


Abstract
The Rev1-Polζ pathway is believed to be the major mechanism of translesion DNA synthesis and base damage-induced mutagenesis in eukaryotes. While it is widely believed that Rev1 plays a non-catalytic function in translesion synthesis, the role of its dCMP transferase activity remains uncertain. To determine the relevance of its catalytic function in translesion synthesis, we separated the Rev1 dCMP transferase activity from its non-catalytic function in yeast. This was achieved by mutating two conserved amino acid residues in the catalytic domain of Rev1, i.e. D467A/E468A, where its catalytic function was abolished but its non-catalytic function remained intact. In this mutant strain, whereas translesion synthesis and mutagenesis of UV radiation were fully functional, those of a site-specific 1,N6-ethenoadenine were severely deficient. Specifically, the predominant A→G mutations resulting from C insertion opposite the lesion were abolished. Therefore, translesion synthesis and mutagenesis of 1,N6-ethenoadenine require the catalytic function of the Rev1 dCMP transferase, in contrast to those of UV lesions, which only require the non-catalytic function of Rev1. These results show that the catalytic function of the Rev1 dCMP transferase is required in a lesion-specific manner for translesion synthesis and base damage-induced mutagenesis.
INTRODUCTION
Translesion synthesis is a major cellular mechanism in response to unrepaired DNA lesions during replication. It directly copies damaged sites of the template during DNA synthesis. Thus, translesion synthesis allows DNA replication to completion in the presence of DNA lesions that block replicative polymerases. Translesion synthesis is signaled by stalled replicative DNA polymerase at the lesion site, leading to mono-ubiquitination of PCNA by the Rad6–Rad18 ubiquitin-conjugating/ligation complex (1). Subsequently, the modified PCNA recruits bypass polymerases such as the Y family polymerases and DNA polymerase ζ (Pol ζ) to the damaged template, replacing the stalled replicative polymerase. Lastly, translesion synthesis occurs by polymerase-catalyzed nucleotide insertion opposite the lesion followed by extension synthesis from opposite the lesion (2). When the correct nucleotide is inserted opposite the lesion, translesion synthesis is error-free; when a wrong base is inserted opposite the lesion, translesion synthesis is error-prone. Error-prone translesion synthesis constitutes the major mechanism of base damage-induced mutagenesis in the yeast model system (3–13). Accumulating evidence also supports the notion that error-prone translesion synthesis is a major mechanism of base damage-induced mutagenesis in higher eukaryotes including mammals (14–18).
In the yeast Saccharomyces cerevisiae, the Rev1-Polζ pathway constitutes the major mechanism of translesion synthesis. Polζ is a B family polymerase and consists of the catalytic subunit Rev3 and the non-catalytic subunit Rev7 (19). Saccharomyces cerevisiae contains two Y family DNA polymerases: Rev1 and Polη. Mammals contain two additional Y family members: Polκ and Polι (20). Whereas Polη functions in translesion synthesis of selected types of DNA lesions such as ultraviolet (UV) lesions and benzo[a]pyrene DNA adducts (7,21,22), Rev1 and Polζ appear to be more generally required in translesion synthesis, and is thus referred to as the Rev1-polζ pathway here (2).
Rev1 is unique in that it is a template-dependant dCMP transferase, rather than a typical DNA polymerase (23–26). In vitro, Rev1 is capable of catalyzing C insertion opposite multiple types of DNA lesions such as AP site (23), 1,N6-ethenoadenine, uracil, 8-oxoguanine, (+)-trans-anti-BPDE-N2-dG, and (−)-trans-anti-BPDE-N2-dG (25). Uniquely, Rev1 uses the Arg324 of the protein, instead of the DNA template G, as the template for choosing dCTP as the incoming base during catalysis (27). Rev1 is required for translesion synthesis and mutagenesis of UV lesions in yeast (5,13,15). However, C insertion is not reflected among the translesion synthesis products of a TT (6–4) UV photoproduct in yeast cells (5). Indeed, the Rev1 dCMP transferase is inactive in response to a template TT (6–4) UV photoproduct or a TT UV dimer in vitro (25). Therefore, it was proposed that Rev1 plays a non-catalytic function in the Rev1-Polζ pathway for translesion synthesis and mutagenesis of UV radiation (5,13), although the precise nature of the non-catalytic function is not known. One hypothesis postulates that Rev1 acts to recruit other Y family DNA polymerases to lesion sites through protein–protein interactions (28,29). It is now generally believed that Rev1 plays a ubiquitous non-catalytic function in translesion synthesis. The role of the Rev1 dCMP transferase in translesion synthesis, however, remains uncertain.
Previously, we found that Rev1 is capable of efficient C insertion opposite 1,N6-ethenoadenine in vitro (25). This lesion is produced by lipid peroxidation products or the carcinogens vinyl chloride and urethane (30,31). Using the yeast model system, we examined the role of the Rev1 dCMP transferase in the bypass of 1,N6-ethenoadenine in vivo. In this report, we show that the dCMP transferase of Rev1 is required for translesion synthesis and mutagenesis of 1,N6-ethenoadenine. Our results show that the catalytic function of the Rev1 dCMP transferase is required in a lesion-specific manner for translesion synthesis and base damage-induced mutagenesis.
MATERIALS AND METHODS
Materials
Purified human Polη, Polι and Polκ in near homogeneity were those preparations described previously (32–34). T4 DNA ligase, the T4 gene 32 protein and T4 polynucleotide kinase were obtained from Enzymax (Lexington, KY, USA). Yeast lytic enzyme (70 000 U/g) was purchased from MP Biomedicals (Irvine, CA, USA). The Wizard PCR Preps DNA Purification Resin was from Promega (Wisconsin, WI, USA). The 29-mer DNA template, 5′-CCATCGCTACCTACCATCCGAATTCGCCC-3′, contained a 1,N6-ethenoadenine at the underlined A, and was used for in vitro translesion synthesis assays. It was synthesized via automated DNA phosphoramidite methods by Operon (Alameda, CA, USA). The 22-mer damaged oligonucleotide, 5′-GTAAGCTAGATCCTCTAGAGCG-3′, contained a site-specific 1,N6-ethenoadenine at the underlined A. This oligonucleotide and the 51-mer uracil-containing scaffold, 5′-CTGUGCCCUCCAUGCGCUCUGGAGGAUCUAGCTUACG AAAAAUCAGTCAAG-3′, were used for construction of site-specifically damaged plasmid DNA. These two oligonucleotides were synthesized via automated DNA phosphoramidite methods by Integrated DNA Technologies (Coralville, IA, USA). Yeast Rev1 protein and the D467A/E468A mutant Rev1 protein were purified by Enzymax according to our previously described method (35).
Yeast strains
Yeast strains used were the wild-type BY4741 (MATa his3 leu2 met15 ura3) and its isogenic mutants BY4741Δrad30 (rad30 deletion), BY4741Δrev1 (rev1 deletion), BY4741Δrev3 (rev3 deletion); the wild-type CL1265-7C (MATα arg4-17 leu2-3,112 his3Δ trp1 ura3-52) and its isogenic strains CL1265-7CΔrev1 (rev1 deletion), CL1265-7CΔrev1/REV1 (rev1 deletion mutant containing the wild-type REV1 gene on the plasmid vector pEAT), and CL1265-7CΔrev1/REV1mt (rev1 deletion mutant expressing the Rev1D467A/E468A mutant protein from the plasmid pEAT). BY4741 was purchased from ATCC (Manassas, VA, USA). BY4741Δrad30 (lacking Polη) was purchased from Research Genetics (Huntsville, AL, USA). BY4741Δrev1 and BY4741Δrev3 (lacking Polζ) were constructed previously (36,37). CL1265-7C was provided by Christopher Lawrence of the University of Rochester (11). CL1265-7CΔrev1 was constructed previously (13). CL1265-7CΔrev1/REV1 and CL1265-7CΔrev1/REV1mt were obtained by transforming the expression plasmid construct pEAT-REV1 and pEAT-REV1mt, respectively, into the CL1265-7CΔrev1 strain. The expression vector pEAT contained the 2 µm origin for multi-copy plasmid replication in yeast, the TRP1 gene for plasmid selection, and the ADH1 promoter for constitutive expression of the REV1 or REV1mt gene.
