


Abstract
Transcription of protein-coding genes in trypanosomes is polycistronic and gene expression is primarily regulated by post-transcriptional mechanisms. Sequence motifs in the untranslated regions regulate mRNA trans-splicing and RNA stability, yet where UTRs begin and end is known for very few genes. We used high-throughput RNA-sequencing to determine the genome-wide steady-state mRNA levels (‘transcriptomes’) for ∼90% of the genome in two stages of the Trypanosoma brucei life cycle cultured in vitro. Almost 6% of genes were differentially expressed between the two life-cycle stages. We identified 5′ splice-acceptor sites (SAS) and polyadenylation sites (PAS) for 6959 and 5948 genes, respectively. Most genes have between one and three alternative SAS, but PAS are more dispersed. For 488 genes, SAS were identified downstream of the originally assigned initiator ATG, so a subsequent in-frame ATG presumably designates the start of the true coding sequence. In some cases, alternative SAS would give rise to mRNAs encoding proteins with different N-terminal sequences. We could identify the introns in two genes known to contain them, but found no additional genes with introns. Our study demonstrates the usefulness of the RNA-seq technology to study the transcriptional landscape of an organism whose genome has not been fully annotated.
INTRODUCTION
Trypanosoma brucei is a protozoan parasite that causes African trypanosomiasis, commonly referred to as Sleeping Sickness in humans and Nagana in livestock. Sleeping Sickness is a vector-borne disease and its transmission among mammalian hosts is mediated by the tsetse (Glossina ssp.). Like other vector-transmitted parasites, T. brucei exists in several life-cycle stages. The two forms most commonly used in laboratory settings are the so-called bloodstream form (BF), which proliferates in the bloodstream of the mammalian host, and the procyclic form (PF), to which the BF differentiates in the midgut of the tsetse. The large environmental differences between its mammalian host and the insect vector require the parasite to undergo extensive cellular remodeling, exchanging the major surface proteins, activating cytochrome-mediated metabolism in the mitochondrion, attenuating endocytic activity and displaying differences in morphology and cell-cycle checkpoints (1,2). Very little is known about how gene expression is regulated in trypanosomes.
Unusually for a eukaryote, genes transcribed by RNA polymerase II (RNA pol II) are arranged in polycistronic transcription units (3). Individual mRNAs are separated post-transcriptionally by coupled splicing and polyadenylation reactions (4,5). A 39-nt leader sequence is trans-spliced onto every mRNA (6,7). The arrangement of functionally unrelated genes in polycistronic transcription units has led to the assumption that there is little regulation of gene expression at the level of transcription initiation. Instead, there is ample evidence that steady-state mRNA levels are strongly influenced by differences in the maturation and stability of individual mRNAs (8).
In yeast and human cells, the rate of mRNA degradation can be greatly influenced by sequence motifs in the 3′ untranslated region (UTR) that are recognized by components of the mRNA degradation machinery or factors contributing to increased RNA stability (9,10). In T. brucei, sequence elements of 16 nt and 26 nt in the 3′ UTR stabilize procyclin mRNAs in PF (11–13). Incorporation of these sequence motifs into the 3′ UTRs of a reporter gene conferred stage-specific regulation, with high expression in PF and low expression in BF (12). Additional regulatory elements have been identified in the 3′ UTRs of genes in T. brucei and related species (14–17).
Analyses of 5′ UTR sequences from several eukaryotes indicate that highly expressed genes have short 5′ UTRs with a low GC content, and contain no ATG codons (18). No such correlations have been described for T. brucei, but two studies demonstrated the role of motifs upstream of the 5′ UTR in trans-splicing efficiency (19,20). In Crithidia fasciculata, a distant relative of T. brucei, an octamer motif in the 5′ UTR regulates cell-cycle-dependent levels of certain mRNA (21,22), and a similar mechanism has been proposed for the cell-cycle regulation of DOT1b in T. brucei (23).
A systematic genome-wide search for sequence motifs in UTRs or a correlation of sequence motifs with RNA stability, as has been done in other eukaryotes, has not been performed in trypanosomes. The reasons for this are 2-fold: the exact UTRs have been determined for very few genes and, with the exception of three publications since the submission of this manuscript (24–26), genome-wide mRNA levels have not been measured. Earlier microarray-based analyses of gene expression in T. brucei either focused on a subset of the genome (27) or used microarrays generated from random genomic clones of unknown sequence (28). In the current study, we used high-throughput sequencing to quantify the transcriptomes for BF and PF and to map 5′ and 3′ UTRs for almost 7000 genes.
MATERIALS AND METHODS
Cell lines and culture conditions
PF of T. brucei strain Lister 427 were cultured in SDM-79 (29) containing 10% fetal bovine serum and hemin (7.5 mg/l). Wild-type BF of Lister 427 (MITat 1.2, clone 221a) and a derivative ‘single marker’ line, which expresses T7 RNA polymerase and the Tet repressor (30), were grown in HMI-9 medium (31).
RNA isolation, mRNA enrichment and synthesis of double-stranded cDNA
For each cDNA library, RNA was isolated from ∼6–10 × 108 BF or PF. Exponentially growing cells were harvested (PF at ∼10 × 106 and BF at ∼1.0 × 106/ml), washed with phosphate-buffered saline (PBS) (PF) or trypanosome dilution buffer (5 mM KCl, 80 mM NaCl, 1 mM MgSO4, 20 mM Na2HPO4, 2 mM NaH2PO4, 20 mM glucose, pH 7.4) (BF) and total RNA was isolated using an RNeasy Mini Kit (Qiagen). Typically, we used one RNeasy mini column per ∼108 cells. Genomic DNA was removed by an on-column DNase treatment according to the manufacturer's instructions. mRNA enrichment was performed using an Oligotex mRNA Mini Kit (Qiagen). The enriched mRNA was ethanol precipitated and resuspended at a concentration of ∼1 µg/µl. Double-stranded cDNA was generated from ∼9 µg mRNA using a SuperScript® Double-Stranded cDNA Synthesis Kit (Invitrogen) according to the manufacturer's instructions except that SuperScript III reverse transcriptase (RT) was used instead of SuperScript II RT.
