

Abstract
Erythrocyte ghosts prepared from fresh blood expressed phosphatidylserine (PS) on the membrane surfaces in a rather stable fashion. The binding of fluorescein-5-isothiocyanate (FITC)-labeled Annexin V (ANV) derivatives to these membranes were studied by titration with proteins and with calcium. Whereas pre-addition of EDTA to reaction mixtures totally prevented membrane binding, Ca++-dependent binding was only partially reversed by EDTA treatment, consistent with an initial Ca++ dependent binding which became partially Ca++ independent. Data derived from saturation titration with ANV derivatives poorly fit simple protein-membrane equilibrium binding equation and showed negative cooperativity of binding with increasing membrane occupancy. In contrast, calcium titration at low binding site occupancy resulted in excellent fit into protein-Ca++-membrane equilibrium binding equation. Calcium titrations of FITC-labeled ANV and ANV-6L15 (a novel ANV-Kunitz protease inhibitor fusion protein) yielded Hill coefficient of approximately 4 in both cases. The apparent dissociation constant for ANV-6L15 was about 4-fold lower than that of ANV at 1.2–2.5 mM Ca++. We propose that ANV-6L15 may provide improved detection of PS exposed on the membrane surfaces of pathological cells in vitro and in vivo.
Keywords: Annexin V, phosphatidylserine, erythrocyte, fluorescein-5-isothiocyanate
INTRODUCTION
Phosphatidylserine (PS3) is one of the four major phospholipids in the plasma membranes of mammalian cells, comprising 8–15 % of the total phospholipids content. In normal cells, PS is exclusively sequestered in the inner layer of the plasma membrane together with most of the phosphatidylethanolamine (PE), whereas the outer layer is mainly composed of phosphatidylcholine (PC) and sphingomyelin [1–3]. Apoptotic cell death, cell senescence, cell activation, oxidative stress, and cell damage can all lead to exposure of PS on cell membrane surfaces and shed microparticles. Ability to detect PS exposure on pathological cells can provide important information in the diagnosis and treatment of various diseases [1].
The Annexins are a family of proteins that share the property of calcium-dependent binding to PS-expressing membranes [4–6]. Annexin V (ANV) is a typical member of the family which has been widely used for detection of PS-expressing cells by confocal microscopy and flow cytometry [7–11]. ANV is also being developed as a diagnostic agent to detect cell death in vivo in cancer chemotherapy, organ transplant rejection, and myocardial infarction [12–15]. Early work showed that ANV bound to model membranes containing 20 % PS-80 % PC with an estimated dissociation constant (Kd) of < 10−10 M at physiological concentration of Ca++ [16,17]. Subsequent studies of ANV binding to various cell types by classical saturation titration assays had produced widely different Kd values ranging from 2.1 × 10−11M to 2.5 × 10−8 M [18–22]. Thus, the real affinity of ANV binding to PS-expressing cells remained imprecisely defined. Tait et al. recently developed a calcium titration method for the measurement of the affinity and cooperativity of ANV-Ca++-membrane binding [23]. The binding of ANV to preservative-treated blood cells was titrated with Ca++ such that < 3% of the membrane binding sites was occupied throughout the titration. This experimental approach circumvented the problems of classical saturation titration in which heterogeneous binding events might occur due to acidic phospholipid segregation [24–26], protein clustering [27,28], and alterations in membrane shape and rigidity [29,30] at high membrane occupancy. Using this method, Tait et al. obtained a drastically different set of binding parameters by non-linear least squares fit of the equilibrium binding equation. However, this original calcium titration method estimated the membrane-bound ANV after washing of cells and treatment with EDTA to release the bound ANV. It was not clear whether cell washing significantly perturbed the binding equilibrium and whether EDTA released the bound ANV completely. In an effort to establish valid methods for quantifying the affinity constants of various ANV derivatives for cell membranes, we revisited the issues and investigated the binding of ANV derivatives to erythrocyte ghosts by classical saturation titration assay and by a modified calcium titration method. We found that erythrocyte ghosts prepared from fresh blood appeared to offer significant advantages over other cell systems since these membranes express PS at higher levels and in a more stable fashion. We discovered that Ca++ dependent binding of ANV derivatives to erythrocyte ghosts was abolished by co-treatment with EDTA, but was only partially reversed by post-treatment with EDTA. This new finding necessitated a modification of the original calcium titration method to measure the membrane-bound ANV derivatives. We further showed that saturation titration data poorly fit simple protein-membrane equilibrium binding equation. In contrast, calcium titration at low membrane binding site occupancy (≤ 2% saturation) provided excellent fit of the ANV-Ca++-membrane equilibrium binding equation and allowed us to calculate various binding parameters. Using this new assay system, we compared the binding parameters of ANV with those of ANV-6L15, a fusion protein consisting of an ANV domain and a Kunitz-type protease inhibitor domain that inhibited tissue factor/factor VIIa with high potency [31]. We found that the Kd for ANV-6L15 was about 4-fold lower than that of ANV at physiological concentrations of ionized Ca++, suggesting that ANV-6L15 bound to PS-expressing cells with stronger affinity than ANV.
MATERIALS AND METHODS
Expression, purification, and FITC labeling of recombinant ANV and ANV-6L15
E. coli BL21(DE3)pLysS and the expression vector pET20b(+) (Novagen, Medison, WI) were used for the expression of recombinant ANV and ANV-6L15, and the recombinant proteins were purified as described before [31]. The purified proteins were labeled with FITC (Pierce, Rockford IL) by the following protocol: ANV or ANV-6L15 (50 μM) was incubated with FITC (250 μM) for 1 h at room temperature (r.t.) in 100 mM Na-borate, pH 9.0. The reaction mixture (1 ml) was quenched by adding 0.1 ml of 1 M glycine and dialyzed extensively against TBS buffer (20 mM Tris, pH 7.4, 150 mM NaCl). The labeled proteins were quantitated by Bradford protein assay (BioRad, Hercules CA) using unlabeled proteins as standards and the amount of fluorescein quantitated by absorbance reading at 494 nm using E494 =80,000. This procedure resulted in FITC:protein (F:P) labeling ratios of 0.37 and 0.76 mol/mol for ANV-FITC and ANV-6L15-FITC, respectively, and the conjugates were designated by subscripts as ANV-FITC0.37 and ANV-6L15-FITC0.76. ANV-FITC with higher F:P ratios (1.3 and 2.4) were obtained by labeling ANV with 1000 μM FITC or N-hydroxysuccinimide-fluorescein. Calcium titrations using ANV-FITC1.3 yielded similar binding parameters as those using ANV-FITC0.37. However, titrations using ANV-FITC2.4 showed higher Kd values, suggesting that the membrane binding affinity decreased when the F:P ratio increased to 2.4. ANV-FITC0.37 and ANV-6L15-FITC0.76 were used for calcium titration in this study.