In vitro translesion synthesis
In vitro translesion synthesis assays were performed at 30°C for 10 min as described before (32–34). The reaction (10 µl) contained 50 µM of dNTPs (dATP, dCTP, dTTP and dGTP individually or together as indicated), 50 fmol of the 29-mer DNA template with a site-specific 1,N6-ethenoadenine and a 20-mer 32P-labeled primer annealed right before the lesion, and a purified DNA polymerase. The reaction products were separated on a 20% polyacrylamide gel containing 8 M urea and visualized by autoradiography. Primer extension was quantitated by scanning densitometry of the autoradiogram using the SigmaGel Software (Sigma, St Louis, MO, USA) for analysis.
Kinetic analysis of nucleotide insertion opposite 1,N6-ethenoadenine was performed as previously described (33,38), using 50 fmol of the primed DNA template, 0.33 ng (4 fmol) of human Polη or 0.4 ng (5 fmol) of human Polι, and increasing concentrations of dATP, dCTP, dTTP or dGTP. Kinetic analysis yielded Vmax and Km values for the incorporation of the correct and the incorrect nucleotides. The misincorporation error rate was calculated from the equation: finc = (Vmax/Km)incorrect/(Vmax/Km)correct.
Construction of the rev1D467A/E468A mutant gene
The rev1D467A/E468A mutant gene was constructed by site-directed mutagenesis using a PCR method (39). Briefly, two PCR amplifications were performed using the wild-type REV1 gene as the template. While one PCR reaction yielded a 360-bp DNA fragment using the primers ACGGATAAGGATACCTACATTATCTTTC (yREV1nd3F) and CAAACAGCTgCAgCAATAGATATAGGTAAAATCAAATTGAATATG; the other reaction produced a 460-bp DNA fragment using the primers CCTATATCTATTGcTGcAGCTGTTTGTGTGAGGATAATCC and GTAAGACTTCTTTGGGATCGTACAG (yREV1R20). The two mutated bases are shown in lower case. The mutations result in D467→A and E468→A double substitutions in protein sequence and a new PstI restriction site in DNA sequence. The two DNA fragments were purified and mixed for PCR amplification using the primers yREV1nd3F and yREV1R20, yielding an 800-bp DNA fragment, which was cloned into the NcoI/HindIII sites of pUC19M1. The insert sequence (648 bp) that contains the rev1D467A/E468A mutations were confirmed by DNA sequencing. The 3′ fragment (1.2 kb) of the REV1 gene was ligated at the 3′ end of the rev1D467A/E468A DNA fragment (HindIII/SalI sites). Lastly, the 5′ fragment (1.2 kb) of the REV1 gene flanked by BglII and NcoI was assembled into the rev1D467A/E468A DNA fragment, yielding the rev1D467A/E468A mutant gene.
The dCMP transferase assay
Assays of dCMP transferase were performed as we previously described (25). Briefly, a standard reaction mixture (10 µl) contained 25 mM KH2PO4 (pH 7.0), 5 mM MgCl2, 5 mM dithiothreitol, 100 µg/ml bovine serum albumin, 10% glycerol, 50 µM of dNTPs (dATP, dCTP, dTTP and dGTP), 50 fmol of a DNA template containing a 5′ 32P-labeled primer, and purified yeast Rev1 protein. After incubation at 30°C for 10 min, reactions were terminated with 7 µl of a stop solution (20 mM EDTA, 95% formamide, 0.05% bromophenol blue and 0.05% xylene cyanol). Reaction products were separated on a 15% polyacrylamide gel containing 8 M urea and visualized by autoradiography.
UV sensitivity and UV mutagenesis assays
Wild-type yeast CL1265-7C and its isogenic strains CL1265-7CΔrev1, CL1265-7CΔrev1/REV1 and CL1265-7CΔrev1/REV1mt were grown at 30°C in minimum medium containing supplemented arginine. Cells were appropriately diluted and plated onto plates of minimum medium containing supplemented arginine. The uncovered plates were irradiated with short wave UV light at the indicated doses. Surviving colonies were scored after 3–4 days of incubation at 30°C. UV survival was calculated by dividing surviving colonies after UV treatment by those without UV treatment.
UV mutagenesis was measured at the chromosomal arg4-17 locus essentially as described (11). Briefly, 4 OD600 cells (4 × 107) were collected by centrifugation in a microcentrifuge at 5000 r.p.m. for 20 s. After washing twice in 1 ml of sterile water, cells were resuspended in 110 µl of sterile water. While 100 µl of cells were plated onto plates of minimum medium lacking arginine for mutagenesis assay, 10 µl of cells were appropriately diluted and plated onto plates of minimum medium supplemented with arginine for measurement of UV survival. The uncovered plates were irradiated with short wave UV light at 20 J/m2. After incubation at 30°C for 2–4 days, colonies were counted.
Construction of plasmid containing a site-specific 1,N6-ethenoadenine DNA adduct
Plasmid containing a site-specific 1,N6-ethenoadenine DNA adduct was constructed by Enzymax using a previously described method (36). Briefly, a 20-mer DNA oligonucleotide, 5′-GTGCCCTCCATGGAAAAATC-3′, was annealed to the single-stranded phagemid pELUf1 vector at its unique NcoI restriction site within the URA3 gene. The phagemid was then linearized by digestion with the NcoI restriction endonuclease. The linear pELUf1, the 51–mer DNA scaffold, and the 5′-phosphorylated 22-mer oligonucleotide containing the lesion were annealed together, in which the mid-region of the scaffold is complementary to the damaged oligonucleotide, while its ends are complementary to the single-stranded pELUf1 ends. The damaged oligonucleotide was ligated into the pELUf1 vector by T4 DNA ligase at 16°C for 20 h, and the DNA was precipitated in ethanol. Finally, the complementary strand of pELUf1 was synthesized with T4 DNA polymerase in the presence of T4 gene 32 protein and 0.5 mM each of dATP, dCTP, dGTP and dUTP, using the scaffold as the primer. The resulting construct was a double-stranded plasmid containing a site-specific 1,N6-ethenoadenine. The undamaged strand contained U in place of T. Formation of double-stranded plasmid pELUf1-ethenoA was confirmed by electrophoresis on a 1% agarose gel.
Translesion synthesis in yeast cells
In vivo translesion synthesis assays were performed as we previously described (7,36). Briefly, yeast cells of various strains were transformed by site-specifically damaged pELUf-ethenoA plasmid (2 µg) using the lithium acetate method (40). Then, cells were collected by centrifugation (20 s at 5000 r.p.m.) in a microcentrifuge, resuspended in 400 µl of sterile water, and plated onto two YNB minimal agar (0.17% yeast nitrogen base, 0.49% ammonium sulfate, 2% glucose and 2% agar) plates lacking leucine but supplemented with 5 mM 5-fluoroorotic acid (5-FOA), 150 µM methionine and 380 µM uracil. Cells containing replicated pELUf1-ethenoA were able to grow into colonies on the 5-FOA plates, whereas cells transformed by the empty vector pELUf1 (a minor fraction of contaminant in the pELUf1-ethenoA preparation) remained URA3 wild-type and thus were unable to grow on the 5-FOA plates. Yeast colonies were counted after incubation at 30°C for 3–4 days. In each experiment with each strain, transformation efficiency was determined by a parallel transformation using the undamaged and double-stranded pELUf1. Translesion synthesis was calculated as transformants per microgram of the damaged plasmid per 106 transformable cells with the undamaged plasmid (i.e. transformants per microgram of the damaged plasmid × 106/transformation efficiency expressed as transformants per microgram of the undamaged plasmid). Relative translesion synthesis was obtained by comparing translesion synthesis in various mutant strains to that in the wild-type strain. To minimize inter-experimental variations and to ensure reproducibility, transformations with all strains were performed side-by-side on the same day using the same batch of pELUf-ethenoA plasmid preparation, and multiple independent transformations were performed for each strain.