cDNA fragmentation
cDNA samples were processed using the gDNA sample preparation kit from Illumina. The cDNA libraries were sheared by nebulization at 35 psi for 6 min, followed by cleanup with QIAquick PCR purification columns (Qiagen). This resulted in a distribution of fragments from ∼100–1000 bp. End repair of the resultant fragments was performed with T4 DNA polymerase, Klenow polymerase, T4 PNK and dNTP’s in T4 ligase buffer for 30 min at 20°C; cleanup of the reactions was performed with QIAquick PCR purification columns (Qiagen). A-tailing of the blunt-ended products was performed using Klenow exo- (3′–5′ exo minus) and dATP in Klenow buffer for 30 min at 37°C; cleanup was performed with QIAquick MinElute columns (Qiagen). Standard gDNA adapters were ligated to the A-tailed fragments with the supplied ligase and buffer for 15 min at 20°C. Cleanup with QIAquick PCR purification column (Qiagen) followed. After ligation, fragments were purified using BioRad Certified Low Range Agarose gel in 1X TAE. A 150- to 200-bp gel band was excised and DNA was extracted with the MinElute Gel Extraction Kit (Qiagen). Eighteen cycles of PCR were performed on the size-selected templates using Phusion DNA polymerase (Finnzymes) and supplied PCR primers with initial denaturation at 98°C for 30 s, subsequent denaturation at 98°C for 10 s, annealing at 65°C for 30 s, elongation at 72°C for 30 s and a final 5 min at 72°C. PCR products were purified using QIAquick PCR purification columns (Qiagen) quantified, and sequenced in accordance with the manufacturer’s protocols.
Alignment of sequence tags and determination of transcript levels
Fragmented and processed cDNA was sequenced using an Ilumina (Solexa) sequencer. The sequenced DNA tags (32, 36 or 76 bp in length) were aligned to the T. brucei genome (version 4) (32) with all members of a gene group masked except one (Supplementary Table S1). Gene groups were generated based on sequence homology and open reading frames (ORFs) within a gene group are ≥90% identical. Tag alignment was performed using the Blast Like Alignment Tool (BLAT) (33) allowing ≤2 mismatches. Default parameters were used except for tileSize (the size of match that triggers an alignment) = 10 bp, stepSize (spacing between tiles) = 5, minScore (sets the minimum score and consists of matches minus mismatches minus a gap penalty) = 30 for 32-bp tags and 34 for 36-bp tags. The number of hits per ORF was determined using custom MATLAB (The MathWorks) scripts.
Statistical analysis
Data from eight sequencing runs were used to compare BF and PF cells. Counts in each data set were scaled uniformly to give the data set a median of 100 counts for a gene. This number is arbitrary and does not affect the outcome of the analysis (raw median counts ranged from 13 to 150). That is, for gene g in biological replicate i and technical replicate j, the scaled count 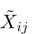 is given by:
is given by:
| (1) |
where 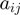 is the scaling coefficient ensuring median
is the scaling coefficient ensuring median 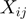 over genes in a data set is 100. The gene index will be dropped in the remainder of the description for clarity.
over genes in a data set is 100. The gene index will be dropped in the remainder of the description for clarity.
We first established that Poisson statistics accurately describe variation between technical replicates (34). This is shown for four technical replicates of PF mRNA in Supplementary Figure S1. We estimate mean and variance over technical replicates as:
and
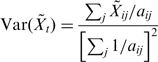 |
(2) |
The estimate of the mean is equivalent to weighting each count by itself, i.e. adding the raw counts, which is optimal for Poisson-distributed samples. In addition, we use the fact that the variance of a Poisson distribution equals its mean. For measurements with 0 count, the variance was estimated based on a single count to prevent underestimation of the variance.
Variation between biological replicates is larger than expected for Poisson noise, with variation between replicates for BF ∼3× larger than expected from counting noise alone and ∼1.15× for PF cells (Supplementary Figure S2). Overdispersion was assumed to affect all genes equally. Excess ‘biological’ variance for each observation was assumed to be additive to counting noise and distributed accordingly over each data set. For example, comparing two biological replicates with technical variance
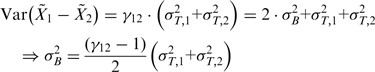 |
with the observed over-dispersion coefficient, and 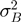 biological variance (in contrast to technical noise).
biological variance (in contrast to technical noise).
To arrive at mean counts for BF and PF, respectively, biological replicates were averaged using weights proportional to 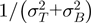 . Finally, statistical significance of differences in counts between PF and BF was assessed using a Student's t-test with the number of degrees of freedom estimated as
. Finally, statistical significance of differences in counts between PF and BF was assessed using a Student's t-test with the number of degrees of freedom estimated as
with 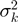 the effective variance for biological replicate k. Because the underlying distributions are Poisson, the variance has the same number of degrees of freedom as the mean.
the effective variance for biological replicate k. Because the underlying distributions are Poisson, the variance has the same number of degrees of freedom as the mean.
Identification of splice-acceptor sites and polyadenylation sites
The last 14 nt (TCTGTACTATATTG) of the spliced leader (SL) sequence (AACTAACGCTATTATTAGAACAGTTTCTGTACTATATTG) are unique. cDNA sequence tags (32, 36 or 76 bp) that contained the 14-nt terminal SL sequence were extracted from the Solexa output of Lister 427 PF and BF samples described earlier, and from one random-primed cDNA sample prepared from PF of strain TREU 927. The SL sequence was found in a minority of the sequenced tags and almost exclusively in the sense direction, because of constraints imposed by the cDNA size-fractionation and sequencing protocols. All of the SL-matching nucleotides were stripped from the sequence tags and the di-nucleotide AG was added to the 5′-end of each tag. All of the genomic matches of 987 777 tag sequences >13-nt long were identified by BLAT analysis against the T. brucei genome sequence. Genes annotated ‘hypothetical protein, unlikely’ were masked; visual inspection suggested most could not encode proteins, as subsequently confirmed for many (35). For short tags, no mismatches were allowed [allowing even one mismatch in these short sequences generated few additional splice-acceptor sites (SAS) and many false hits]. Two mismatches were allowed when aligning the 408 305 tags derived from 76-bp sequences. The BLAT output from a total of 15 sequencing runs of the three datasets were cleaned up using Perl scripts, then imported into FileMakerPro databases that were configured to allow various filters and calculations to be applied for initial curation of the matched sequences, with the ultimate aim of identifying a highly curated set of high-confidence major SAS for each gene. All imperfect matches, or matches to the wrong strand in relation to the relevant adjacent gene, or tags that did not match any relevant gene, probably because we were analyzing mostly Lister 427 strain data against the TREU 927 genome sequence, were deleted, as were all hits linked to RNA genes, retrotransposon hot spot proteins, pseudogenes and non-chromosome-internal expression-site-associated genes. Certain features of the data were more easily assessed in spreadsheet format, which was performed by exchanging data between FileMakerPro and Excel. The three different datasets were merged during curation, but their individual source-identifier information and data were retained. This allows any differences between SASs used for the same gene in PF versus BF trypanosomes to be evaluated, although none was identified.