Preparation of erythrocyte ghosts
Erythrocyte ghosts were prepared from ACD blood or preserved blood (4C Plus Control or 5C control, Beckman-Coulter, Miami, FL) by repeated hypo-osmotic lysis. Ten ml of blood was dispensed into 2-ml microfuge tubes in 0.5 ml aliquots and centrifuged at 13,000 rpm for 5 min at r.t. The supernatant plasma was discarded. De-ionized water (1.8 ml) was then added to each tube containing the cell pellet. The tubes were vortexed for 50 sec., incubated in a 37°C bath for 45 min, and centrifuged at 13,000 rpm for 5 min at r.t. TBSA (TBS buffer containing 1 mg/ml BSA and 0.02% NaN3) was added to each cell pellet to a final volume of 2 ml to re-suspend the cells. The tubes were vortexed, incubated at 37°C, and centrifuged as above. De-ionized water (1.8 ml) was added to each cell pellet to lyse the cells again. The tubes were vortexed, incubated at 37°C, and centrifuged as above. TBSA (1.8 ml) was then added to each cell pellet to re-suspend the cells. The cells were then left at 4 °C overnight. After centrifugation, the cell ghosts were pooled, re-suspended in a final volume of 10 ml TBSA, and stored at 4°C. A Coulter Z1 dual threshold model was used to determine cell count using the setup [TU35fl, TL30fl, >TU] for red cell ghosts. Stocks of erythrocyte ghosts (2.8 × 108 cells/ml) were stored at 4 °C in TBSA. The cell counts of the stocks did not change over a 3-month storage period.
Confocal microscopy of preserved blood cells and erythrocyte ghosts
4C Plus Control, 5C Control, and erythrocyte ghosts were diluted 200 fold into HBSA buffer [10 mM Hepes, pH 7.4, 137 mM NaCl, 4 mM KCl, 0.5 mM MgCl2, 0.5 mM NaH2PO4, 0.1 % D(+)-glucose, 0.02 % NaN3, 0.1 % BSA] containing 2.5 mM CaCl2 and 50 nM ANV-FITC. The mixtures were incubated at r.t. for 1 h followed by centrifugation at 13,000 rpm for 3 min to pellet the cells. The cell pellets were washed once by suspending in an equal volume of HBSA-2.5 mM CaCl2. The washed cell samples were drop onto Lab-Tek 8-chamber slides (Nunc Inc. Rochester, NY) and sealed with a coverslip and nail polish. The samples were examined under a Olympus confocal laser scanning microscope (FV1000) using a 20× objective lens and excitation/emission filters 488/510–560 nm.
Flow cytometric analysis of preserved blood cells and erythrocyte ghosts
5C Control cells or erythrocyte ghosts were diluted to about 5 × 106 cells/ml in HBSA buffer containing 2.5 mM CaCl2 and 50 nM ANV-FITC. The samples were incubated at r.t. for 30 min and aliquots were aspirated into a FACSCalibur (Becton-Dikinson) for flow cytometric analysis (excitation 488 nm, emission filter 530±30 nm).
Saturation binding of ANV derivatives to erythrocyte ghosts
A 1:200 dilution of the ghosts was mixed with 1–256 nM FITC-labeled ANV derivatives at 2-fold increasing concentrations in 1.5-ml microfuge tubes containing 0.4 ml of TBSA buffer supplemented with 1.25 mM, 2.5 mM or 5 mM CaCl2. A separate set of microfuge tubes contained 0.4 ml of the same mixtures but were supplemented with 50 μl of 0.5 M EDTA to prevent the binding of FITC-labeled proteins to the erythrocyte ghosts. After incubation for 40 min at r.t., all the reaction mixtures were centrifuged at 13,000 rpm for 10 min to separate the free- and the cell-bound FITC-labeled proteins. Duplicate samples of supernatants (160 μl from mixtures with Ca++ and 180 μl from mixtures with EDTA) were transferred into a black 96-well plate. The wells with 160 μl supernatants were added 20 μl of 0.5 M EDTA so that all the sample wells had the same buffer composition. Fluorescence measurements were carried out using a fmax microplate reader (Molecular Device, Sunnyvale, CA) with excitation/emission at 485/538 nm. Fluorescence was read 4 times and the average values were used for calculating the concentrations of free- and bound- FITC-labeled ligands.
The binding reaction between the proteins and the membrane binding sites was analyzed according to the following model:
| eq. (1) |
where Kd was the equilibrium constant of the dissociation reaction.
Equation (1) could be transformed into equation (2)
| eq. (2) |
where [Protein]bound was the concentration of protein bound to the membrane, [Protein]free was the concentration of free protein, and Bmax was the maximum concentration of membrane-bound protein at saturation. Non-linear least-squares analysis using the Solver function of Microsoft Excel [32] was performed to determine the fit of equation (2).
Effects of Ca++ and EDTA on the binding of ANV-FITC and ANV-6L15-FITC to erythrocyte ghosts
The distribution of fluorescence after incubation of erythrocyte ghosts with ANV-FITC or ANV—6L15-FITC was determenied as follows. Erythrocyte ghosts (7.1×106 cells/ml in TBSA-1.25mM CaCl2) were centrifuged at 13,000 rpm for 10 min. Aliquot of the supernatant (380 μl) was mixed with 20 μl of 40 nM ANV-FITC or ANV-6L15-FITC so that the final concentration of ANV-FITC was 2 nM. Duplicate samples (160 μl) of the supernatant were each mixed with 20 μl of 0.5 M EDTA for fluorescence reading. The reading was taken as 100% fluorescence in solution phase control. ANV-FITC or ANV-6L15-FITC (2 nM) and erythrocyte ghosts (7.1×106 cells/ml) in 400 μl TBSA-1.25 mM CaCl2 was mixed with 50 μl of 0.5 M EDTA, incubated for 40 min at r.t., and centrifuged at 13,000 rpm for 10 min. Duplicate samples (180 μl) of the supernatant were taken for fluorescence reading. The reading was taken as the fluorescence remained in the solution phase after incubation in the presence of EDTA. A mixutre of ANV-FITC or ANV-6L15-FITC (2 nM) and erythrocyte ghosts (7.1×106 cells/ml) in 400 μl of TBSA-1.25 mM CaCl2 was incubated at r.t. for 40 min (c), 80 min (d), 4 h (e), and 22 h (f), respectively. The reaction mixtures were centrifuged as above and duplicate samples (160 μl) of the supernatant were each mixed with 20 μl of 0.5 M EDTA for fluorescence reading. The reading was taken as the unbound flourescence. Each cell pellet was re-suspended in 400 μl TBSA plus 50μl 0.5 M EDTA, incubated and centrifuged as above. Duplicate samples (180 μl) of the supernatant were taken for fluorescence reading. The reading was taken as the flourescence in the EDTA-extracted phase. The flourecence in the EDTA-resistant membrane phase was calulated by sustracting the flourescence in solution phase and flourescence in EDTA extracted phase from the 100% fluorescence in solution phase control.
Calcium titration assay: The binding model
The binding reaction between calcium ions, ANV derivatives, and membrane binding sites was analyzed according to the model described by Tait et al. [23].
| eq. (3) |
where K was the equilibrium constant of the dissociation reaction.
When the concentrations of free and bound protein were equal, and Equation (3) reduced to
| eq. (4) |
| eq. (5) |
where EC50 is the free calcium concentration at which half of the protein was bound to the membrane and [Membrane] was the concentration of membrane binding sites [m]. [m] could be estimated by measuring the amount of ANV derivatives bound at saturating concentration of Ca++ (15 mM). If the reaction were highly cooperative with respect to calcium, the Hill coefficient (N) would be nearly the same as the calcium binding stoichiometry n. The binding parmeters EC50 and N could be determined by fitting the calcium titration data to the following equation, which could be derived from Equations (3) and (4):
| eq. (6) |
where B is the amount of protein bound at a given calcium concentration, Bmax is the amount of protein bound at saturating calcium concentrations. Fits were performed by non-linear least-squares analysis using the Solver function of Microsoft Excel [32]. The apparent dissociation constant at a fixed calcium concentration, Kapp, can be calculated from the following equation: .