Yeast colonies on the 5-FOA plates were further processed to determine the specificity of translesion synthesis. Individual colonies were resuspended each in 10 µl of a solution containing 1 mg/ml yeast lytic enzyme in sterile water, followed by incubation at 37°C for 1.5–2 h. An aliquot of 1 µl was used for PCR amplification of a 670-bp plasmid region containing the original lesion site, using the primers 5′-CCCGCAGAGTACTGCAATTTGAC and 5′-GAGCGGATAACAATTTCACACAGG. After heating the PCR reaction mixture (20 µl) at 94°C for 4 min, 35 cycles of amplification were performed according to the following conditions: 30 s denaturation at 94°C, 30 s annealing at 65°C and 45 s extension at 72°C. After the last cycle, the reaction was continued for seven more minutes at 72°C. An aliquot of 2 µl PCR products was separated by electrophoresis on a 1% agarose gel containing 0.5 µg/ml ethidium bromide. Then, two aliquots of 5 µl PCR products each were used for DNA digestions with BamHI and HindIII restriction endonucleases, respectively. PCR products that were sensitive to BamHI cleavage are indicative of C insertion opposite the 1,N6-ethenoadenine DNA adduct; while those sensitive to HindIII cleavage are indicative of A insertion opposite the lesion. All translesion synthesis products analyzed in this study were either sensitive to BamHI or HindIII cleavage.
RESULTS
Translesion synthesis of the 1,N6-ethenoadenine DNA adducts by Y family DNA polymerases in vitro
The 1,N6-ethenoadenine is an important type of exocyclic DNA adduct. To understand molecular mechanisms of bypass and mutagenesis of this lesion, we examined its translesion synthesis in vitro by the human Y family DNA polymerases REV1, Polη, Polι and Polκ. Translesion synthesis was performed with the purified polymerases on a 29-mer DNA template containing a 32P-labeled 20-mer primer annealed right before the lesion (Figure 1A). As shown in Figure 1B (lane 1), human Polη efficiently bypassed 1,N6-ethenoadenine. To identify the base incorporated opposite the lesion, we performed translesion synthesis assays with only one deoxyribonucleoside triphosphate: dATP, dCTP, dGTP or dTTP individually. As shown in Figure 1B (lanes 2–5), human Polη preferred A insertion opposite the lesion, although the other three nucleotides were also inserted. To more accurately compare the efficiency of nucleotide insertion, we performed kinetic analysis using increasing concentrations of a single dNTP. From these assays, the kinetic parameters Vmax and Km were obtained (Table 1). While the efficiency of nucleotide insertion is indicated by the Vmax/Km value, the accuracy of nucleotide insertion is indicated by the finc value, i.e. (Vmax/Km)incorrect/(Vmax/Km)correct. These kinetic measurements show that human Polη is error-prone in catalyzing translesion synthesis of 1,N6-ethenoadenine in vitro, with nucleotide insertion efficiency in the order of A > T > C ∼ G (Table 1).
Figure 1.

Translesion synthesis of 1,N6-ethenoadenine DNA adducts by human Polη, Polκ, and Polι. (A) The DNA template for translesion synthesis. A 20-mer primer was labeled with 32P at its 5′-end (*) and annealed right before a template 1,N6-ethenoadenine. (B) Translesion synthesis reactions were performed with purified human Polη (lanes 1–5), Polκ (lanes 6–10), and Polι (lanes 11–15) as indicated in the presence of a single deoxyribonucleoside triphosphate dATP (A), dCTP (C), dTTP (T) or dGTP (G), or all four dNTPs (N4). Reaction products were separated by electrophoresis on denaturing polyacrylamide gel and visualized by autoradiography. Quantitation of extended primers is shown at the bottom of the gel. DNA size markers in nucleotides are indicated on the left.
Table 1.
Kinetic measurement of nucleotide insertion by human Polη and Polι opposite a template 1,N6-ethenoadenine
| dNTP | Vmax (fmol/min) | Km (µM) | Vmax/Km | finca | |
|---|---|---|---|---|---|
| Polη | dATP | 2.39 ± 0.09 | 4.91 ± 0.68 | 0.49 | 3.5 |
| dGTP | 2.33 ± 0.15 | 24.7 ± 4.31 | 0.094 | 0.67 | |
| dCTP | 1.94 ± 0.14 | 17.1 ± 5.12 | 0.11 | 0.79 | |
| dTTP | 2.19 ± 0.12 | 15.9 ± 3.44 | 0.14 | 1 | |
| Polι | dCTP | 2.31 ± 0.04 | 25.6 ± 2.00 | 0.090 | 0.18 |
| dTTP | 2.16 ± 0.07 | 4.30 ± 0.60 | 0.50 | 1 |
afinc = (Vmax/Km)incorrect/(Vmax/Km)correct.
Human Polι was capable of efficient translesion synthesis opposite 1,N6-ethenoadenine in vitro (Figure 1B, lane 11). However, DNA synthesis was aborted after nucleotide insertion opposite the lesion. As shown in Figure 1B (lanes 12–15), the correct T was preferentially inserted opposite the lesion. Less frequently, C was also inserted. A or G insertion was not detected. Kinetic analysis indicates that the correct T was preferred over C insertion by ∼6-fold (Table 1). These results show that Polι performs efficient and error-free translesion synthesis in response to 1,N6-ethenoadenine in vitro.
Polκ was unable to perform effective translesion synthesis across from 1,N6-ethenoadenine. Only when excessive Polκ was used in the reaction, limited translesion synthesis was observed with nucleotide insertion in the order of T > G > C ∼ A (Figure 1B, lanes 6–10). Based on the results of Figure 1B, we estimated that Polκ was ∼48- and ∼44-fold less efficient than Polη and Polι, respectively, in catalyzing translesion synthesis using the 1,N6-ethenoadenine DNA template.
Our results of in vitro translesion synthesis opposite 1,N6-ethenoadenine by human REV1 were reported previously (25). Together, these results reveal the intrinsic biochemical activities of the human Y family DNA polymerases in response to 1,N6-ethenoadenine. That is, at the site of a template 1,N6-ethenoadenine DNA adduct, Polι is capable of efficient error-free translesion synthesis; Polη and REV1 are capable of efficient error-prone translesion synthesis by preferentially inserting A and C, respectively; and Polκ is incapable of effective translesion synthesis.
The Rev1-Polζ pathway is the major mechanism for translesion synthesis of the 1,N6-ethenoadenine DNA adducts in yeast cells
While in vitro biochemistry is highly informative in elucidating the intrinsic activities of potential bypass DNA polymerases in response to 1,N6-ethenoadenine DNA adduct, whether any or all of these polymerases play a role in translesion synthesis and mutagenesis in vivo can only be determined by genetic analysis in cells. Additionally, we are particularly interested in determining whether REV1 plays a catalytic function by employing its dCMP transferase activity in translesion synthesis and mutagenesis of 1,N6-ethenoadenine in cells. Therefore, we examined in vivo translesion synthesis and mutagenesis of this lesion in yeast cells with the help of a genetic system we established (7,36).