Potential polyadenylation site (PAS)-spanning tags were extracted from the raw sequence data from 15 Solexa runs, of which six were from cDNA samples specifically primed with oligo(dT), including four 76-bp sequence runs. For the same reasons described for the SAS tag sequences, the vast majority of sequences corresponding to the mRNA 3′-end were in the antisense direction. Sequences that started with at least eight consecutive Ts were identified, of which 80% came from the oligo(dT)-primed samples. After removing all of the terminal T residues, 392 095 tag sequences >15-nt long (3′ UTR sequences are much less distinct than 5′ UTRs, so a longer minimum aligned length was used) were aligned by BLAT against the T. brucei genome. As with the SAS data, the PAS BLAT output was evaluated by applying various filters to generate a curated set of high-confidence PAS. After deleting records where the number of hits per tag was >14 (the maximum copy number of any reiterated gene) and where the predicted UTR length was >5000 nt, the number of records was further reduced by manual curation, which principally involved deleting tags that did not yield unique genome hits.
Identification of introns
The 76-bp sequence tags were aligned to the genome using BLAT and allowing three mismatches. Gapped hits within genes were retrieved using a Perl script and imported into a Filemaker Pro database. Alignments that spanned <90 bp, representing <15-bp insertions or deletions between the sample and genome sequence strains, were deleted. An apparent anomaly in the BLAT algorithm was encountered that occasionally assigned erroneous start or end positions to an alignment, suggesting the presence of a long gap (potential intron) that was incorrect. In some cases, the incorrect alignments were attributable to repeats within a gene. To refine the identification of true intron-spanning tag sequences, all of the tags that BLAT had aligned to supposedly discontinuous regions were re-aligned with the genome using the non-gapping Bowtie algorithm (36) and spurious gapped BLAT matches were thereby identified and eliminated, leaving a small set of potential intron-spanning alignments for further evaluation.
RESULTS
Genome-wide analysis of RNA transcript levels by RNA-seq
Traditional microarray-based transcriptome analyses have several disadvantages, including the high initial cost of high-quality genome-wide tiled microarrays for a novel organism and the lack of precision required for the identification of 5′ SAS and 3′ PAS. Other problems commonly associated with hybridization-based approaches include cross-hybridization artifacts (37) and a limited dynamic range (38). We decided, therefore, to adopt high-throughput cDNA sequencing (RNA-seq), which has several advantages. RNA-seq can be used for the genome-wide analysis of RNA levels and for identifying 5′ SAS and 3′ PAS, it has low background noise, and it has a large dynamic range that appears to be limited only by the depth of the sequencing (39).
Total RNA was isolated from PF or BF cells and enriched for polyA-tailed mRNA. A ∼20-fold decrease in 18s rRNA transcript levels compared to β-tubulin RNA, measured by real time PCR, was typically achieved after one round of enrichment. mRNA was transcribed into double-stranded cDNA, which was fragmented to a narrow size range, adapters were ligated to both ends, and the processed cDNA was amplified and sequenced. The 32- or 36-bp sequence ‘tags’ were mapped to the genome and the number of tags aligned to each gene was summed, yielding relative transcript levels for individual genes. We prepared four cDNA libraries for PF and three for BF, two from wild-type and one from ‘single-marker’ (SM) BF cells (30) that express T7 polymerase and a Tet repressor. Two of the PF libraries were generated using oligo(dT) primers, but all other cDNA libraries were primed with random hexamers and only these were used for expression analyses. The two cDNA libraries primed with oligo(dT) over-represented the 3′-ends of transcripts by up to 10-fold, and were mainly used to identify 3′ PAS. Most of the libraries were sequenced multiple times for a total of 16 sequence runs (5 BF and 11 PF) (Table 1).
Table 1.
Enumeration of sequenced tags
| Bloodstream form | Procyclic form | |
|---|---|---|
| Biological replicates | 3 | 4 |
| Technical replicates | 5 | 11 |
| Total bases sequenced | 727 822 044 | 5 816 182 540 |
| Tag length | 36 bp | 32, 36 or 76 bp |
| Unique tags | 11 108 029 | 12 661 997* |
| Tags used for expression analyses | 5 592 775 | 7 334 554* |
*Excluding the 65 691 627 76-bp tags. Although the majority of these were unique, because of their length, they were poly(A)-primed and not used for expression analysis.
Sequence tags were aligned to the published T. brucei genome using the BLAT alignment tool, a BLAST-like alignment algorithm well suited for the alignment of short tags to genomes of small and intermediate size (33). To evaluate reproducibility of the RNA-seq technology for transcriptome analysis, we determined the number of sequenced cDNA tags for each ORF in each technical and biological replicate and compared the values. The scatter plots and Pearson coefficients of >0.97 indicate a high degree of reproducibility (Figure 1A and B). Differences between SM and wild-type BF were not larger on average than between the two wild-type BF biological replicates (Figure 1B). Therefore, sequence tags from BF wild-type and SM cells were combined for the transcript analysis.
Figure 1.

Evaluation of the reproducibility and coverage of the RNA-seq data. (A) Comparison of two RNA-seq technical replicates (top panel) and two biological replicates (lower panel), expressed as sequence tags per ORF. The number of sequence tags is normalized, based on the median tag count per ORF, to adjust for differences in total tag number between runs. (B) Pearson Coefficients of all sequencing runs used for the transcriptome analysis. Relevant Pearson Coefficients are shaded as follows: dark green, technical replicate BF; light green, biological replicate BF; dark blue, technical replicate PF; light blue, biological replicate PF. (C) Average relative RNA-seq tag coverage of all ORFs from the 5′ (left panel) and 3′ (right panel) ends for a representative sequencing run. (D) RNA-seq tag coverage (one data set) of two representative ORFs, Tb927.6.3740 (heat shock 70 protein), which is highly expressed, and Tb927.5.3510 (structural maintenance of chromosome 3 protein), whose abundance is about 80-fold lower. The blue line indicates the number of sequence tags per nucleotide and the red line represents a moving average of sequence tags per 50 nt. The length of the ORF is indicated by a green bar on the x-axis. Orange and blue bars indicate 5′ UTR and 3′ UTR respectively (left panel).
