M. Mayer (National Institute of Child Health and Human Development, Bethesda, MD). GluK4 and GluK5 plasmids were generously provided by S. Heinemann (Salk Institute for Biological Studies, La Jolla, CA). GluN1 and GluN2 were generously provided by S. Nakanishi (Osaka Bioscience Institute, Osaka, Japan). CP-465,022 was a gift from F. Menniti (Pfizer, Groton, CT). Kainate, carbachol, pilocarpine and glycine were purchased from Sigma Chemical Company (St. Louis, MO). Domoate, s-AMPA, d-APV, bicuculline methochloride, CNQX, GYKI 52466, TTX and NMDA were purchased from Tocris Bioscience (Ellisville, MO). All tissue culture reagents were obtained from Invitrogen (Carlsbad, CA).
Acknowledgments
The authors would like to thank Renee Shaw for expert technical assistance in cRNA production and oocyte preparation. This work was supported by grants from the National Institute of Neurological Disease and Stroke [Grants NS065869 to DDM, NS055883 to MB, NS036604 to RJD]; the Epilepsy Foundation (DDM) and NARSAD (DDM).
Abbreviations
- ACh
acetylcholine
- AChR
acetylcholine receptor
- ACSF
artificial cerebrospinal fluid
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionate
- AMPAR
AMPA receptor
- ANOVA
analysis of variance
- D-APV
D-(-)-2-Amino-5-phosphonopentanoic acid
- CCh
carbachol
- CNQX
6-Cyano-7-nitroquinoxaline-2,3-dione
- CP-465,022
3-(2-Chlorophenyl)-2-[2-[6-[(diethylamino)methyl]-2-pyridinyl]ethenyl]-6-fluoro-4(3H)-quinazolinone hydrochloride
- Con A
concanavalin A
- EPSC
excitatory postsynaptic current
- GluK1-3
kainate GluR5-7 subunits
- GluK4-5
kainate KA1-2 subunits
- GluN1
NMDA NR1 subunit
- GluN2B
NMDA NR2B subunits
- GYKI 52466
4-(8-Methyl-9H-1,3-dioxolo[4,5-h][2,3]benzodiazepin-5-y l)-benzenamine hydrochloride
- KAR
kainate receptor
- LDH
lactate dehydrogenase
- mAChR
muscarinic acetylcholine receptor
- MF
mossy fiber
- NMDA
N-methyl-D-aspartate
- NMDAR
NMDA receptor
- PKC
protein kinase C
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acquas E, Wilson C, Fibiger HC. Conditioned and unconditioned stimuli increase frontal cortical and hippocampal acetylcholine release: effects of novelty, habituation, and fear. J Neurosci. 1996;16:3089–3096. doi: 10.1523/JNEUROSCI.16-09-03089.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach JM, Segal M. A novel cholinergic induction of long-term potentiation in rat hippocampus. J Neurophysiol. 1994;72:2034–2040. doi: 10.1152/jn.1994.72.4.2034. [DOI] [PubMed] [Google Scholar]
- Bah J, Quach H, Ebstein RP, Segman RH, Melke J, Jamain S, Rietschel M, Modai I, Kanas K, Karni O, Lerer B, Gourion D, Krebs MO, Etain B, Schurhoff F, Szoke A, Leboyer M, Bourgeron T. Maternal transmission disequilibrium of the glutamate receptor GRIK2 in schizophrenia. Neuroreport. 2004;15:1987–1991. doi: 10.1097/00001756-200408260-00031. [DOI] [PubMed] [Google Scholar]
- Barberis A, Sachidhanandam S, Mulle C. GluR6/KA2 kainate receptors mediate slow-deactivating currents. J Neurosci. 2008;28:6402–6406. doi: 10.1523/JNEUROSCI.1204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguin P, Beggah A, Cotecchia S, Geering K. Adrenergic, dopaminergic, and muscarinic receptor stimulation leads to PKA phosphorylation of Na-K-ATPase. Am J Physiol. 1996;270:C131–C137. doi: 10.1152/ajpcell.1996.270.1.C131. [DOI] [PubMed] [Google Scholar]
- Blokland A. Acetylcholine: a neurotransmitter for learning and memory? Brain Res Brain Res Rev. 1995;21:285–300. doi: 10.1016/0165-0173(95)00016-x. [DOI] [PubMed] [Google Scholar]
- Cho K, Francis JC, Hirbec H, Dev K, Brown MW, Henley JM, Bashir ZI. Regulation of kainate receptors by protein kinase C and metabotropic glutamate receptors. J Physiol. 2003;548:723–730. doi: 10.1113/jphysiol.2003.040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutar P, Bassant MH, Senut MC, Lamour Y. The septohippocampal pathway: structure and function of a central cholinergic system. Physiol Rev. 1995;75:393–427. doi: 10.1152/physrev.1995.75.2.393. [DOI] [PubMed] [Google Scholar]