Using a 22-mer oligonucleotide containing a site-specific 1,N6-ethenoadenine adduct, a site-specifically damaged plasmid was constructed by ligating the oligonucleotide into a single-stranded plasmid and subsequently converting it into the double-stranded form by in vitro synthesis of the complementary strand using dUTP instead of dTTP. Then, an in vivo genetic assay (7,36) was used to examine translesion synthesis of the site-specific 1,N6-ethenoadenine adduct in yeast cells. In this assay, the site-specifically damaged plasmid was transformed into cells, in which the complementary strand was degraded as a result of extensive DNA strand cleavage at sites of uracil by the sequential actions of a uracil-DNA glycosylase and an AP endonuclease, converting the plasmid DNA back into the single-stranded form (36). Thus, this assay specifically reflects translesion synthesis without interference from DNA repair and template switching mechanisms (36), both of which require double-stranded DNA. The transformation efficiency was determined by using undamaged and double-stranded plasmids in the same experiment. After normalizing for transformation efficiency, the translesion synthesis efficiency in various strains relative to that in the wild-type was calculated. Lastly, the site of translesion synthesis in each colony was recovered by PCR within a 670-bp DNA fragment of the replicated plasmid. The specificity of translesion synthesis was determined by digestions with the BamHI and HindIII restriction endonucleases. PCR products that were sensitive to BamHI cleavage are indicative of C insertion opposite the lesion; while those sensitive to HindIII cleavage are indicative of A insertion.
To examine roles of the Rev1-Polζ pathway and Polη in bypassing the 1,N6-ethenoadenine adduct, we performed in vivo translesion synthesis assays in yeast rev3 (lacking Polζ), rev1 and rad30 (lacking Polη) deletion mutant cells. As shown in Figure 2, translesion synthesis was not affected in the absence of Polη. In contrast, translesion synthesis was reduced to 14 and 15% of the wild-type level in rev1 and rev3 mutant cells, respectively (Figure 2). These results show that the Rev1-Polζ pathway is the major mechanism for translesion synthesis of the 1,N6-ethenoadenine DNA adduct in yeast cells.
Figure 2.

Relative frequencies of translesion synthesis (TLS) in various yeast strains. Using the plasmid pELUf1-ethenoA containing a site-specific 1,N6-ethenoadenine DNA adduct, in vivo translesion synthesis assays were performed as described in ‘Materials and Methods’ section. Relative TLS was obtained by comparing translesion synthesis in various mutant strains to that in the wild-type cells. Slightly different transformation efficiencies as determined with the undamaged pELUf1 were taken into account in calculating the relative efficiencies. Standard deviations are shown as error bars. The genetic background of all strains was isogenic to the BY4741 strain. WT, wild-type; rad30Δ, lacking Polη; rev1Δ, lacking Rev1; rev3Δ, lacking Polζ.
The 1,N6-ethenoadenine DNA adducts are extremely mutagenic
Following in vivo translesion synthesis assays using site-specifically damaged plasmid pELUf1-ethenoA, individual yeast colonies containing replicated plasmid were analyzed to determine the fidelity of translesion synthesis across from the 1,N6-ethenoadenine DNA adduct. In wild-type cells, 100% of the replicated plasmid contained mutations at the lesion site (Table 2). The vast majority resulted from C insertion opposite the lesion, yielding A→G transition mutation (Table 2). A small fraction of the replicated plasmid resulted from A insertion opposite the lesion, yielding A→T transversion mutation (Table 2). Neither the mutation frequency nor the specificity of translesion synthesis was affected by deleting the RAD30 gene that codes for Polη (Table 2). In contrast, mutation frequency was greatly reduced in rev1 and rev3 mutant cells (Table 2). Furthermore, C insertion opposite the lesion was abolished. All residual translesion synthesis products in both mutant strains resulted from A insertion opposite the lesion (Table 2). These results show that the 1,N6-ethenoadenine adduct is extremely mutagenic in yeast cells, resulting in predominantly A→G transition mutation and less frequently A→T transversion mutation; and that mutagenesis, specifically the predominant A→G transition mutation, requires the function of both Rev1 and Polζ.
Table 2.
Specificity of translesion synthesis opposite 1,N6-ethenoadenine in various yeast strains
| Straina | Clones analyzedb | Base incorporation |
|
|---|---|---|---|
| C | A | ||
| Wild-type | 79 | 76 (96%) | 3 (4%) |
| rad30Δ | 80 | 75 (94%) | 5 (6%) |
| rev1Δ | 9c | – | 9 (100%) |
| rev3Δ | 9c | – | 9 (100%) |
arad30Δ, lacking Polη and rev3Δ, lacking Polζ. The genetic background of all strains was isogenic to the BY4741 strain.
bNumber of independent clones analyzed following in vivo translesion synthesis assays using the pELUf1-ethenoA plasmids containing a site-specific 1,N6-ethenoadenine.
cOnly nine colonies of translesion synthesis were detected in transformations with this strain.
Separation of the Rev1 dCMP transferase activity from its non-catalytic function in translesion synthesis
In vitro, the Rev1 dCMP transferase is catalytically active in response to a template 1,N6-ethenoadenine adduct, inserting a C opposite the lesion (25). In vivo, C was predominantly inserted opposite this lesion during translesion synthesis (Table 2). Thus, we asked whether Rev1 acts catalytically in cells by inserting C opposite 1,N6-ethenoadenine DNA adduct. Our approach was to first separate the Rev1 dCMP transferase from its non-catalytic function in translesion synthesis; and then determine if inactivating the dCMP transferase activity alone (while leaving the non-catalytic function intact) affects translesion synthesis.
In order to separate the dCMP transferase activity of Rev1 from its non-catalytic function, we mutated two conserved amino acid residues in the catalytic domain, i.e. D467A/E468A. Whereas the dCMP transferase was readily detected with the wild-type Rev1 protein in vitro (Figure 3B, lanes 2, 3, 5 and 6), this activity was not detectable with the purified mutant Rev1 (Rev1D467A/E468A) (Figure 3B, lanes 8, 9, 11 and 12). To determine whether the mutant Rev1D467A/E468A retains its non-catalytic function in translesion synthesis, we transformed the mutant rev1 gene on a plasmid vector into the rev1 deletion mutant strain and assayed for translesion synthesis and mutagenesis following UV radiation. Loss of translesion synthesis in yeast cells as in the case of rev1 deletion mutant cells results in moderate UV sensitivity (10,13). Thus, cellular UV sensitivity was measured as an indication for the translesion synthesis function of various yeast strains. As shown in Figure 4, UV sensitivity of the rev1 deletion mutant strain was complemented by the mutant rev1D467A/E468A gene on a plasmid vector, as efficient as complementation by the wild-type REV1 gene on the same vector. Thus, the mutant Rev1D467A/E468A has lost its dCMP transferase activity, but retained its non-catalytic activity for translesion synthesis. These results show that the Rev1 dCMP transferase activity is separable from its non-catalytic function in translesion synthesis.
Figure 3.

In vitro assays for the dCMP transferase of Rev1 and the Rev1D467A/E468A mutant protein. (A) Purified mutant Rev1D467A/E468A protein, which was visualized by staining the 10% polyacrylamide gel with Coomassie blue. The full-length mutant Rev1 is indicated by the arrowhead. (B) Standard translesion synthesis assays were performed with purified wild-type (lanes 1–6) or mutant Rev1D467A/E468A (lanes 7–12) protein using either a G template or an AP template as indicated. The AP site in the template sequence is indicated by the X. The 17-mer DNA band is indicative of the dCMP transferase activity.