Because uniform sequence coverage is important for accurate quantification of transcript levels and detection of low-abundance transcripts, we calculated the average distribution of sequence tags along all ORFs (Figure 1C) and for a selection of representative individual genes (Figure 1D). These analyses suggested a uniform distribution of sequence tags throughout the transcript, except for an ∼2-fold drop at the 5′- and 3′-ends. The reason for the drop is that only unidirectional 32- to 36-bp tag sequences will be obtained from the ends of the transcripts. Close to the 5′-end of the RNA transcript, only the forward direction of a 150- to 200-bp cDNA fragment will be represented in the sequence tags; close to the 3′-end of the RNA transcript, only the reverse direction of a cDNA fragment will be represented in the sequence tags.
Comparing transcript levels among genes within each life-cycle stage and between life-cycle stages
Eukaryotic genomes typically contain large regions of repetitive elements and numerous families of paralogous genes. Such situations present a challenge when aligning short sequence tags to the genome. We will refer to tags that align to multiple sites in the genome as non-unique sequence tags. In the past, this problem has been dealt with by either disregarding genes that contribute to non-unique sequence tags (38) or by specifically analyzing the unique regions of those genes (40). Even though the T. brucei genome is only ∼30 Mb, it contains ∼100 gene families. For example, there are 14 copies of histone H2A and at least 10 for α-tubulin. Because many of the non-unique sequence tags align to different members of multi-gene families, we grouped genes based on homology and then determined the total number of sequence tags for these different groups of genes. ‘Gene groups’ were generated by BLAST alignments of ORFs against each other and then grouping ORFs with >90% homology together into a gene group (for a list of grouped genes see Supplementary Table S1). For each gene group, all but one member was masked, thus allowing sequence tags only to align to the unmasked member of a gene group. This approach significantly reduced the problem of non-unique sequence tags. All remaining non-unique sequence tags and all corresponding genes were excluded from the transcript analyses.
To compare transcript levels among genes within a life-cycle stage, we calculated the number of sequence tags per kilobase of ORF. For genes with UTRs longer than 150–200 bp (the average length of the cDNA fragments) the drop in sequence coverage (see above) will fall into the UTR and will have no effect on the sequence coverage across the ORF. Because a drop in sequence coverage would disproportionally affect short genes and genes with short UTRs, we excluded sequence tags that aligned to the first or last 150 bp of each ORF and did not analyze RNA transcript levels of genes shorter than 400 bp (1058 genes). When comparing transcript levels between BF and PF, where reduced sequence coverage at the termini of the ORF will have the same effect in both life cycle stages, we included all genes regardless of their length.
When comparing RNA abundance among genes within one life-cycle stage, we were able to determine RNA transcript levels for 6798 (BF) or 6840 (PF) ORFs. These numbers exclude masked paralogous ORFs, ORFs containing repetitive elements, or ORFs shorter than 400 bp. The distribution of highly expressed and weakly expressed ORFs was similar between BF and PF (Figure 2A and B). One drawback of hybridization-based DNA techniques, like microarrays, is the limited dynamic range—usually 10- to 100-fold. For the current RNA-seq experiments, the dynamic range was almost 106 (Figure 2B). There were many regions where we detected no transcripts, including at divergent strand-switch regions, which are sites of probable transcription initiation (41).
Figure 2.

Distribution of transcripts by tag abundance. (A) Transcript levels were determined for 6798 (BF) or 6840 (PF) ORFs and grouped by the number of unique sequence tags per ORF. ‘Gene groups’ of paralogous genes are counted as one ORF. For 229 (BF) or 322 (PF) ORFs we identified less than five combined unique and non-unique sequence tags. Not included in the graph are 555 (BF) and 513 (PF) ORFs for which the number of non-unique sequence tags was >20% of the unique sequence tags, and 1058 ORFs shorter than 400 bp. All remaining ORFs were grouped based on the number of unique sequence tags per ORF. (B) Distribution of tag abundance by ORF. Only one copy of a gene is included per gene family, n = 6798 (BF) or 6840 (PF). Sequence tags aligning to the first or last 150 bp of each ORF are not counted. ORFs shorter than 400 bp and ORFs for which the number of non-unique sequence tags was more than 20% of the unique sequence tags were omitted.
Based on analyses of selections of genes, previous studies estimated the number of life-cycle-regulated genes at 1–14% of the genome (27,28,42). We compared transcript levels from BF and PF for 7571 genes (excluding paralogous ORFs and ORFs for which the number of non-unique sequence tags was >20% of the number of unique sequence tags). For P ≤ 0.01 (per gene, one-sided t-test), we found 221 genes upregulated >2-fold in BF and 204 upregulated more than 2-fold in PF (Figure 3A and Supplementary Table S2). Thus, 425 genes (5.6% of all genes) are life-cycle regulated by 2-fold or more (P ≤ 0.01), which falls within the range reported previously.
Figure 3.

Comparative analysis of PF and BF RNA levels. (A) Scatter plot showing the normalized abundance of sequence tags per ORF for BF and PF cells. Sequence tag abundance was normalized among replicates based on the median number of tags per ORF. Blue and green squares represent genes upregulated in PF and BF, respectively, with P ≤ 0.01. Gray squares represent genes for which P > 0.01. Diagonal orange lines indicate 2-fold upregulation. Groups of paralogous genes are counted as one ORF and ORFs for which the number of non-unique sequence tags was >20% of the unique sequence tags were omitted. Significance was determined using one-sided t-tests. Error bars indicate standard errors of the mean. (B) Tag abundance per kb of ORF along two representative PCUs (41). Orange arrows indicate the direction of transcription. For Tb10.70.2890, the number of non-unique tags was >20% of the number of unique tags, thus tags per kb of ORF could not be reliably determined.
A detailed comparison of our data with previously published microarray-based transcription data is not possible for a lack of a quantitative analysis of fold-regulation (43) or for a lack of genome-wide analysis (27). However, comparing our data to a set of 113 genes that were found to be stage-specifically regulated in a microarray analysis (43), we observed striking similarities. There were only nine genes where our results appeared to differ, in the degree of regulation, but these differences were not statistically significant (Supplementary Table S3).
Next, we compared our RNA-seq data with microarray and real-time PCR data previously obtained for a selection of genes involved in the ‘strongly regulated membrane trafficking system’ (27). The RNA-seq data correlated weakly with the microarray data (n = 92, R2 = 0.43) but showed good correspondence with the real-time PCR data (n = 26, R2 = 0.68) (Supplementary Figure S3).