Calcium titration was carried out as follows. Stock solutions of TBSA, TBSA-8 mM CaCl2, TBSA-40 nM ANV- FITC or TBSA-40 nM ANV-6L15-FITC, and TBSA-erythrocyte ghosts were mixed in appropriate proportions in sixteen 1.5-ml microfuge tubes such that each reaction mixture contained 0.4 ml TBSA, 2 nM FITC-proteins, 7.1 × 106 cells/ml erythrocyte ghosts, and varied concentrations of CaCl2 (0–3 mM). Under this reaction condition, less than 2.1 % of membrane binding sites [m] were occupied throughout titration. A separate set of microfuge tubes contained 0.4 ml of the same mixtures but were supplemented with 50 μl of 0.5 M EDTA to prevent the binding of FITC-labeled proteins to erythrocyte ghosts. After incubation for 40 min at r.t., all the reaction mixtures were centrifuged at 13,000 rpm for 10 min to separate the free- and the cell-bound FITC-labeled proteins. Duplicate samples of supernatants (160 μl from mixtures with Ca++ and 180 μl from mixtures with EDTA) were transferred into a black 96-well plate. The wells with 160 μl supernatants were added 20 μl of 0.5 M EDTA so that all the sample wells had the same buffer composition. Fluorescence measurements were carried out using a SpectraMax® M5 microplate reader (Molecular Device, Sunnyvale, CA) with excitation/emission/cutoff at 485/538/530 nm). Fluorescence was read 4 times and the average values were used for calculating the B/Bmax values according to the following equation:
where FE was the fluorescence intensity remained in the supernatant of EDTA-supplemented reaction mixtures; FCa was the fluorescence intensity remained in the supernatants of reaction mixtures with varying concentration of Ca++; and Fm was the fluorescence intensity remained in the supernatants of reaction mixtures that showed maximal binding. Under the above experimental condition, maximal binding was reached at 1.5–3 mM Ca++ for both ANV-FITC and ANV-6L15-FITC.
RESULTS
Comparison of the binding of ANV-FITC to preservative-treated blood cells and erythrocyte ghosts
A preservative-treated human blood, 4C Plus Cell Control (Beckman-Coulter) was proposed for routine study of ANV-Ca++-membrane binding [23]. We compared the binding of ANV-FITC to 4C Plus Control, 5C Control (Beckman-Coulter), and erythrocyte ghosts prepared from preservative-treated blood and freshly collected ACD blood using fluorescence confocal microscopy. Fig. 1(A) showed that relatively few blood cells in un-expired 4C Plus Control were fluorescently labeled by ANV-FITC, suggesting that only a small number of erythrocytes in the sample expressed PS. Fig. 1(B) showed that a larger percentage of erythrocytes were fluorescently labeled by ANV-FITC after 5C Control had been stored at 4 °C for 6 months. Fig. 1(C) showed that erythrocyte ghosts prepared from 5C Control were fluorescently labeled by ANV-FITC more intensely than those in Fig. (A) and (B). However, many fluorescently labeled microparticles, possibly derived from fragmented cells, were present. Fig. 1(D) showed that virtually all the erythrocyte ghosts prepared from fresh ACD blood were fluorescently labeled by ANV-FITC, and very few cell fragments were present. Flow cytometric analysis of various preparations of blood cells and ghosts was also carried out. Fig. 2(A) showed that 5C Control that had been aged 11 months had a normal FSC-H/SSC-H plot (erythrocytes in R1; lymphocytes in R2; neutrophils in R3) [panel a], and had moderate cell-associated fluorescence in 2.5 mM Ca++ [panel b] and low cell-associated fluorescence in 1.25 mM Ca++ [panel c]. Fig. 2(B) showed that cell ghosts prepared from 5C Control that had been aged 1 month had large amount of cell fragments in FSC-H/SSC-H plot [panel a], and had high cell-associated fluorescence in 2.5 mM Ca++ [panel b] and moderate cell-associated fluorescence in 1.25 mM Ca++ [panel c]. Fig. 2(C) showed that cell ghosts prepared from 5C Control that had been aged 11 month had even larger amount of cell fragments in FSC-H/SSC-H plot [panel a], and had moderate cell-associated fluorescence in 2.5 mM Ca++ [panel b] and very low cell-associated fluorescence in 1.25 mM Ca++ [panel c]. Fig. 2(D) showed that ghosts prepared from fresh ACD blood had very few cell fragments in FSC-H/SSC-H plot [panel a], and had higher cell-associated fluorescence [panels b and c] than those in Fig. 2(A), (B), and (C). Thus, results from fluorescence confocal microscopy and flow cytometry analysis together indicated that erythrocyte ghosts prepared from fresh ACD blood expressed PS to higher levels and were less heterogeneous in size and binding of ANV-FITC compared with preserved blood cells and ghosts prepared from them. Therefore, erythrocyte membranes prepared from fresh blood appeared better suited for ANV binding studies.
Fig. 1. Imaging of ANV-FITC-treated blood cells and ghosts using confocal microscopy.
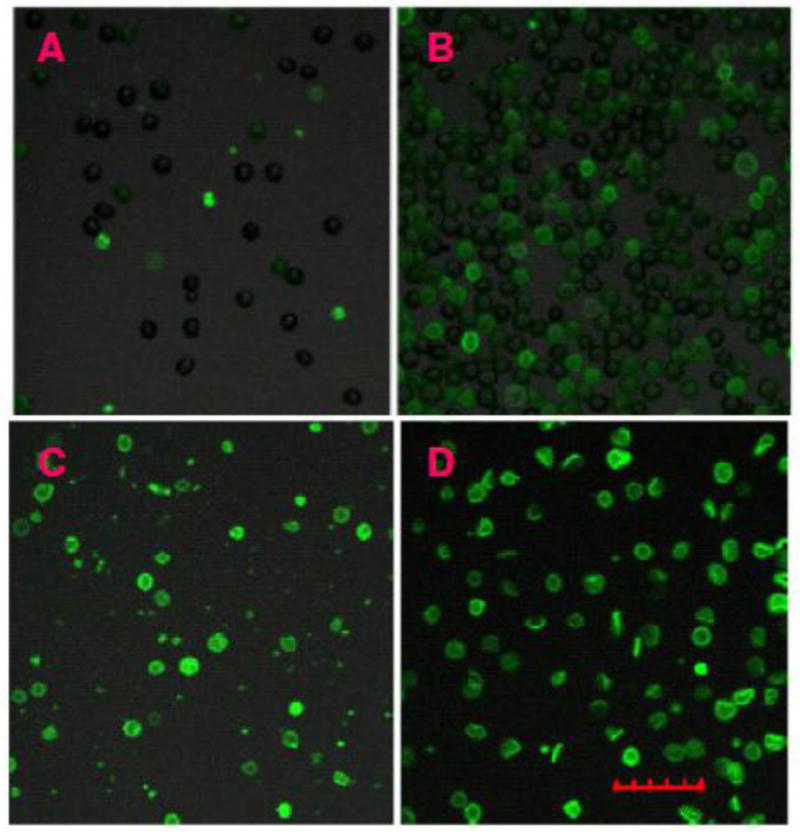
Preserved blood cells and ghosts were treated with 50 nM ANV-FITC in HBSA-2.5 mM CaCl2 and examined under a Olympus confocal laser scanning microscope (FV1000) using a 20× objective lens and excitation/emission filters 488/510–560 nm as described in Materials and Methods. (A) 4C Plus Control; (B) 5C Control, aged 6 months at 4°C; (C) Ghosts prepared from 5C Control that had been aged 1 month at 4° after expiration date; and (D) Ghosts prepared from fresh ACD blood. The scale was 30-μm.