Figure 4.

UV sensitivity of various yeast strains. Yeast cells were grown in minimum medium. After appropriate dilution, cells were plated onto minimum medium plates. The uncovered plates were irradiated with UV light at the indicated doses. Surviving colonies were counted after incubation at 30°C for 3–4 days. Survival rates are expressed relative to those of non-irradiated cells. Results are averages of triplicate experiments with the standard deviations shown as error bars. The genetic background of all strains was isogenic to the CL1265-7C strain. Strains rev1Δ/REV1 and rev1Δ/REV1mt are rev1 deletion mutant (rev1Δ) cells containing the wild-type REV1 gene and the mutant rev1D467A/E468A gene, respectively, on a plasmid under the ADH1 promoter control.
Translesion synthesis and mutagenesis of UV lesions require a non-catalytic function of Rev1 but are independent of its dCMP transferase activity
The Rev1-Polζ translesion synthesis pathway is the major mechanism of UV-induced mutagenesis in yeast (10,11,13). The UV sensitivity results (Figure 4) suggest that translesion synthesis of UV lesions is not affected by the D467A/E468A mutations in Rev1 protein. To provide further evidence supporting this conclusion, we assayed for UV mutagenesis at the arg4-17 genetic locus of various yeast strains. As expected, mutagenesis induced by UV radiation at 20 J/m2 was reduced by ∼253-fold in the rev1 deletion mutant strain as compared to that of the wild-type strain (Table 3). This deficiency in UV mutagenesis was fully complemented by either the wild-type REV1 gene or the mutant rev1D467A/E468A gene (Table 2). Thus, the mutant Rev1D467A/E468A protein is fully functional in translesion synthesis and mutagenesis following UV radiation. These results show that translesion synthesis and mutagenesis of UV lesions require a non-catalytic function of Rev1 but are independent of its dCMP transferase activity.
Table 3.
UV-induced mutagenesis in various yeast strains
| Straina | UV survival (% at 20 J/m2) | Mutants/108 viable cells |
|---|---|---|
| Wild-type | 50 ± 2 | 2279 ± 147 |
| rev1Δ | 7 | <9 b |
| rev1Δ/REV1 | 52 ± 1 | 2472 ± 271 |
| rev1Δ/REV1mt | 44 ± 5 | 3357 ± 349 |
arev1Δ/REV1, rev1 deletion mutant expressing wild-type Rev1 protein from the plasmid pEAT-REV1; rev1Δ/REV1mt, rev1 deletion mutant expressing the mutant Rev1D467A/E468A protein from the plasmid pEAT-Rev1D467A/E468A. The genetic background of all strains was isogenic to the CL1265-7C strain.
bLimit of detection. No mutant colonies were detected.
Translesion synthesis and mutagenesis of 1,N6-ethenoadenine DNA adducts require the dCMP transferase activity of Rev1
The mutant Rev1D467A/E468A lacks the dCMP transferase activity, but retains its non-catalytic function for translesion synthesis (see ‘Results’ section above). This allowed us to determine whether C insertion opposite 1,N6-ethenoadenine DNA adduct by the Rev1 dCMP transferase as we observed in vitro (25) is biologically relevant for translesion synthesis and mutagenesis in cells. Using site-specifically damaged plasmids pELUf1-ethenoA containing a single 1,N6-ethenoadenine adduct, we performed in vivo translesion synthesis assays with various yeast strains. In rev1 deletion mutant cells, translesion synthesis was greatly reduced, leaving a residual activity of ∼16% of the wild-type level (Figure 5). This deficiency in translesion synthesis was complemented by the wild-type REV1 gene carried on a plasmid vector, but could not be complemented by the rev1D467A/E468A mutant gene (Figure 5). That is, cells containing the Rev1D467A/E468A mutant protein instead of the wild-type Rev1 remain as deficient as the rev1 deletion mutant cells for translesion synthesis of 1,N6-ethenoadenine adduct (Figure 5).
Figure 5.

Relative frequencies of translesion synthesis (TLS) in yeast strains expressing the mutant Rev1D467A/E468A protein. Using the plasmid pELUf1-ethenoA containing a site-specific 1,N6-ethenoadenine, in vivo translesion synthesis assays were performed as described in ‘Materials and Methods’ section. Relative TLS was obtained by comparing translesion synthesis in various mutant strains to that in the wild-type cells. Slightly different transformation efficiencies as determined with the undamaged pELUf1 were taken into account in calculating the relative efficiencies. Standard deviations are shown as error bars. The genetic background of all strains was isogenic to the CL1265-7C strain. WT, wild-type; rev1Δ, rev1 deletion mutant; rev1Δ/REV1 and rev1Δ/REV1mt, rev1 deletion mutant cells containing the wild-type REV1 gene and the mutant rev1D467A/E468A gene, respectively, on a plasmid under the ADH1 promoter control.
Products of translesion synthesis were analyzed to determine the in vivo specificity of nucleotide insertion opposite 1,N6-ethenoadenine adduct. As we observed before (Table 2), C was predominantly inserted opposite this lesion, which was abolished in rev1 deletion mutant cells (Table 4). Transformation of the wild-type REV1 gene on a plasmid vector into the mutant cells restored the specificity of translesion synthesis with predominant C insertion opposite the lesion (Table 4). In contrast, the yeast rev1 deletion mutant strain containing the rev1D467A/E468A mutant gene on the same plasmid vector showed no C insertion opposite the lesion. Instead, 100% of the residual translesion synthesis products contained A insertion opposite the lesion, as in the case of the rev1 deletion mutant strain (Table 4). These results show that translesion synthesis and mutagenesis of 1,N6-ethenoadenine DNA adducts require the dCMP transferase activity of Rev1.
Table 4.
Specificity of translesion synthesis opposite 1, N6-ethenoadenine in cells expressing the mutant Rev1D467A/E468A protein
| Straina | Clones analyzedb | Base incorporation |
|
|---|---|---|---|
| C | A | ||
| Wild-type | 64 | 54 (84%) | 10 (16%) |
| rev1Δ | 27 | – | 27 (100%) |
| rev1Δ/REV1 | 69 | 65 (94%) | 4 (6%) |
| rev1Δ/REV1mt | 21 | – | 21 (100%) |
arev1Δ/REV1, rev1 deletion mutant expressing wild-type Rev1 protein from the plasmid pEAT-REV1; rev1Δ/REV1mt, rev1 deletion mutant expressing the mutant Rev1D467A/E468A protein from the plasmid pEAT-REV1D467A/E468A. The genetic background of all strains was isogenic to the CL1265-7C strain.
bNumber of independent clones analyzed following in vivo translesion synthesis assays using the pELUf1-ethenoA plasmids containing a site-specific 1,N6-ethenoadenine.
DISCUSSION
The 1,N6-ethenoadenine lesion is an exocyclic DNA adduct induced by the carcinogens vinyl chloride and urethane. It is also formed spontaneously in cells from lipid peroxidation products (30,31). In mammalian cells, 1,N6-ethenoadenine DNA adducts are mutagenic (31,41,42), which induce mainly A→G transitions in simian kidney cells (42). In another study with human cells, it was reported that slightly more A→T than A→G mutations and least frequently A→C mutations were induced (41). To understand the molecular mechanisms of translesion synthesis and mutagenesis of this important DNA base damage, we determined the intrinsic biochemical activities of the human Y family DNA polymerases in response to a template 1,N6-ethenoadenine in vitro. Furthermore, we identified the major mechanism of in vivo translesion synthesis and mutagenesis of a site-specific 1,N6-ethenoadenine DNA adduct in the yeast model system, and examined the roles of Polζ, Polη and Rev1 in these cellular processes.