Transcription in T. brucei is polycistronic, so life-cycle-dependent differences in RNA abundance could be due to differences in transcription of individual PCUs or to differences in post-transcriptional processing and RNA stability. The former mechanism would presumably lead to a uniform regulation of genes within a given PCU while the latter would allow regulation of individual genes. To identify the predominant type of gene regulation, we plotted RNA transcript levels from BF and PF for individual PCUs (Figure 3B). This revealed a large degree of variability in terms of life-cycle-dependent regulation for genes within the same PCU, suggesting that gene expression is indeed predominantly regulated by post-transcriptional mechanisms.
Identification of trans-splicing sites
Although the majority of inter-transcript regions are characterized by a sharp reduction in RNA-seq tags, and mapping these ‘boundary regions’ was used to define transcript boundaries to a precision of ∼50 nt for many genes in yeast (38), the trans-splicing of all trypanosome mRNAs offered the possibility of precisely mapping the 5′ UTRs and quantifying the use of alternative splice sites. From all of the sequencing runs, we retrieved 987 800 sequence tags (497 427 unique) that spanned the trans-splicing junction and retained at least 14 nt of gene-specific sequence after the SL sequence was removed. The main criteria used to filter the genome alignment results from these 5′ UTR-defining tag sequences were that they gave unique (in the sense that the number of hits matched the appropriate number of genes) perfect matches (or allowing two mismatches for the 76-bp-derived sequences), on the correct strand, with at least two hits per SAS. Pseudogenes were ignored, as were hits that led to UTRs longer than 1999 nt—an arbitrary cutoff, but longer predicted 5′ UTRs probably represent persistent splicing intermediates, or, in some cases, they could indicate that an unidentified ORF was present between the SAS and the downstream gene to which it was assigned. Minor SAS of very abundant transcripts were omitted from the curated dataset.
Using this approach, we were able to identify 10 857 SAS for 6959 genes (Supplementary Table S4). The average number of significant SAS per gene was 1.6 (2.6 before very minor hits were edited out of the curated set). The average 5′ UTR length, based on the predominant SAS when more than one SAS was identified, was 184 bp (median 89 bp), and 80% of 5′ UTRs were shorter than 248 bp (Figure 4A). For most genes, it was impossible to determine whether any SAS were used differentially between life-cycle stages due to limited data for minor SAS sites and for bloodstream-stage cells. In the single TREU 927 sample, 298 SAS were identified for 288 genes that were not represented in the Lister 427 samples, which was presumably due to sequence polymorphisms or to different patterns of gene expression between the two strains. It would have been preferable to be able to analyze data only or mainly from TREU 927, but this was not possible for technical reasons. It would have been ideal for the Lister 427 genome sequence to be available, as this is the most widely used and conveniently manipulated laboratory strain of T. brucei, but we do not yet have that option.
Figure 4.

Length of 5′ and 3′ UTRs. (A) Histogram showing the length distribution of 5′ UTR (left panel; n = 6 644; window 50 nt) and 3′ UTR (right panel; n = 5911; window 100 nt) for the predominant SAS and PAS. If multiple SAS or PAS occurred at the same frequency the length of the shortest UTR was used for this histogram. (B) Quantification of multiple PAS used by β-tubulin (left panel) and α-tubulin (right panel). Nucleotides labeled in red indicate PAS. For β-tubulin, additional sequence tags indicated PAS at 318 bp (1 tag), 323 bp (3), 334 bp (1) and 476 bp (1) downstream of the ORF.
There were 587 SAS that predicted 5′ UTRs <10 nt in length. The 39-nt SL was not included in the UTR length calculation, but it is still possible that translation does not start at the first ATG in these transcripts. There is no way of predicting this, without a body of experimental data.
Although an SAS must obviously precede the start of an ORF, the tagging of a splice site downstream of the originally predicted ATG indicated that a downstream in-frame ATG must form the N-terminus of the protein. There are 488 genes for which 609 SAS predict ORF start sites internal to the originally predicted ORF. Of these 488, there are 321 genes where >95% of the predicted SAS (2–1986 sequence hits per gene) predict the same in-frame ATG inside the originally predicted ORF. There are another 167 genes where between 5 and 95% of the SAS tags (5–642 hits per gene) align downstream of the originally predicted ATG, indicating that alternative proteins can be produced from the same gene, one with the originally predicted (first in-frame) ATG and one that presumably starts at the next in-frame (or a further-downstream) ATG (Supplementary Table S5).
In addition, proteomics data (35) suggested that some trypanosome genes have in-frame ATG codons upstream of the ones that were assigned during genome annotation. The existence of an upstream in-frame ATG that was downstream of at least 90% of the predicted SAS for 178 genes for which we have SAS data suggests that the ORF for these genes should be revised (Supplementary Table S5). For the 12 genes where proteomics data were available, the upstream ATG predicted by the SAS location was consistent with the proteomics data. There were another 23 genes where multiple SAS predict the existence of two alternative forms of the encoded protein: one as originally predicted and one longer. Examination of 1768 genes for which we had no SAS data identified a further 47 genes where the ATG that would give the longest ORF was upstream of the originally predicted one. Nine of these are very unlikely to encode protein extensions because clear polyY tracts that are probably splicing signals were apparent by visual inspection of the predicted extension sequences.
Identification of introns
Two T. brucei genes have been reported to contain introns: Tb927.3.3160, a poly(A) polymerase (44), and Tb927.8.1510, a DNA/RNA helicase (32).The 35 76-bp tags that gave gapped alignments for these two genes (13 for Tb927.3.3160 and 22 for Tb927.8.1510) exactly spanned their introns (Supplementary Figure S4). There were 12 additional tags that identified the trans-splicing site of the first exon and 3 that represented trans-splicing to the second exon of Tb927.3.3160. There were 18 tags that defined the correct trans-splicing site and none that indicated trans-splicing of the second exon of Tb927.8.1510.
All of the potential intron sequences in other genes were retrieved and three criteria were applied to identify true introns: they must terminate in the consensus dinucleotides GT/AG (CT/AC for tags matching the antisense strand), the majority of tags must accurately predict the same intron sequence, and the length of the predicted protein (with the intron removed) must be an integer. These criteria were only satisfied for Tb927.3.3160 and Tb927.8.1510.
Although we could accurately identify the introns in the two genes known to contain them, we did not identify new examples of cis-splicing in T. brucei. The RNA-seq data displayed a significant but incomplete fall-off in the intron region of Tb927.8.1510, but not for Tb927.3.3160, so this method does not provide a reliable way to identify introns. Tb927.3.3160 and Tb927.8.1510 are not abundantly expressed but, by comparing their RNA-seq tag abundance to the tag abundance for all 6979 genes for which we identified the trans-splicing site, we calculated that we should have been able to identify any introns occurring in 85% of the tagged genes. We would not have identified introns within the 5′ regions of ORFs longer than 3000 bp, where the signal from the poly-A-primed cDNA used for the 76-bp runs fell off rapidly, or in genes that are only expressed in the three life-cycle stages that we could not examine.