Fig. 2. Flow cytometric analysis of preserved blood cells and blood cell ghosts.

5C Control and blood cell ghosts were diluted to concentrations of about 5 ×106 cells/ml in HBSA buffer containing 50 nM ANV-FITC and 2.5 mM CaCl2 or 1.25 mM CaCl2. The samples were incubated at r.t. for 30 min and aliquots were aspirated into a FACSCalibur (Becton-Dikinson) for flow cytometric analysis (excitation 488 nm, emission filter 530±30 nm). (A) 5C Control cells aged for 11 months at 4°C; (B) Erythrocyte ghosts prepared from 5C Control that had been aged for 1 month at 4°C; (C) Erythrocyte ghosts prepared from 5C Control that had been aged for 11 month at 4°C; and (D) Erythrocyte ghosts prepared from fresh ACD blood. Panel a, forward scatter (FSC-H) vs. side scatter (SSC-H) plot. Panels b and c, histograms of cells incubated with 50 nM ANV-FITC in HBSA-2.5 mM CaCl2, and HBSA-1.25 mM CaCl2, respectively.
Determination of maximum membrane binding sites [m] on erythrocyte ghosts
FITC-labeled proteins were incubated with erythrocyte ghosts at varying ratios in TBSA-15 mM Ca++ to determine the concentration of membrane binding sites on the erythrocyte ghosts Following incubation and centrifugation, the amount of labeled proteins bound to erythrocyte ghosts was calculated from the decrease of fluorescence in the supernatants compared with the control. Fig. 3 showed the concentration of bound ANV-FITC (A) and ANV-6L15-FITC (B) as a function of protein/cell ratio. The protein/cell ratio corresponding to 100% occupancy of the binding sites on erythrocyte ghosts was determined from the ratio at which the ascending line intersected with the horizontal line. Saturation was reached at ratios of approximately 7.1 ×106 and 8.5 × 106 molecules/cell, respectively for ANV-FITC (A) and ANV-6L15-FITC (B). The average [m] values of the erythrocyte ghosts (7.1 × 106 cells/ml) were determined to be 76 ± 7 nM and 91 ± 9 nM, respectively for ANV-FITC and ANV-6L15-FITC (n = 7).
Fig. 3. Determination of maximum membrane binding sites [m] on erythrocyte ghosts.

ANV-FITC or ANV-6L15-FITC (10 nM) and erythrocyte ghosts (8.88 × 104–1.14 × 107 cells/ml) were mixed in 0.4 ml of TBSA supplemented with 15 mM CaCl2 to provide the indicated protein/cell ratio. The reaction mixtures were incubated at r.t. for 40 min. Following centrifugation at 13,000 rpm for 5 min, duplication samples (160 μl) of the supernatants were mixed with 20 μl of 0.5 M EDTA for fluorescence reading. No-binding controls were prepared by mixing 50 μl of 0.5 M EDTA with 0.4 ml of the above reaction mixtures, incubated and centrifuged as above. Duplicate samples (180 μl) of the supernatants were taken for fluorescence reading. The concentrations of the membrane-bound FITC-proteins were calculated by the following equation: [(FT − FS)/FT] × 10 nM, where FT is the relative fluorescence units of the no-binding control; FS is the relative fluorescence units of the supernatant of the reaction mixture. (A) Concentration of bound ANV-FITC as a function of ANV:cell ratio. (B) Concentration of bound ANV-6L15-FITC as a function of ANV-6L15:cell ratio. The protein-cell ratios corresponding to 100 % saturation of binding sites as determined from the intersecting lines were approximately 7.1 × 106 ANV-FITC molecules/cell (A), and 8.5 × 106 ANV-6L15-FITC molecules/cell (B), respectively.
Effects of Ca++ and EDTA on the binding of ANV-FITC and ANV-6L15-FITC to erythrocyte ghosts
ANV-FITC or ANV-6L15-FITC (2 nM) was incubated with erythrocyte ghosts (7.1×106 cells/ml) in TBSA buffer containing 1.25 mM CaCl2 in the presence and absence of excess EDTA for 40 min at r.t. followed by EDTA extraction of the cell pellets. The percentages of unbound FITC-labeled proteins in the solution phase, the EDTA-extracted phase, and the EDTA-resistant membrane phase were calculated. Fig. 4(A) showed the percentage distribution of ANV-FITC in various phases. Column (a) was 100% fluorescence in solution phase control obtained by adding ANV-FITC to the cell-free, ANV-FITC-free supernatant of the mixture. Column (b) was the percentage distribution of fluorescence after incubation of ANV-FITC with erythrocyte ghosts in the presence of excess EDTA. Note that pre-addition of EDTA in the reaction mixture completely prevented the binding of ANV-FITC to erythrocyte ghosts since 100% of fluorescence was recovered in the solution phase as compared to that in column (a). Columns (c), (d), (e) and (f) were percentage distributions of fluorescence in solution phase, EDTA-extracted phase, and EDTA-resistant membrane phase after incubation of ANV-FITC and erythrocyte ghosts for 40 min, 80 min, 4 h, and 22 h, respectively, followed by EDTA extraction of the cell pellets. Note that significant fractions (20–36%) of the fluorescence initially added to the reaction mixtures remained bound to membranes (EDTA-resistant membrane phase) and were not released by EDTA treatment. The EDTA resistant fraction was larger when ANV-FITC was bound to the membranes for a prolonged period (4 h and 22 h). As shown in Fig. 4(B), similar results were obtained when the experiment was carried out using ANV-6L15-FITC instead of ANV-FITC. Thus, these experiments clearly indicated: a) Ca++-mediated the binding of ANV and ANV-6L15 to erythrocyte ghosts; b) an excess of EDTA over Ca++ completely prevented the binding of these proteins to the erythrocyte ghosts; and c) once membrane-bound in the presence of Ca++, ANV and ANV-6L15 were only partially released by EDTA treatment.
Fig. 4. Effects of Ca++ and EDTA on the binding of ANV-FITC and ANV-6L15-FITC to erythrocyte ghosts.

(A) Distribution of fluorescence after incubation of erythrocyte ghosts with ANV-FITC and extraction of cell pellets with EDTA. Column (a), 100% fluorescence in solution phase control. Erythrocyte ghosts (7.1×106 cells/ml in TBSA-1.25mM CaCl2) were centrifuged at 13,000 rpm for 10 min. Aliquot of the supernatant (380 μl) was mixed with 20 μl of 40 nM ANV-FITC so that the final concentration of ANV-FITC was 2 nM. Duplicate samples (160 μl) of the supernatant were each mixed with 20 μl of 0.5 M EDTA for fluorescence reading. Column (b), distribution of fluorescence after incubation of erythrocyte ghosts with ANV-FITC in the presence of excess EDTA. ANV-FITC (2 nM) and erythrocyte ghosts (7.1×106 cells/ml) in 400 μl TBSA-1.25 mM CaCl2 was mixed with 50 μl of 0.5 M EDTA, incubated for 40 min at r.t., and centrifuged at 13,000 rpm for 10 min. Duplicate samples (180 μl) of the supernatant were taken for fluorescence reading. Columns (c), (d), (e) and (f) were distributions of fluorescence in solution phase (gray), EDTA-extracted phase (dotted), and EDTA-resistant membrane phase (open) after incubation of ANV-FITC (2 nM) and erythrocyte ghosts (7.1×106 cells/ml) in 400 μl of TBSA-1.25 mM CaCl2 at r.t. for 40 min (c), 80 min (d), 4 h (e), and 22 h (f), respectively, followed by EDTA extraction of the cell pellets. The reaction mixtures were centrifuged as above and duplicate samples (160 μl) of the supernatant were each mixed with 20 μl of 0.5 M EDTA for fluorescence reading. Each cell pellet was re-suspended in 400 μl TBSA plus 50 μl 0.5 M EDTA, incubated and centrifuged as above. Duplicate samples (180 μl) of the supernatant were taken for fluorescence reading. (B) Distribution of fluorescence after incubation of erythrocyte ghosts with ANV-6L15-FITC followed by extraction of cell pellets with EDTA. The experimental protocols were identical to (A).