In response to a template 1,N6-ethenoadenine DNA adduct in vitro, human Polι is capable of efficient error-free translesion synthesis. Human REV1 (25) and Polη are capable of efficient error-prone translesion synthesis by preferentially inserting C and A, respectively. In contrast, human Polκ is incapable of effective translesion synthesis. These results illustrate again that there are no uniformed rules to predict the behavior of the various bypass polymerases in response to different types of DNA lesions. Such intrinsic biochemical activities of the polymerases for translesion synthesis need to be experimentally determined. Translesion synthesis can be error-free or error-prone, depending on the specific bypass polymerase and the specific type of DNA lesion. Our results are generally in good agreement with those of Levine et al. (43) who reported in vitro translesion synthesis of 1,N6-ethenoadenine by human Polη and Polκ in a different sequence context. A notable difference is the preferred nucleotide insertion by human Polη opposite the lesion. Whereas A was preferred by 3.5-fold over T in our studies, T was preferred by 2-fold in the studies of Levine et al. (43). This difference likely resulted from the different sequence contexts used in these two studies. In addition to A insertion opposite the lesion, our sequence context, 3′-CXTC-5′, may promote A incorporation via base paring with the 5′ T by looping out the lesion on the template (X). In contrast, the sequence context of Levine et al. (43), 3′-AXCT-5′, may promote T incorporation via base paring with the 3′ A by −1 slippage of the primer end, followed by template-primer realignment to copy the next template base C 5′ of the lesion.
In vitro biochemistry is highly informative in elucidating the intrinsic activities of potential bypass DNA polymerases in response to a DNA lesion. However, due to the presence of multiple bypass polymerases in cells, not every bypass polymerase is equally utilized for in vivo translesion synthesis. A polymerase with intrinsic bypass capability may not be utilized at all in cells for translesion synthesis of the same lesion. This underscores the importance of performing genetic analysis of translesion synthesis in cells. When such genetic analysis is combined with in vitro biochemistry, precise mechanism of translesion synthesis and mutagenesis can often be elucidated.
Human REV1 efficiently inserts a C opposite a template 1,N6-ethenoadenine in vitro (25). This raised the possibility that A→G mutations induced by 1,N6-ethenoadenine might result from REV1-catalyzed C insertion opposite the lesion during translesion synthesis. Using the eukaryotic model system S. cerevisiae, we have demonstrated that this hypothesis in indeed correct.
In yeast cells, 1,N6-ethenoadenine induces A→G transition mutations as the major event and A→T tansversion mutations as a minor event. Such a mutagenesis specificity is similar to that in the mammalian system (42). In the absence of the Rev1-Polζ translesion synthesis pathway, as in the rev3 or rev1 deletion mutant cells, replication of the damaged plasmid was dramatically reduced and the predominant A→G transition mutations were abolished. Thus, mutations induced by 1,N6-ethenoadenine DNA adducts result from replication of the damaged site via translesion synthesis by the Rev1-Polζ pathway. In a unique strain where the Rev1 protein had been mutated (Rev1D467A/E468A) inactivating its dCMP transferase but retaining its non-catalytic function in lesion bypass, translesion synthesis of 1,N6-ethenoadenine DNA adducts was dramatically reduced and A→G transition mutations were abolished, just like the rev1 deletion mutant strain. Therefore, the Rev1 dCMP transferase activity is exclusively responsible for inserting C opposite the lesion as the major mechanism of translesion synthesis and mutagenesis of 1,N6-ethenoadenine DNA adducts in yeast cells. Since Rev1 is unable to perform extension synthesis from opposite 1,N6-ethenoadenine (25), another bypass polymerase is needed to complete translesion synthesis. Without an extension polymerase, translesion synthesis and A→G transition mutations would not occur. This was indeed the case in rev3 deletion mutant cells that lack Polζ function, suggesting that Polζ acts as the extension polymerase following C insertion by Rev1 during translesion synthesis and mutagenesis of 1,N6-ethenoadenine DNA adducts. Therefore, 1,N6-ethenoadenine DNA adducts are predominantly replicated in yeast cells by Rev1-catalyzed C insertion followed by Polζ-catalyzed extension, a two-polymerase two-step mechanism of translesion synthesis (Figure 6).
Figure 6.
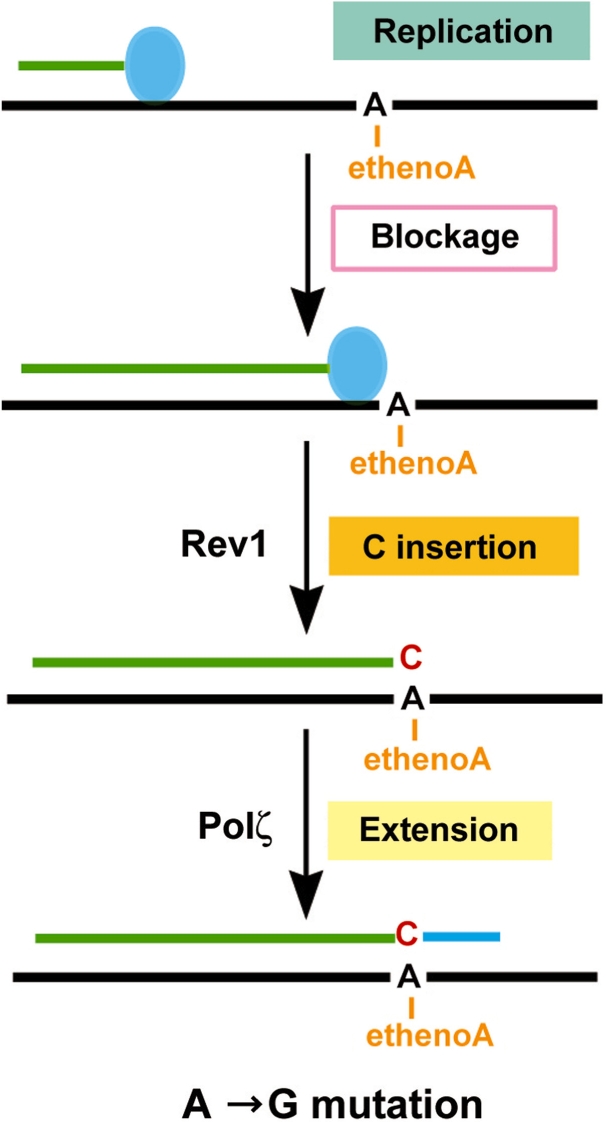
A mechanistic model for translesion synthesis of the 1,N6-ethenoadenine DNA adduct in yeast cells. The replication complex (represented by the filled blue oval) is blocked by the lesion, signaling translesion synthesis. Translesion synthesis is mediated predominantly by C insertion opposite the lesion catalyzed by the Rev1 dCMP transferase. Extension synthesis by Polζ completes the lesion bypass. This major mechanism of translesion synthesis results in A→G transition mutations.
In addition to the major A→G mutagenesis mechanism catalyzed by the Rev1-Polζ pathway, a very minor mechanism of translesion synthesis was also observed, in which A was inserted opposite the 1,N6-ethenoadenine DNA adduct, resulting in A→T transversion mutation. Since A→T mutations were still observed in the absence of either Rev1 or Polζ (Table 2), A insertion opposite the lesion is likely catalyzed by another polymerase. Human Polη is capable of efficient translesion synthesis by preferentially inserting A opposite 1,N6-ethenoadenine in vitro (Figure 1B and Table 1). However, both translesion synthesis and mutagenesis of 1,N6-ethenoadenine are not significantly affected in mutant cells (rad30) lacking this polymerase. Therefore, Polη unlikely plays a role in translesion synthesis and mutagenesis of 1,N6-ethenoadenine in yeast. We speculate that a replicative polymerase such as Polα could be responsible for A insertion in the minor translesion synthesis pathway of 1,N6-ethenoadenine DNA adducts.