Identification of polyadenylation sites
Alternative polyadenylation sites (PAS) are common in Leishmania major (4) but contradictory data exist for T. brucei (3,45). To identify PAS, we identified 392 095 sequence tags (366 829 unique sequences) abutting polyA8 tails. Using short sequence tags, we were able to assign PAS sites with high confidence for only 429 abundantly expressed genes, because of the low complexity of 3′ UTR sequences. We therefore performed four additional sequencing runs to obtain 76-bp tag sequences. These allowed us to identify a total of 16 863 PAS for 5948 genes (Supplementary Table S6). For genes that had >20 PAS tag hits, the average number of PAS was ∼10 per gene, indicating the promiscuity of polyadenylation, although the PAS were generally grouped in one or more clusters. All sequence tags predicting 3′ UTRs longer than 5000 bp were disregarded, as they probably represent stable intergenic transcripts or the existence of unpredicted intergenic ORFs. The average predominant 3′ UTR length is 604 bp (median: 400 bp) (Figure 4A). The alternative PAS used by the highly expressed tubulin genes are quantified in Figure 4B.
DISCUSSION
In this study, we present a genome-wide analysis of RNA transcript levels in T. brucei. Our data were highly reproducible over a large dynamic range and sensitive enough to detect RNA transcripts (≥5 sequence tags) from over 7282 genes or gene groups, representing ∼90% of the T. brucei genome. Based on estimates that a single BF trypanosome contains ∼20 000 mRNA molecules (46), detection of five sequence tags per ORF means that the RNA transcript for that particular ORF is present for ∼10% of the BF cell cycle or in 1 of 10 cells. Furthermore, we compared BF and PF transcript datasets and found 425 to be regulated by 2-fold or more. However, a potential problem associated with comparative genome-wide analyses of transcript levels is the lack of a reference point—a gene known to be expressed at equal levels in both life-cycle stages. For this analysis we scaled the BF and PF data sets such that the median of counts per gene is the same in both life cycle stages. A second challenge is that we do not know the natural variability of biological replicates, although, as shown in Supplementary Figure S2, our assumption that biological variance is proportional to average gene expression level (as is technical noise) appears reasonable. Seventy-two genes are common to the top 100 genes expressed in either the BF or PF life-cycle stages (Supplementary Table S7).
Since our manuscript was submitted, three microarray-based analyses of the T. brucei transcriptome were published (24–26), yielding additional insights into gene regulation throughout the parasite’s life cycle. Two of these studies focus on changes in gene expression during differentiation between slender (dividing) BF, stumpy (cell-cycle arrested) BF and PF (25,26). The third study (24) is the most relevant to ours, and reports that 10–25% of genes are regulated among life-cycle stages. Our lower estimate of genes expressed differentially can be attributed to the higher fold-change used as criterion for regulation and the lower P-value chosen as threshold for significant regulation in our study. Of the 50 most highly expressed genes listed (24), 29 were derived from life-cycle stages or growth states (stationary-phase cultures) that we did not examine. A total of 12 of the other 21 that were highly expressed either in BF or PF were in the top 100 genes expressed in one or both of the life-cycle stages in our study. None of the microarray-based analyses could provide UTR or intron information.
mRNA maturation in T. brucei is accompanied by coupled trans splicing and polyadenylation reactions that add a SL RNA and a polyA tail to the ends of the 5′ UTR and 3′ UTR, respectively. Because UTRs contain important regulatory motifs, delineating UTRs will be pivotal for understanding post-transcriptional gene regulation in T. brucei. So far, SAS and PAS have been determined only for a relatively small number of genes, in scattered studies of gene structure and function, and not in any large-scale systematic study.
In yeast and human cells, high-throughput sequencing of cDNA has proven to be a powerful tool not only to determine transcript levels but also to identify novel splice junctions or to confirm known splice sites (38–40). In the current study, we mapped sequence tags spanning splice sites, which enabled us to identify potential SAS for almost 7000 genes. SAS analysis revealed 321 genes in which essentially all of the SAS mapped downstream of the previously assigned initiator ATG, indicating that the original assignment of the coding sequence to the first in-frame ATG of the ORF was erroneous for those genes, which could not have been known without having identified the SAS. There were about 167 genes where the identification of alternative SAS would predict the synthesis of varying amounts of the originally annotated proteins and to alternative versions truncated or extended at the amino terminus.
Life-cycle-specific alternative SAS used by a reporter gene expressed from an rRNA locus has been reported (47), but our SAS data, although not sufficiently extensive to be conclusive on this point, suggest that life-cycle-dependent alternative splicing is not a widespread phenomenon in T. brucei. However, we did observe extensive usage of alternative SAS. It remains to be seen if the use of alternative SAS simply results from sloppiness of the splicing machinery or if it contributes to gene regulation.
ATG codons in the 5′ UTR, creating so-called upstream ORFs, decrease mRNA translation efficiency in many organisms including T. brucei (20,48,49). The fraction of 5′ UTRs containing upstream ATGs ranges from 26% in Saccharomyces sp. (average length of 5′ UTR: 136 bp) and 37% in humans (160 bp), to 52% in Drosophila sp. (288 bp) (50), yet we found an upstream ATG in only 19.5% of T. brucei 5′ UTRs (average length 232 bp, median 104, range 0–2000 bp). This is much less than would occur by chance (almost four per average-length UTR), suggesting that there is strong active selection against ATGs in the UTR.
Identification of PAS was more challenging because the abundance of low-complexity sequences in the 3′ UTR initially made it difficult to identify unique sequence tags spanning potential PAS. Longer (76 bp) sequencing tags allowed us to determine potential PAS for almost 6000 transcripts. The data indicate widespread use of alternative PAS (Figure 4B and Supplementary Table S6). For histone H3, our PAS data indicate a 3′ UTR of 972 bp (∼2.5 times the length of the ORF). This means that transcripts attributed to the gene annotated as ‘hypothetical protein, unlikely’ (Tb927.1.2540) in a recent study (24) most likely stem from the 3′ UTR of histone H3, as we have experimentally confirmed by RT-PCR (J. Wright and G.A.M. Cross, unpublished data). By visual inspection of their sequences, it seems very probable that few if any of the genes annotated as ‘hypothetical protein, unlikely’ are real, which is why we masked them in assigning SAS and PAS to adjacent genes.