- Felder CC. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J. 1995;9:619–625. [PubMed] [Google Scholar]
- Fernandez de Sevilla D, Nunez A, Borde M, Malinow R, Buno W. Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons. J Neurosci. 2008;28:1469–1478. doi: 10.1523/JNEUROSCI.2723-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner CR, Webster RA. Convulsant-anticonvulsant interactions on seizure activity and cortical acetylcholine release. Eur J Pharmacol. 1977;42:247–256. doi: 10.1016/0014-2999(77)90291-6. [DOI] [PubMed] [Google Scholar]
- Giardina SF, Beart PM. Excitotoxic profiles of novel, low-affinity kainate receptor agonists in primary cultures of murine cerebellar granule cells. Neuropharmacology. 2001;41:421–432. doi: 10.1016/s0028-3908(01)00086-7. [DOI] [PubMed] [Google Scholar]
- Hao ZB, Pei DS, Guan QH, Zhang GY. Calcium/calmodulin-dependent protein kinase II (CaMKII), through NMDA receptors and L-Voltage-gated channels, modulates the serine phosphorylation of GluR6 during cerebral ischemia and early reperfusion period in rat hippocampus. Brain Res Mol Brain Res. 2005;140:55–62. doi: 10.1016/j.molbrainres.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Herb A, Burnashev N, Werner P, Sakmann B, Wisden W, Seeburg PH. The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron. 1992;8:775–785. doi: 10.1016/0896-6273(92)90098-x. [DOI] [PubMed] [Google Scholar]
- Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Coussen F, Mulle C, Dev KK, Coutinho V, Meyer G, Isaac JT, Collingridge GL, Henley JM. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron. 2003;37:625–638. doi: 10.1016/s0896-6273(02)01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JB, Schousboe A, Pickering DS. AMPA receptor mediated excitotoxicity in neocortical neurons is developmentally regulated and dependent upon receptor desensitization. Neurochem Int. 1998;32:505–513. doi: 10.1016/s0197-0186(97)00130-7. [DOI] [PubMed] [Google Scholar]
- Jensen JB, Schousboe A, Pickering DS. Role of desensitization and subunit expression for kainate receptor-mediated neurotoxicity in murine neocortical cultures. J Neurosci Res. 1999;55:208–217. doi: 10.1002/(SICI)1097-4547(19990115)55:2<208::AID-JNR8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Jope RS, Gu X. Seizures increase acetylcholine and choline concentrations in rat brain regions. Neurochem Res. 1991;16:1219–1226. doi: 10.1007/BF00966699. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S. Kainate receptor-mediated presynaptic inhibition at the mouse hippocampal mossy fibre synapse. J Physiol. 2000;523(Pt 3):653–665. doi: 10.1111/j.1469-7793.2000.t01-1-00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Goldberg MP, Hartley DM, Choi DW. Non-NMDA receptor-mediated neurotoxicity in cortical culture. J Neurosci. 1990;10:693–705. doi: 10.1523/JNEUROSCI.10-02-00693.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larm JA, Beart PM, Cheung NS. Neurotoxin domoic acid produces cytotoxicity via kainate- and AMPA-sensitive receptors in cultured cortical neurones. Neurochem Int. 1997;31:677–682. doi: 10.1016/s0197-0186(97)00030-2. [DOI] [PubMed] [Google Scholar]
- Lazzaro JT, Paternain AV, Lerma J, Chenard BL, Ewing FE, Huang J, Welch WM, Ganong AH, Menniti FS. Functional characterization of CP-465,022, a selective, noncompetitive AMPA receptor antagonist. Neuropharmacology. 2002;42:143–153. doi: 10.1016/s0028-3908(01)00170-8. [DOI] [PubMed] [Google Scholar]
- Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci. 2003;4:481–495. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci USA. 1998;95:11465–11470. doi: 10.1073/pnas.95.19.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Segal M. The inositol 1,4,5-trisphosphate pathway mediates cholinergic potentiation of rat hippocampal neuronal responses to NMDA. J Physiol. 1992;447:513–533. doi: 10.1113/jphysiol.1992.sp019015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrosu F, Portas C, Mascia MS, Casu MA, Fa M, Giagheddu M, Imperato A, Gessa GL. Microdialysis measurement of cortical and hippocampal acetylcholine release during sleep-wake cycle in freely moving cats. Brain Res. 1995;671:329–332. doi: 10.1016/0006-8993(94)01399-3. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Kornblum HI, Mendoza D, Lozada A, Leite JP, Chimelli L, Born DE, Fried I, Sakamoto AC, Assirati JA, Peacock WJ, Ojemann GA, Adelson PD. Altered hippocampal kainate-receptor mRNA levels in temporal lobe epilepsy patients. Neurobiol Dis. 1998;5:151–176. doi: 10.1006/nbdi.1998.0200. [DOI] [PubMed] [Google Scholar]
- Mott DD, Benveniste M, Dingledine RJ. pH-dependent inhibition of kainate receptors by zinc. J Neurosci. 2008;28:1659–1671. doi: 10.1523/JNEUROSCI.3567-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, Traynelis SF, Dingledine R. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- Mott DD, Rojas A, Fisher JL, Dingledine RJ, Benveniste M. Subunit-specific desensitization of heteromeric kainate receptors. J Physiol. 2010;588:683–700. doi: 10.1113/jphysiol.2009.185207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott DD, Washburn MS, Zhang S, Dingledine RJ. Subunit-dependent modulation of kainate receptors by extracellular protons and polyamines. J Neurosci. 2003;23:1179–1188. doi: 10.1523/JNEUROSCI.23-04-01179.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W, Petrozzino JJ, Griffith LC, Danho W, Connor JA. Specific involvement of Ca(2+)-calmodulin kinase II in cholinergic modulation of neuronal responsiveness. J Neurophysiol. 1992;68:2264–2269. doi: 10.1152/jn.1992.68.6.2264. [DOI] [PubMed] [Google Scholar]
- O'Neill MJ, Bogaert L, Hicks CA, Bond A, Ward MA, Ebinger G, Ornstein PL, Michotte Y, Lodge D. LY377770, a novel iGlu5 kainate receptor antagonist with neuroprotective effects in global and focal cerebral ischaemia. Neuropharmacology. 2000;39:1575–1588. doi: 10.1016/s0028-3908(99)00250-6. [DOI] [PubMed] [Google Scholar]
- Park Y, Jo J, Isaac JT, Cho K. Long-term depression of kainate receptor-mediated synaptic transmission. Neuron. 2006;49:95–106. doi: 10.1016/j.neuron.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Rebola N, Sachidhanandam S, Perrais D, Cunha RA, Mulle C. Short-term plasticity of kainate receptor-mediated EPSCs induced by NMDA receptors at hippocampal mossy fiber synapses. J Neurosci. 2007;27:3987–3993. doi: 10.1523/JNEUROSCI.5182-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Frerking M, Nicoll RA. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 2000;27:327–338. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- Selak S, Paternain AV, Aller IM, Pico E, Rivera R, Lerma J. A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron. 2009;63:357–371. doi: 10.1016/j.neuron.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Feldmeyer D, Kaneda M, Cull-Candy SG. Effect of RNA editing and subunit co-assembly single-channel properties of recombinant kainate receptors. J Physiol. 1996;492(Pt 1):129–142. doi: 10.1113/jphysiol.1996.sp021295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wahl P. Control of rat GluR6 glutamate receptor open probability by protein kinase A and calcineurin. J Physiol. 1997;503(Pt 3):513–531. doi: 10.1111/j.1469-7793.1997.513bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent P, Mulle C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience. 2009;158:309–323. doi: 10.1016/j.neuroscience.2008.02.066. [DOI] [PubMed] [Google Scholar]