Although REV1 is a member of the Y family DNA polymerases, its role in translesion synthesis and mutagenesis is widely believed to be non-catalytic, i.e. its dCMP transferase activity is not required for biological function. REV1 interacts with other Y family DNA polymerases (28). Thus, it was postulated that REV1 functions non-catalytically by recruiting other Y family bypass polymerases for translesion synthesis (28). Regardless of how REV1 fulfils its non-catalytic function in translesion synthesis, a key mechanistic question is whether this protein functions catalytically in translesion synthesis at all. In this study, we have now resolved this question.
By mutating two conserved amino acid residues in the catalytic domain of yeast Rev1, i.e. D467A/E468A, we were able to separate the catalytic function from its non-catalytic function. This mutant Rev1 has lost the dCMP transferase activity as demonstrated by in vitro biochemistry. Its non-catalytic function, however, remains intact, as demonstrated by the fact that this mutant Rev1 protein is fully functional in translesion synthesis and mutagenesis of UV radiation in vivo. Based on the mutation spectra and the fact that REV1 is unable to catalyze C insertion opposite the major UV lesions in vitro, it was concluded that Rev1 functions non-catalytically in translesion synthesis and mutagenesis of UV radiation (5,25). Our results with the rev1D467A/E468A mutant cells provided direct and unequivocal evidence for this conclusion. Our in vitro biochemical and in vivo genetic results together show that the catalytic function of Rev1 is separable from its non-catalytic function in translesion synthesis.
Using the rev1D467A/E468A mutant cells in which the Rev1 catalytic function is inactivated whereas its non-catalytic function remains intact, we found that the dCMP transferase activity is required for translesion synthesis and mutagenesis of 1,N6-ethenoadenine DNA adducts. In fact, this catalytic function of Rev1 is responsible for the predominant mechanism of translesion synthesis and mutagenesis of this lesion in yeast cells. Therefore, the catalytic function of the Rev1 dCMP transferase is required in a lesion-specific manner for translesion synthesis and base damage-induced mutagenesis. We have shown here that 1,N6-ethenoadenine is clearly one such DNA lesion whose replication and mutagenesis depend on the catalytic function of Rev1. AP site is probably another lesion that belongs to this category of translesion synthesis. Several pieces of evidence from multiple laboratories support the involvement of the Rev1 dCMP transferase in the bypass and mutagenesis of AP sites (5,36,44). Definitive prove of this conclusion about AP site bypass could be performed now using the yeast rev1D467A/E468A mutant strain.
Replication of DNA containing the 1,N6-ethenoadenine lesion is heavily dependent on the Rev1-Polζ translesion synthesis pathway. In the absence of this pathway as in rev3 or rev1 deletion mutant cells, the efficiency of replicating the 1,N6-ethenoadenine site is reduced by ∼7-fold. Thus, 1,N6-ethenoadenine is a strong blocker to normal DNA replication. A strong blocking effect was also observed by Tolentino et al. (45) in human cell extracts for replicating plasmids containing a site-specific 1,N6-ethenoadenine. Remarkably, all replicated products in yeast cells contain mutations at the lesion site, i.e. T insertion opposite the lesion was never recovered (Tables 2 and 4). The mutation frequency is 100%. Every time this lesion is replicated, a mutation is produced at the lesion site. Thus, the 1,N6-ethenoadenine DNA adducts are extremely mutagenic. In mammals, the mutagenecity of this lesion may be somewhat reduced by Polι-catalyzed error-free translesion synthesis as suggested by our in vitro results (Figure 1 and Table 1). Whether this hypothesis is correct will have to wait for genetic analysis in mammalian mutant cells lacking Polι. The 1,N6-ethenoadenine DNA adducts can be formed spontaneously in cells from lipid peroxidation products (30,31). Under excessive oxidative stress conditions, such as chronic inflammation in colitis, hepatitis and pancreatitis, higher levels of 1,N6-ethenoadenine are produced (46). These conditions are associated with increased risk of cancer (47). The extreme mutagenic property of 1,N6-ethenoadenine raises the possibility that this lesion might play an important role in carcinogenesis associated with a chronic inflammation disease.
FUNDING
NIH grant CA92528 and a Kentucky Lung Cancer Research grant. Funding for open access charge: the Kentucky Lung Cancer Research Program.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Christopher Lawrence for providing us with the yeast strain CL1265-7C.
REFERENCES
- 1.Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z. Mechanism of bypass polymerases in eukaryotes. In: Siede W, Kow YW, Doetsch PW, editors. DNA Damage Recognition. New York, NY: Taylor & Francis Group; 2006. pp. 507–528. [Google Scholar]
- 3.Xie Z, Zhang Y, Guliaev AB, Shen H, Hang B, Singer B, Wang Z. The p-benzoquinone DNA adducts derived from benzene are highly mutagenic. DNA Repair. 2005;4:1399–1409. doi: 10.1016/j.dnarep.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Xie Z, Braithwaite E, Guo D, Zhao B, Geacintov NE, Wang Z. Mutagenesis of benzo[a]pyrene diol epoxide in yeast: requirement for DNA polymerase ζ and involvement of DNA polymerase η. Biochemistry. 2003;42:11253–11262. doi: 10.1021/bi0346704. [DOI] [PubMed] [Google Scholar]
- 5.Nelson JR, Gibbs PE, Nowicka AM, Hinkle DC, Lawrence CW. Evidence for a second function for Saccharomyces cerevisiae Rev1p. Mol. Microbiol. 2000;37:549–554. doi: 10.1046/j.1365-2958.2000.01997.x. [DOI] [PubMed] [Google Scholar]
- 6.Gibbs PE, McDonald J, Woodgate R, Lawrence CW. The relative roles in vivo of Saccharomyces cerevisiae Pol η, Pol ζ, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6-4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics. 2005;169:575–582. doi: 10.1534/genetics.104.034611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao B, Wang J, Geacintov NE, Wang Z. Polη, Polζ and Rev1 together are required for G to T transversion mutations induced by the (+)- and (-)-trans-anti-BPDE-N2-dG DNA adducts in yeast cells. Nucleic Acids Res. 2006;34:417–425. doi: 10.1093/nar/gkj446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson RE, Torres-Ramos CA, Izumi T, Mitra S, Prakash S, Prakash L. Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1, and its role in the repair of abasic sites. Genes. Dev. 1998;12:3137–3143. doi: 10.1101/gad.12.19.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawrence CW, Das G, Christensen RB. REV7, a new gene concerned with UV mutagenesis in yeast. Mol. Gen. Genet. 1985;200:80–85. doi: 10.1007/BF00383316. [DOI] [PubMed] [Google Scholar]
- 10.Larimer FW, Perry JR, Hardigree AA. The REV1 gene of Saccharomyces cerevisiae: isolation, sequence, and functional analysis. J. Bacteriol. 1989;171:230–237. doi: 10.1128/jb.171.1.230-237.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison A, Christensen RB, Alley J, Beck AK, Bernstine EG, Lemontt JF, Lawrence CW. REV3, a Saccharomyces cerevisiae gene whose function is required for induced mutagenesis, is predicted to encode a nonessential DNA polymerase. J. Bacteriol. 1989;171:5659–5667. doi: 10.1128/jb.171.10.5659-5667.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawrence CW, Hinkle DC. DNA polymerase ζ and the control of DNA damage induced mutagenesis in eukaryotes. Cancer Surveys. 1996;28:21–31. [PubMed] [Google Scholar]