The SAS and PAS data are publicly available in three formats. Genome browser tracks indicating the co-ordinates of all curated SASs and PASs can be viewed on the TriTrypDB component of EuPathDB (http://tritrypdb.org/tritrypdb). The alignments of all 987 777 SAS-associated and all 392 095 PAS-associated sequence tags are mapped on parallel tracks, so minor SAS, unexplored potential SAS, and alignments that were excluded during the curation described above can be compared and could lead to novel discoveries. High-confidence SAS and PAS data are being used to reannotate and redefine genes via the TrypDB component of GeneDB (http://www.genedb.org/genedb/tryp/index.jsp). Files containing original data, formatted curated data, and interpretative notes, are available at http://tryps.rockefeller.edu/trypsru2_genome_analyses.html.
In the current study, we explored several possibilities of RNA-seq with respect to transcriptome analysis and generated genome-wide data on transcript levels and splice sites. Although RNA-seq data should be able to identify cases of cis-splicing in trypanosomes (44), we could independently identify introns only in the two genes in which they had previously been reported. These two examples were clear, which leads us to the conclusion that introns are rare in trypanosome genes, unless they are over-represented in genes expressed at very low abundance or in life-cycle stages that were not accessible for the current study. We hope that our data will encourage others to perform comprehensive searches for regulatory motifs in UTRs, and will help to elucidate the secrets of post-transcriptional regulation of gene expression in T. brucei.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Boehringer Ingelheim Fonds and the David Rockefeller Graduate Program (T.N.S.); National Institute of Allergy and Infectious Diseases (NIAID) of the US National Institutes of Health (Grant No. R01AI021729). Funding for open access charge: The Rockefeller University.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Cross lab for helpful discussions, Luisa M. Figueiredo for valuable comments on the manuscript, and Brian Brunk of the TriTrypDB for expeditious development of pipelines for genome-wide displays. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the National Institutes of Health.
REFERENCES
- 1.Matthews KR, Ellis JR, Paterou A. Molecular regulation of the life cycle of African trypanosomes. Trends Parasitol. 2004;20:40–47. doi: 10.1016/j.pt.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 2.Hammarton TC. Cell cycle regulation in Trypanosoma brucei. Mol. Biochem. Parasitol. 2007;153:1–8. doi: 10.1016/j.molbiopara.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tschudi C, Ullu E. Polygene transcripts are precursors to calmodulin mRNAs in trypanosomes. EMBO J. 1988;7:455–463. doi: 10.1002/j.1460-2075.1988.tb02833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LeBowitz JH, Smith HQ, Rusche L, Beverley SM. Coupling of poly(A) site selection and trans-splicing in Leishmania. Genes Dev. 1993;7:996–1007. doi: 10.1101/gad.7.6.996. [DOI] [PubMed] [Google Scholar]
- 5.Matthews KR, Tschudi C, Ullu E. A common pyrimidine-rich motif governs trans-splicing and polyadenylation of tubulin polycistronic pre-mRNA in trypanosomes. Genes Dev. 1994;8:491–501. doi: 10.1101/gad.8.4.491. [DOI] [PubMed] [Google Scholar]
- 6.Campbell DA, Thornton DA, Boothroyd JC. Apparent discontinuous transcription of Trypanosoma brucei variant surface antigen genes. Nature. 1984;311:350–355. doi: 10.1038/311350a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milhausen M, Nelson RG, Sather S, Selkirk M, Agabian N. Identification of a small RNA containing the trypanosome spliced leader: a donor of shared 5′ sequences of trypanosomatid mRNAs? Cell. 1984;38:721–729. doi: 10.1016/0092-8674(84)90267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clayton CE. Life without transcriptional control? From fly to man and back again. EMBO J. 2002;21:1881–1888. doi: 10.1093/emboj/21.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zubiaga AM, Belasco JG, Greenberg ME. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol. Cell. Biol. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JEJ. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003;13:1863–1872. doi: 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hehl A, Vassella E, Braun R, Roditi I. A conserved stem-loop structure in the 3′ untranslated region of procyclin mRNAs regulates expression in Trypanosoma brucei. Proc. Natl Acad. Sci. USA. 1994;91:370–374. doi: 10.1073/pnas.91.1.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hotz HR, Hartmann C, Huober K, Hug M, Clayton C. Mechanisms of developmental regulation in Trypanosoma brucei: a polypyrimidine tract in the 3′-untranslated region of a surface protein mRNA affects RNA abundance and translation. Nucleic Acids Res. 1997;25:3017–3026. doi: 10.1093/nar/25.15.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furger A, Schurch N, Kurath U, Roditi I. Elements in the 3′ untranslated region of procyclin mRNA regulate expression in insect forms of Trypanosoma brucei by modulating RNA stability and translation. Mol. Cell. Biol. 1997;17:4372–4380. doi: 10.1128/mcb.17.8.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotz HR, Lorenz P, Fischer R, Krieger S, Clayton C. Role of 3′-untranslated regions in the regulation of hexose transporter mRNAs in Trypanosoma brucei. Mol. Biochem. Parasitol. 1995;75:1–14. doi: 10.1016/0166-6851(95)02503-0. [DOI] [PubMed] [Google Scholar]
- 15.Di Noia JM, D'Orso I, Sanchez DO, Frasch AC. AU-rich elements in the 3′-untranslated region of a new mucin-type gene family of Trypanosoma cruzi confers mRNA instability and modulates translation efficiency. J. Biol. Chem. 2000;275:10218–10227. doi: 10.1074/jbc.275.14.10218. [DOI] [PubMed] [Google Scholar]
- 16.Boucher N, Wu Y, Dumas C, Dube M, Sereno D, Breton M, Papadopoulou B. A common mechanism of stage-regulated gene expression in Leishmania mediated by a conserved 3′-untranslated region element. J. Biol. Chem. 2002;277:19511–19520. doi: 10.1074/jbc.M200500200. [DOI] [PubMed] [Google Scholar]