Saturation binding of ANV-FITC and A6L15-FITC to erythrocyte ghosts
Erythrocyte ghosts (1:200 dilution of the preparation) was incubated with increasing concentrations of FITC-labeled ANV or ANV-6L15 in TBSA containing 1.25 mM, 2.5 mM, or 5 mM CaCl2. After incubation, the concentrations of free- and membrane bound- labeled proteins were quantitated by fluorescence measurement as described in Material and Methods. Figure 5(A) showed that the binding of ANV-FITC to the erythrocyte ghosts reached increasing saturation levels with increasing Ca++ concentration. The full-range saturation titration data, however, poorly fit the equilibrium binding equation, [Protein]bound = {Bmax × [Protein]free}/(Kd + [Protein]free), derived for simple binding reaction between a protein and a membrane. Inset to Fig. 5(A) showed that nonlinear least-squares regression analysis using the first 6 titration points produced a reasonable fit (R2 = 0.991, Bmax=177 nM, Kd=32 nM) of the equation (solid and dotted lines). This predicted a much higher saturation level than the actual saturation data points (solid triangles). The result suggested negative cooperativity of ANV-FITC binding to the membrane when the membrane binding sites were increasingly occupied. Negative cooperativity of binding at high protein density on membranes had been demonstrated for protein kinase C and other Ca++-dependent phospholipid-binding proteins [33]. These proteins induced clustering of acidic phospholipids in membranes and reduced the membrane’s ability to bind later-binding proteins [25]. The results of saturation titration study suggested a sequential binding model described previously in which the affinity of proteins for membranes progressively decreased with increasing occupancy of membrane binding sites [33]. Therefore, the classical saturation titration method could not be used for determination of the equilibrium binding constant with precision. Fig. 5(B) showed that the binding of ANV-6L15-FITC to the erythrocyte ghosts also reached increasing saturation levels with increasing Ca++ concentration. At each Ca++ concentration, however, saturation level was higher for ANV-6L15-FITC than ANV-FITC [compare Fig. 5(A) and (B)]. Similarly, saturation titration data for ANV-6L15-FITC did not fit the equilibrium binding equation.
Fig. 5. Saturation binding of ANV-FITC and ANV-6L15-FITC to erythrocyte ghosts.

Saturation binding was carried out as described in Materials and Methods. Erythrocyte ghosts (1:200 dilution of the ghost preparation) were mixed with 1-256 nM ANV-FITC (A) or ANV-6L15-FITC (B) at 2-fold increasing concentrations in TBSA buffer supplemented with 1.25 mM (●), 2.5 mM (■) or 5 mM (▲) CaCl2. A separate set of tubes containing the same reaction mixtures were supplemented with EDTA to prevent the binding of FITC-labeled proteins to the erythrocyte ghosts as controls. After incubation for 40 min at r.t., all the reaction mixtures were centrifuged at 13,000 rpm for 10 min to separate the free- and the cell-bound FITC-labeled proteins. Supernatants were taken for measurement of florescence with excitation/emission wavelengths at 485 nm/538 nm. The concentrations of the free- and membrane bound- proteins were then calculated from the fluorescence reading. Inset to Fig. 5(A): Non-linear least-squares regression analysis of the binding of ANV-FITC to erythrocyte ghosts in TBSA buffer supplemented with 5 mM CaCl2 using the first 6 titration data points (0–32 nM ANV-FITC) to fit the saturation binding equation, [Protein]bound = {Bmax × [Protein]free}/(Kd + [Protein]free). Solid line was the fit based on the first 6 experimental data points. Dotted lines were the 95% confidence intervals around the fit. The correlation coefficient of the fit based on the 6 data points was R2=0.991 with Bmax=177 nM and Kd=32 nM.
Calcium titration of the binding of ANV-FITC and A6L15-FITC to erythrocyte ghosts
Titration of the binding of ANV-FITC or A6L15-FITC to erythrocyte ghosts with increasing concentration of Ca++ was carried out as described in Methods. The experimental titration data were fit to the binding equation B/Bmax = [Ca]N/( [Ca]N + EC50N) by nonlinear least-squares regression analysis [32] to determine the binding parameters, EC50 (the [Ca++] at which half of the protein is bound to the membranes), N (Hill coefficient), and R2 (correlation coefficient). Fig. 6 showed excellent non-linear least-squares fits for the binding of ANV-FITC (A) and ANV-6L15-FITC (B) to the erythrocyte ghosts. Solid lines were the fits of the experimental data points (solid circles). Dotted lines were the 95% confidence intervals around the fits. Table 1 summarized the binding parameters for ANV-FITC and ANV-6L15-FITC. Note that the Hill coefficient (N) was approximately 4 for both ANV-FITC and ANV-6L15-FITC, suggesting that about 4 Ca++ ions participated in the binding of each ANV-FITC or ANV-6L15-FITC molecule to the membranes. The mean values of N and EC50 from six calcium titrations were used for calculation of the dissociation constants (Kd, pK) and apparent dissociation constants (Kapp) at 1.2 mM and 2.5 mM Ca++ concentrations. The Kapp values at 1.2 mM and 2.5 mM Ca++ were approximately 4-fold lower for ANV-6L15-FITC compared to those for ANV-FITC, suggesting that ANV-6L15 had higher binding affinity for the erythrocyte ghosts than ANV.
Fig. 6. Calcium titration of the binding of ANV-FITC and ANV-6L15-FITC to erythrocyte ghosts.

Calcium titration was carried out as described in Materials and Methods. The binding parameters, EC50 (the [Ca++] at which half of the protein is bound to the membrane), N (Hill coefficient), and R2 (correlation coefficient) were determined by fitting the experimental calcium titration data to the following equation: B/Bmax = [Ca]N/([Ca]N + EC50N). Fits were performed by nonlinear least-squares analysis with the Solver function of Microsoft Excel33. Solid lines were the fits of the experimental data points (solid circles). Dotted lines were the 95% confidence intervals around the fits. (A) Calcium titration of the binding of ANV-FITC to erythrocyte ghosts. The following parameters were obtained from this titration: N = 3.954; EC50 = 0.704; and R2 = 0.995. (B) Calcium titration of the binding of ANV-6L15-FITC to erythrocyte ghosts. The following parameters were obtained from this titration: N = 4.157; EC50 = 0.441; and R2 = 0.997.
Table 1.