- 13.Rajpal DK, Wu X, Wang Z. Alteration of ultraviolet-induced mutagenesis in yeast through molecular modulation of the REV3 and REV7 gene expression. Mutat. Res. 2000;461:133–143. doi: 10.1016/s0921-8777(00)00047-1. [DOI] [PubMed] [Google Scholar]
- 14.Gibbs PE, McGregor WG, Maher VM, Nisson P, Lawrence CW. A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase ζ. Proc. Natl Acad. Sci. USA. 1998;95:6876–6880. doi: 10.1073/pnas.95.12.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbs PE, Wang XD, Li Z, McManus TP, McGregor WG, Lawrence CW, Maher VM. The function of the human homolog of saccharomyces cerevisiae REV1 is required for mutagenesis induced by UV light. Proc. Natl Acad. Sci. USA. 2000;97:4186–4191. doi: 10.1073/pnas.97.8.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhopadhyay S, Clark DR, Watson NB, Zacharias W, McGregor WG. REV1 accumulates in DNA damage-induced nuclear foci in human cells and is implicated in mutagenesis by benzo[a]pyrenediolepoxide. Nucleic Acids Res. 2004;32:5820–5826. doi: 10.1093/nar/gkh903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dumstorf CA, Mukhopadhyay S, Krishnan E, Haribabu B, McGregor WG. REV1 is implicated in the development of carcinogen-induced lung cancer. Mol. Cancer Res. 2009;7:247–254. doi: 10.1158/1541-7786.MCR-08-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poltoratsky V, Horton JK, Prasad R, Wilson SH. REV1 mediated mutagenesis in base excision repair deficient mouse fibroblast. DNA Repair. 2005;4:1182–1188. doi: 10.1016/j.dnarep.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Nelson JR, Lawrence CW, Hinkle DC. Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science. 1996;272:1646–1649. doi: 10.1126/science.272.5268.1646. [DOI] [PubMed] [Google Scholar]
- 20.Burgers PM, Koonin EV, Bruford E, Blanco L, Burtis KC, Christman MF, Copeland WC, Friedberg EC, Hanaoka F, Hinkle DC, et al. Eukaryotic DNA polymerases: proposal for a revised nomenclature. J. Biol. Chem. 2001;276:43487–43490. doi: 10.1074/jbc.R100056200. [DOI] [PubMed] [Google Scholar]
- 21.Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- 22.Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson JR, Lawrence CW, Hinkle DC. Deoxycytidyl transferase activity of yeast REV1 protein. Nature. 1996;382:729–731. doi: 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]
- 24.Lin W, Xin H, Zhang Y, Wu X, Yuan F, Wang Z. The human REV1 gene codes for a DNA template-dependent dCMP transferase. Nucleic Acids Res. 1999;27:4468–4475. doi: 10.1093/nar/27.22.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Wu X, Rechkoblit O, Geacintov NE, Taylor JS, Wang Z. Response of human REV1 to different DNA damage: preferential dCMP insertion opposite the lesion. Nucleic Acids Res. 2002;30:1630–1638. doi: 10.1093/nar/30.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masuda Y, Takahashi M, Fukuda S, Sumii M, Kamiya K. Mechanisms of dCMP transferase reactions catalyzed by mouse Rev1 protein. J. Biol. Chem. 2002;277:3040–3046. doi: 10.1074/jbc.M110149200. [DOI] [PubMed] [Google Scholar]
- 27.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- 28.Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, Kisker C, Friedberg EC. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, Ulrich HD, Friedberg EC. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell. 2006;23:265–271. doi: 10.1016/j.molcel.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 30.Bartsch H, Barbin A, Marion MJ, Nair J, Guichard Y. Formation, detection, and role in carcinogenesis of ethenobases in DNA. Drug Metabolism Rev. 1994;26:349–371. doi: 10.3109/03602539409029802. [DOI] [PubMed] [Google Scholar]
- 31.Nair J, Barbin A, Velic I, Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutat. Res. 1999;424:59–69. doi: 10.1016/s0027-5107(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Yuan F, Wu X, Talor J-S, Wang Z. Error-prone lesion bypass by human DNA polymerase η. Nucleic Acids Res. 2000;28:4717–4724. doi: 10.1093/nar/28.23.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Yuan F, Wu X, Wang Z. Preferential incorporation of G opposite template T by the low fidelity human DNA polymerase ι. Mol. Cell. Biol. 2000;20:7099–7108. doi: 10.1128/mcb.20.19.7099-7108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Yuan F, Wu X, Wang M, Rechkoblit O, Taylor J-S, Geacintov NE, Wang Z. Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo D, Xie Z, Shen H, Zhao B, Wang Z. Translesion synthesis of acetylaminofluorene-dG adducts by DNA polymerase ζ is stimulated by yeast Rev1 protein. Nucleic Acids Res. 2004;32:1122–1130. doi: 10.1093/nar/gkh279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao B, Xie Z, Shen H, Wang Z. Role of DNA polymerase η in the bypass of abasic sites in yeast cells. Nucleic Acids Res. 2004;32:3984–3994. doi: 10.1093/nar/gkh710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie Z, Braithwaite E, Guo D, Bo Z, Geacintov NE, Wang Z. Mutagenesis of benzo[a]pyrene diol epoxide in yeast: Requirement for DNA polymerase ζ and involvement of DNA polymerase η. Biochemistry. 2003;42:11253–11262. doi: 10.1021/bi0346704. [DOI] [PubMed] [Google Scholar]
- 38.Creighton S, Bloom LB, Goodman MF. Gel fidelity assay measuring nucleotide misinsertion, exonucleolytic proofreading, and lesion bypass efficiencies. Methods Enzymol. 1995;262:232–256. doi: 10.1016/0076-6879(95)62021-4. [DOI] [PubMed] [Google Scholar]
- 39.Ausubel FM, Brent R, Kinston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. New York, NY: Greene Publishing Associates and Wiley-Interscience; 1987. [Google Scholar]
- 40.Becker DM, Guarente L. High-efficiency transformation of yeast by electroporation. Methods Enzymol. 1991;194:182–187. doi: 10.1016/0076-6879(91)94015-5. [DOI] [PubMed] [Google Scholar]
- 41.Levine RL, Yang IY, Hossain M, Pandya GA, Grollman AP, Moriya M. Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res. 2000;60:4098–4104. [PubMed] [Google Scholar]
- 42.Pandya GA, Moriya M. 1,N6-ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry. 1996;35:11487–11492. doi: 10.1021/bi960170h. [DOI] [PubMed] [Google Scholar]
- 43.Levine RL, Miller H, Grollman A, Ohashi E, Ohmori H, Masutani C, Hanaoka F, Moriya M. Translesion DNA synthesis catalyzed by human pol η and pol κ across 1,N6-ethenodeoxyadenosine. J. Biol. Chem. 2001;276:18717–18721. doi: 10.1074/jbc.M102158200. [DOI] [PubMed] [Google Scholar]
- 44.Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, de Wind N. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Experimental Med. 2006;203:319–323. doi: 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tolentino JH, Burke TJ, Mukhopadhyay S, McGregor WG, Basu AK. Inhibition of DNA replication fork progression and mutagenic potential of 1, N6-ethenoadenine and 8-oxoguanine in human cell extracts. Nucleic Acids Res. 2008;36:1300–1308. doi: 10.1093/nar/gkm1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbeck's Archives of Surgery / Deutsche Gesellschaft fur Chirurgie. 2006;391:499–510. doi: 10.1007/s00423-006-0073-1. [DOI] [PubMed] [Google Scholar]
- 47.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]