- 17.Mayho M, Fenn K, Craddy P, Crosthwaite S, Matthews K. Post-transcriptional control of nuclear-encoded cytochrome oxidase subunits in Trypanosoma brucei: evidence for genome-wide conservation of life-cycle stage-specific regulatory elements. Nucleic Acids Res. 2006;34:5312–5324. doi: 10.1093/nar/gkl598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochetov AV, Ischenko IV, Vorobiev DG, Kel AE, Babenko VN, Kisselev LL, Kolchanov NA. Eukaryotic mRNAs encoding abundant and scarce proteins are statistically dissimilar in many structural features. FEBS Lett. 1998;440:351–355. doi: 10.1016/s0014-5793(98)01482-3. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Estrano C, Tschudi C, Ullu E. Exonic sequences in the 5′ untranslated region of alpha-tubulin mRNA modulate trans splicing in Trypanosoma brucei. Mol. Cell. Biol. 1998;18:4620–4628. doi: 10.1128/mcb.18.8.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siegel TN, Tan KS, Cross GAM. Systematic study of sequence motifs for RNA trans splicing in Trypanosoma brucei. Mol. Cell. Biol. 2005;25:9586–9594. doi: 10.1128/MCB.25.21.9586-9594.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasion SG, Hines JC, Ou X, Mahmood R, Ray DS. Sequences within the 5′ untranslated region regulate the levels of a kinetoplast DNA topoisomerase mRNA during the cell cycle. Mol. Cell. Biol. 1996;16:6724–6735. doi: 10.1128/mcb.16.12.6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown LM, Ray DS. Cell cycle regulation of RPA1 transcript levels in the trypanosomatid Crithidia fasciculata. Nucleic Acids Res. 1997;25:3281–3289. doi: 10.1093/nar/25.16.3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janzen CJ, Hake SB, Lowell JE, Cross GAM. Selective di- or trimethylation of histone H3 lysine 76 by two DOT1 homologs is important for cell cycle regulation in Trypanosoma brucei. Mol. Cell. 2006;23:497–507. doi: 10.1016/j.molcel.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 24.Jensen BC, Sivam D, Kifer CT, Myler PJ, Parsons M. Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei. BMC Genomics. 2009;10:482. doi: 10.1186/1471-2164-10-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabani S, Fenn K, Ross A, Ivens A, Smith TK, Ghazal P, Matthews K. Genome-wide expression profiling of in vivo-derived bloodstream parasite stages and dynamic analysis of mRNA alterations during synchronous differentiation in Trypanosoma brucei. BMC Genomics. 2009;10:427. doi: 10.1186/1471-2164-10-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Queiroz R, Benz C, Fellenberg K, Hoheisel JD, Clayton C. Transcriptome analysis of differentiating trypanosomes reveals the existence of multiple post-transcriptional regulons. BMC Genomics. 2009;10:495. doi: 10.1186/1471-2164-10-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koumandou VL, Natesan SK, Sergeenko T, Field MC. The trypanosome transcriptome is remodelled during differentiation but displays limited responsiveness within life stages. BMC Genomics. 2008;9:298. doi: 10.1186/1471-2164-9-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diehl S, Diehl F, El-Sayed NM, Clayton C, Hoheisel JD. Analysis of stage-specific gene expression in the bloodstream and the procyclic form of Trypanosoma brucei using a genomic DNA-microarray. Mol. Biochem. Parasitol. 2002;123:115–123. doi: 10.1016/s0166-6851(02)00138-x. [DOI] [PubMed] [Google Scholar]
- 29.Brun R, Schonenberger M. Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop. 1979;36:289–292. [PubMed] [Google Scholar]
- 30.Wirtz E, Leal S, Ochatt C, Cross GAM. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 31.Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989;75:985–989. [PubMed] [Google Scholar]
- 32.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, et al. The genome of the African trypanosome. Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 33.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panigrahi AK, Ogata Y, Zikova A, Anupama A, Dalley RA, Acestor N, Myler PJ, Stuart KD. A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics. 2009;9:434–450. doi: 10.1002/pmic.200800477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casneuf T, Van de Peer Y, Huber W. In situ analysis of cross-hybridisation on microarrays and the inference of expression correlation. BMC Bioinformatics. 2007;8:461. doi: 10.1186/1471-2105-8-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bahler J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 40.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 41.Siegel TN, Hekstra DR, Kemp LE, Figueiredo LM, Lowell JE, Fenyo D, Wang X, Dewell S, Cross GAM. Four histone variants mark the boundaries of polycistronic transcription units in. Trypanosoma brucei. Genes Dev. 2009;23:1063–1076. doi: 10.1101/gad.1790409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Sayed NM, Hegde P, Quackenbush J, Melville SE, Donelson JE. The African trypanosome genome. Int. J. Parasitol. 2000;30:329–345. doi: 10.1016/s0020-7519(00)00015-1. [DOI] [PubMed] [Google Scholar]
- 43.Brems S, Guilbride DL, Gundlesdodjir-Planck D, Busold C, Luu VD, Schanne M, Hoheisel J, Clayton C. The transcriptomes of Trypanosoma brucei Lister 427 and TREU927 bloodstream and procyclic trypomastigotes. Mol. Biochem. Parasitol. 2005;139:163–172. doi: 10.1016/j.molbiopara.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 44.Mair G, Shi H, Li H, Djikeng A, Aviles HO, Bishop JR, Falcone FH, Gavrilescu C, Montgomery JL, Santori MI, et al. A new twist in trypanosome RNA metabolism: cis-splicing of pre-mRNA. RNA. 2000;6:163–169. doi: 10.1017/s135583820099229x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hug M, Hotz HR, Hartmann C, Clayton C. Hierarchies of RNA-processing signals in a trypanosome surface antigen mRNA precursor. Mol. Cell. Biol. 1994;14:7428–7435. doi: 10.1128/mcb.14.11.7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haanstra JR, Stewart M, Luu VD, van Tuijl A, Westerhoff HV, Clayton C, Bakker BM. Control and regulation of gene expression: quantitative analysis of the expression of phosphoglycerate kinase in bloodstream form Trypanosoma brucei. J. Biol. Chem. 2008;283:2495–2507. doi: 10.1074/jbc.M705782200. [DOI] [PubMed] [Google Scholar]
- 47.Helm JR, Wilson ME, Donelson JE. Different trans RNA splicing events in bloodstream and procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 2008;159:134–137. doi: 10.1016/j.molbiopara.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kozak M. New ways of initiating translation in eukaryotes? Mol. Cell. Biol. 2001;21:1899–1907. doi: 10.1128/MCB.21.6.1899-1907.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kozak M. Do the 5′ untranslated domains of human cDNAs challenge the rules for initiation of translation (or is it vice versa)? Genomics. 2000;70:396–406. doi: 10.1006/geno.2000.6412. [DOI] [PubMed] [Google Scholar]
- 50.Rogozin IB, Kochetov AV, Kondrashov FA, Koonin EV, Milanesi L. Presence of ATG triplets in 5′ untranslated regions of eukaryotic cDNAs correlates with a ‘weak' context of the start codon. Bioinformatics. 2001;17:890–900. doi: 10.1093/bioinformatics/17.10.890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.