Parameters for the binding of ANV-FITC (A) and ANV-6L15-FITC (B) to erythrocyte ghosts.
| a N | a EC50 (mM) | a R2 | bKd | b pK | bKapp (1.2 mM) (M) | bKapp (2.5 mM) (M) | |
|---|---|---|---|---|---|---|---|
| (A) | 3.903±0.334 | 0.683±0.050 | 0.995±0.003 | 3.35E-20 | 19.48 | 7.15E-9 | 4.78E-10 |
| (B) | 3.802±0.319 | 0.443±0.044 | 0.997±0.001 | 1.62E-20 | 19.79 | 1.77E-9 | 1.27E-10 |
N, EC50, and R2 were mean ± SD (n=6) from calcium titrations performed over a 4-week period using the same preparation of erythrocyte ghosts stored at 4°.
Kd, pK, and Kapp were calculated values based on the mean values of N and EC50 determined by calcium titration using the following equations: Kd = (EC50)N × [m]; pK = − log K = − (N log EC50 + log [m]); and Kapp = Kd/[Ca]N
DISCUSSION
ANV has been extensively used for detection of PS-expression on the membrane surfaces of cells such as activated platelets and leukocytes, abnormal red cells, and a variety of cells undergoing programmed cell death. Extent of PS expression is cell type-dependent and varies with time and experimental conditions. This makes it difficult to precisely define the binding parameters and to compare the Ca++-dependent binding of ANV derivatives to different cell membranes. Tait et al. proposed to use aged 4C Plus Cell Control (Beckman-Coulter) for routine comparative study of the binding of ANV derivatives [23]. However, we found that only a small fraction of red blood cells in the unexpired preserved blood bound ANV-FITC and the expression of PS on erythrocytes varied over time on prolonged storage. These irregularities render the assay ineffective for side-by-side and longitudinal comparisons of the binding properties of ANV derivatives. Erythrocyte ghosts prepared from preserved blood expressed increased amount of PS but such preparations contained numerous cell fragments. In contrast, erythrocyte ghosts prepared from fresh ACD blood described here offered a number of advantages compared with other membrane systems: (a) Most of the erythrocyte membranes bound ANV-FITC and the preparation contained fewer cell fragments; (b) PS expression on the erythrocyte ghosts was highly stable and could be used for calcium titration experiments over weeks with consistent results; (c) The erythrocyte ghosts were roughly the same size (diameter ~6 μm) as the erythrocytes in normal blood. Simple microcentrifugation afforded easy separation and quantification of free- and membrane bound- ligands; and (d) Erythrocyte ghosts might simulate membranes of pathological cells more closely than artificial phospholipids vesicles. We propose that erythrocyte ghosts prepared by the procedure outlined in this study may be used as a reference material for the study of PS exposure on stored, sickle and thalassemia red blood cells, and for routine quantitative assessment of the affinities of phospholipid-binding proteins to biological membranes.
Calcium-dependent binding to PS-expressing membranes is a characteristic property of annexins. It has been widely assumed that EDTA chelation of Ca++ completely released the membrane-bound ANV. The experiments summarized in Fig. 4 contradicted such assumption. ANV-FITC and ANV-6L15-FITC bound to erythrocyte ghosts in the presence of 1.2 mM Ca++ were not completely released into solution phase after treatment with EDTA. This suggested the existence of two pools of bound proteins - an EDTA-releasable pool; and an EDTA-resistant pool. The percentage of the bound proteins released by EDTA decreased when the proteins were allow to stay membrane-bound for longer periods, suggesting that there was a transition of part of the proteins from the EDTA-releasable to the EDTA-resistant pool. However, when the proteins were incubated with erythrocyte ghosts in the presence of EDTA, the binding of the proteins to the membranes was prevented completely. To account for the partial release of ANV derivatives by EDTA treatment, we proposed a two-step binding model: Initial binding of the proteins to the PS-exposed membranes was dependent on the formation of protein-calcium-phosphate chelates; this was followed by increased interactions between the proteins and the phospholipid bilayer, the latter being resistant to dissociation by EDTA. This model would be consistent with a previous isothermal microcalorimetry study [34] in which protein-calcium-phosphate chelates was found to accounted for about 70 % of the free energy of binding while dehydration of the hydrophobic region of the protein surface as they entered the interfacial region contributed to the rest of overall binding energy. Hydrophobic interaction might contribute to the EDTA resistance observed in the present study.
In previous studies, the affinity constant of ANV-membrane interaction was mostly determined by saturation titration method in which a fixed concentration of membranes was titrated with increasing concentrations of ANV at a specific Ca++ concentration [16,18–22]. Our study showed that saturation titration data poorly fit the equilibrium binding equation and suggested heterogeneity of binding events over the full range of titration (Fig. 5). Tait et el. developed the original calcium titration method in which ANV derivatives and preservative-treated erythrocytes were titrated with increasing concentrations of Ca++ such that the membrane binding site occupancy was 1–3% throughout the titration [23]. In theory, this would minimize the heterogeneity of binding events and allow measurement of the parameters of a single binding equilibrium. However, this method involved washing cells with buffers followed by releasing cell-bound ANV derivatives by treatment with EDTA. Cell washing could significantly disturb the binding equilibrium, and EDTA was found to release membrane-bound ANV derivatives incompletely (Figure 4). Thus, Tait’s original calcium titration method might not yield true equilibrium binding parameters. In our modified calcium titration assay, no cell washing was involved and cell-bound ANV derivatives was directly derived from the difference between the total and the free without EDTA release step. This approach allowed equilibrium to be maintained throughout the assay and more accurate equilibrium binding parameters might be obtained. Our modified calcium titration method yielded Hill coefficients (N) of 3.9 ± 0.3 and 3.8 ± 0.3, respectively, for the binding of ANV-FITC and ANV-6L15-FITC to erythrocyte ghosts (Table 1). This results correlated well with the structure of the core domains of ANV which were composed of a 4-fold repeat of conserved amino acid sequence, with each repeat containing a type II Ca++ binding site that was thought to mediate Ca++-dependent binding to the membrane by a ‘Ca++-bridge’ mechanism [35]. In contrast, Tait et al reported N ~8 for the binding of ANV to 4C Plus Cell Control [23]. Furthermore, the Kd value for the binding of ANV to erythrocyte ghosts as determined by our modified method was 3.4 × 10−20 (Table 1), in contrast to 10−30 reported by Tait et al.[23]. The large discrepancies in binding parameters may be accounted for by methodological differences of the assays.
The number of Ca++ involved in the binding of annexins to membranes had been extensively investigated by others before, yet the reported stoichiometries had differed widely ranging from 3 to 12 [35,36 and references therein]. Based on data from crystal structures, 45Ca++ copelleting assay, and isothermal titration calorimetry study, Patel et al. proposed a molecular model to account for up to 12 Ca++-binding sites on ANV and annexin XII that mediated the binding to vesicles containing 2:1 PS-PC [35]. In this model, the footprint of ANV monomer would cover approximately 26 phospholipids on the monolayer. A high-affinity type II Ca++ binding site and two “low affinity” carboxylate side chains on each of the 4-fold repeat of annexin core domain were postulated to form “Ca++-bridges” with the phospholipids in a complementary manner, so 12 of the 26 phospholipids were anchored to the protein via Ca++ bridges. However, since the PS content of human erythrocyte was only approximately 15% of total phospholipids [37], the erythrocyte ghost membranes might not be able to form 12 Ca++-bridges under each ANV monomer. In stead, 3.9 Ca++-bridges (26 phospholipids × 15 %) per ANV monomer could be involved, assuming random distribution of phospholipids across the bilayer of the ghost membranes. Thus, our experimentally determined Hill coefficient (N) of approximately 4 for the binding to erythrocyte ghosts was consistent with Patel’s model, albeit with lower Ca++ stoichiometry because of much lower PS content compared with 2:1 PS-PC vesicles. According to the equilibrium binding equation (3), N value affected the dissociation constant exponentially. Decreases of Ca++ concentration and N value could translate into greatly diminished binding affinity. PS expressions on the membrane surface of pathological cells were generally in the range of 0–15% of total phospholipids. In contrast, vesicles containing 20–100% PS were commonly used for study of ANV-Ca++-phospholipid interactions [17,33,36,38]. This might be a major reason for the large discrepancies of reported binding parameters. ANV has been used for detection of PS expression during platelet activation, cell senescence, pathological changes, and apoptosis. Thrombin-treated platelets and cells in early stages of apoptosis have been shown to express low levels of PS which was poorly detected by ANV at physiological concentration of Ca++, but was easily detected by the Ca++-independent PS binding protein, lacadherin [39]. Thus, the sensitivity of detecting PS-expressing cells by ANV was critically dependent on the exoplasmic PS content and the extracellular Ca++ concentration.
The affinity of ANV binding to erythrocyte ghosts declines sharply over a narrow range of Ca++ concentration with Kapp increased 15-fold from 4.78 × 10−10 M to 7.15 × 10−9 M when the ionized Ca++ decreased from 2.5 mM to 1.2 mM. In vitro imaging of ANV binding is usually made at 2.5 mM Ca++. Under such condition, the binding signal can be detected using 10–100 nM concentrations of labeled ANV without difficulties. For in vivo imaging applications, suboptimal detection sensitivity and low signal-to- background ratio have been important problems that continue to hamper the progress of ANV imaging [15.16]. ANV binding occurs at a Ca++ concentration of 1.2 mM (the typical ionized Ca++ in circulating plasma) under in vivo conditions in which the binding affinity is not very high. Furthermore, pathological cells may express lower levels of PS in vivo. It is also possible that PS-exposed sites may also be partly occupied by endogenous ANV in circulating blood [40] and released locally from apoptotic, injured, and activated cells. Thus, a relatively high blood concentration of labeled ANV may be needed to allow sensitive detection of the target cells. This in turn may compromise the signal-to-background ratio. ANV-6L15, an ANV derivative with a 4-fold lower Kapp at physiological concentration of Ca++, may displace endogenous ANV from the PS sites more effectively and afford detection of PS-exposed cells with greater sensitivity in in vivo imaging applications. This will be the subject of a future study.
The mechanism for the increased binding affinity of ANV-6L15 for erythrocyte membranes compared to ANV is currently unknown. Calcium titration study indicated that it was not due to increased Ca++-bridging since the Hill coefficient for the binding of ANV-6L15 was almost identical to that of ANV. ANV-6L15 was previously found to require much lower concentration of Ca++ than ANV to bind to homogenized E. coli membranes. PS is virtually absent in E. coli, whereas PE constitutes about 69 % of the total phospholipids [41]. Therefore, the PE co-expressed on the erythrocyte membranes might contribute importantly to the increased affinity of ANV-6L15 compared to ANV.
Acknowledgments
This study was supported in part by the Chang Gung Memorial Hospital grants to T-C Yen [BMRPG360011] and T-C Wun [CMRPG270281]; the National Institutes of Health grants to S. Achilefu [R01 CA109754 and U01 HL080729]; and T-C. Wun [R43 HL077061 and R43 HL093843].
The authors would like to thank Ya-Fen Lu for flow cytometry support.
Footnotes
Abbreviations used: ANV, annexin V; FITC, fluorescein 5-isothiocyanate; PS, phosphatidylserine; ACD, acid-citrate-dextrose; TBS, Tris buffered saline; EDTA, ethylenediaminetetraacetic acid.
TW has declared financial interest in EVAS Therapeutics, LLC. All other authors have declared no financial conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zwaal RFA, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–988. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
- 2.Balasubramanian K, Schroit A. Aminophospholipid asymmetry: A matter of life and death. Annu Rev Physiol. 2003;65:701–734. doi: 10.1146/annurev.physiol.65.092101.142459. [DOI] [PubMed] [Google Scholar]
- 3.Daleke DL. Regulation of phospholipid asymmetry in the erythrocyte membrane. Curr Opin Hematol. 2008;15:191–195. doi: 10.1097/MOH.0b013e3282f97af7. [DOI] [PubMed] [Google Scholar]
- 4.Raynal P, Pollard HB. Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim Biophys Acta. 1994;1197:63–93. doi: 10.1016/0304-4157(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 5.Swairjo MA, Seaton BA. Annexin structure and membrane interactions: a molecular perspective. Annu Rev Biophys Biomol Struct. 1994;23:193–213. doi: 10.1146/annurev.bb.23.060194.001205. [DOI] [PubMed] [Google Scholar]
- 6.Gerke SE, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 7.Dachary-Prigent J, Freyssinet JM, Pasquet JX, Carron JC, Nurden AT. Annexin V as a probe of aminophospholipid exposure and platelet membrane vesiculation: a flow cytometry study showing a role for free sulfhydryl group. Blood. 1993;81:2554–2565. [PubMed] [Google Scholar]
- 8.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- 9.Wood BL, Gibson DF, Tait JF. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: flow-cytometric measurement and clinical association. Blood. 1996;88:1873–1880. [PubMed] [Google Scholar]
- 10.Tait JF, Smith C, Wood BL. Measurement of phosphatidylserine exposure in leukocytes and platelets by whole-blood flow cytometry with annexin V. Blood Cells Mol Dis. 1999;25:271–278. doi: 10.1006/bcmd.1999.0254. [DOI] [PubMed] [Google Scholar]
- 11.van Genderen H, Kenis H, Lux P, Ungeth L, Maassen C, Deckers N, Narula J, Hofstra L, Reutelingsperger C. In vitro measurement of cell death with the annexin A5 affinity assay. Nat Protoc. 2006;1:363–367. doi: 10.1038/nprot.2006.55. [DOI] [PubMed] [Google Scholar]
- 12.Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Naumovski L, Ohtsuki K, Kopiwoda S, Abrams MJ, Darkes M, Robbins RC, Maecker HT, Strauss HW. In vivo detection and imaging of phosphatidylserine expression during programmed cell death. Proc Natl Acad Sci USA. 1998;95:6349–6354. doi: 10.1073/pnas.95.11.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boersma HH, Kietselaer BL, Stolk LM, Bennaghmouch A, Hofstra L, Narula J, Heidendal GA, Reutelingsperger CP. Past, present, and future of annexin A5: from protein discovery to clinical applications. J Nucl Med. 2005;46:2035–2050. [PubMed] [Google Scholar]
- 14.Belhocine TZ, Blankenberg FG. The imaging of apoptosis with the radiolabeled annexin A5: a new tool in translational research. Curr Clin Pharmacol. 2006;1:129–137. doi: 10.2174/157488406776872541. [DOI] [PubMed] [Google Scholar]
- 15.Korngold EC, Jaffer FA, Weissleder R, Sosnovik DE. Noninvasive imaging of apoptosis in cardiovascular disease. Heart Fail Rev. 2008;3:163–173. doi: 10.1007/s10741-007-9068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tait JF, Gibson D, Fujikawa K. Phospholipid binding properties of human placental anticoagulant protein-I, a member of the lipocortin family. J Biol Chem. 1989;264:7944–7949. [PubMed] [Google Scholar]
- 17.Andree HAM, Reutelingsperger CPM, Hauptmann R, Hemker HC, Hermens WT, Willems GM. Binding of vascular anticoagulant α (VAC α) to planar phospholipids bilayers. J Biol Chem. 1990;265:4923–4928. [PubMed] [Google Scholar]
- 18.Thiagarajan P, Tait JF. Binding of Annexin V/Placental anticoagulant protein I to platelets. J Biol Chem. 1990;265:17420–17423. [PubMed] [Google Scholar]
- 19.Tait JF, Gibson D. Measurement of membrane phospholipids asymmetry in normal and sickle-cell erythrocytes by means of Annexin V binding. J Lab Clin Med. 1994;123:741–748. [PubMed] [Google Scholar]
- 20.Rao LVM, Tait JF, Hoang AD. Binding of Annexin V to a human ovarian carcinoma cell line (OC-2008): contrasting effects on cell surface factor VIIa/tissue factor activity and prothrombinase activity. Thromb Res. 1992;67:517–531. doi: 10.1016/0049-3848(92)90013-z. [DOI] [PubMed] [Google Scholar]
- 21.Sun J, Bird P, Salem HH. Interaction of Annexin V and platelets: effects on platelet function and protein S bind. Thromb Res. 1993;69:289–296. doi: 10.1016/0049-3848(93)90026-k. [DOI] [PubMed] [Google Scholar]
- 22.van Heerde WL, Poort S, van’t Veer C, Reutelingsperger CPM, de Groot PhG. Binding of recombinant Annexin V to endothelial cells: effect of Annexin V binding on endothelial-cell mediated thrombin formation. Biochem J. 1994;302:305–312. doi: 10.1042/bj3020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tait JF, Gibson DF, Smith C. Measurement of the affinity and cooperativity of annexin V-membrane binding under low membrane occupancy. Anal Biochem. 2004;329:112–119. doi: 10.1016/j.ab.2004.02.043. [DOI] [PubMed] [Google Scholar]
- 24.Haverstick DM, Glaser M. Visualization of Ca2+-induced phospholipid domains. Proc Natl Acad Sci USA. 1987;84:4475–4479. doi: 10.1073/pnas.84.13.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bazzi MD, Nelsestuen GL. Extensive segregation of acidic phospholipids in membranes induced by protein kinase C and related proteins. Biochemistry. 1991;30:7961–7969. doi: 10.1021/bi00246a013. [DOI] [PubMed] [Google Scholar]
- 26.Gilmanshin R, Creutz CE, Tamm LK. Annexin IV reduces the rate of lateral lipid diffusion and changes the fluid phase structure of the lipid bilayer when it binds to negatively charged membranes in the presence of calcium. Biochemistry. 1994;33:8225–8232. doi: 10.1021/bi00193a008. [DOI] [PubMed] [Google Scholar]
- 27.Concha NO, Head JF, Kaetzel MA, Dedman JR, Seaton BA. Annexin V forms calcium-dependent trimeric units on phospholipids vesicles. FEBS Lett. 1999;314:159–162. doi: 10.1016/0014-5793(92)80964-i. [DOI] [PubMed] [Google Scholar]
- 28.Pigault C, Follenius-Wund A, Schmutz M, Freyssinet JM, Brisson A. Formation of two-dimensional arrays of annexin V on phosphatidylserine-containing liposomes. J Mol Biol. 1994;236:199–208. doi: 10.1006/jmbi.1994.1129. [DOI] [PubMed] [Google Scholar]
- 29.Andree HA, Stuart MC, Hermens WT, Reutelinsperger CP, Hemker HC, Frederik PM, Willem GM. Clustering of lipid bound annexin V may explain its anticoagulant effect. J Biol Chem. 1992;267:17907–12. [PubMed] [Google Scholar]
- 30.Megli FM, Selvaggi M, Liemann S, Quagliariello E, Huber R. The calcium-dependent binding of annexin V to phospholipid vesicles influences the bilayer inner fluidity gradient. Biochemistry. 1998;37:10540–10546. doi: 10.1021/bi9801255. [DOI] [PubMed] [Google Scholar]
- 31.Chen HH, Vincente CP, He L, Tollefsen DM, Wun TC. Fusion proteins comprising annexin V and Kunitz protease inhibitors are highly potent thrombogeneic site-directed anticoagulants. Blood. 2005;105:3902–3909. doi: 10.1182/blood-2004-11-4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown AM. A step-by-step guide to non-linear regression analysis of experimental data using a Microsoft Excel spreadsheet. Comput Methods Programs Biomed. 2001;65:191–200. doi: 10.1016/s0169-2607(00)00124-3. [DOI] [PubMed] [Google Scholar]
- 33.Bazzi MD, Nelsestuen GL. Highly sequential binding of protein kinase C and related proteins to membranes. Biochemistry. 1991;30:7970–7977. doi: 10.1021/bi00246a014. [DOI] [PubMed] [Google Scholar]
- 34.Jeppesen B, Smith C, Gibson DF, Tait JF. Entropic and enthalpic contributions to annexin V-membrane binding: a comprehensive quantitative model. J Biol Chem. 2008;283:6126–6135. doi: 10.1074/jbc.M707637200. [DOI] [PubMed] [Google Scholar]
- 35.Patel DR, Jao CC, Mailliard WS, Isas JM, Langen R, Haigler HT. Calcium-dependent binding of Annexin 12 to phospholipid bilayers: Stoichiometry and implications. Biochemistry. 2001;40:7054–7060. doi: 10.1021/bi010345+. [DOI] [PubMed] [Google Scholar]
- 36.Patel DR, Isas JM, Ladokhin AS, Jao CC, Kim YE, Kirsch T, Langen R, Haigler HT. The conserved core domains of annexins A1, A2, A5, and B12 can be divided into two groups with different Ca++-dependent membrane-binding properties. Biochemistry. 2005;44:2833–2844. doi: 10.1021/bi047642+. [DOI] [PubMed] [Google Scholar]
- 37.Virtanen JA, Cheng KH, Somerharju P. Phospholipid composition of the mammalian red cell membrane can be rationalized by a superlattice model. Proc Natl Acad Sci USA. 1998;95:4964–4969. doi: 10.1073/pnas.95.9.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tait JF, Gibson D. Phospholipid binding of annexin V: Effects of calcium and membrane phosphatidylserine content. Arch Biochem Biophys. 1992;298:187–191. doi: 10.1016/0003-9861(92)90111-9. [DOI] [PubMed] [Google Scholar]
- 39.Dasgupta SK, Guchhait P, Thiagarajan P. Lactadhrin binding and phosphatidylserine expression on cell surface - comparison with annexin A5. Transl Res. 2006;148:19–25. doi: 10.1016/j.lab.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Rand JH, Arslan AA, Wu XX, Wein R, Mulholland J, Shah M, van Heerde WL, Reutelinsperger CP, Lockwood CJ, Kuczynski E. Reduction of circulating annexin A5 levels and resistance to annexin A5 anticoagulant activity in women with recurrent spontaneous pregnancy losses. Am J Obst Gyn. 2006;194:182–188. doi: 10.1016/j.ajog.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 41.Ames GF. Lipids of Salmonella typhimurium and Escherichia coli: Structure and metabolism. J Bacteriol. 1968;95:833–843. doi: 10.1128/jb.95.3.833-843.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]