


Abstract
Accurate tools for multiple sequence alignment (MSA) are essential for comparative studies of the function and structure of biological sequences. However, it is very challenging to develop a computationally efficient algorithm that can consistently predict accurate alignments for various types of sequence sets. In this article, we introduce PicXAA (Probabilistic Maximum Accuracy Alignment), a probabilistic non-progressive alignment algorithm that aims to find protein alignments with maximum expected accuracy. PicXAA greedily builds up the multiple alignment from sequence regions with high local similarities, thereby yielding an accurate global alignment that effectively grasps the local similarities among sequences. Evaluations on several widely used benchmark sets show that PicXAA constantly yields accurate alignment results on a wide range of reference sets, with especially remarkable improvements over other leading algorithms on sequence sets with local similarities. PicXAA source code is freely available at: http://www.ece.tamu.edu/∼bjyoon/picxaa/.
INTRODUCTION
Multiple sequence alignment (MSA) has become an essential tool in various areas of molecular biology research, including the reconstruction of phylogenetic trees, predicting the structure of biomolecules, detection of key functional regions, identification of conserved sequence motifs and homology modeling (1–8). The main goal of an MSA algorithm can be viewed as detecting and aligning the homologous regions across different sequences, which have equivalent structures and/or similar functional roles. This is typically achieved by optimizing an objective function that measures the quality of the alignment, either explicitly or implicitly. One of the primary challenges in sequence alignment is to find a biologically meaningful objective function. A common choice of many alignment algorithms has been the ‘sum-of-pairs’ (SP) score, which simply takes the sum of the scores of all pairwise alignments in a given multiple alignment. The optimal alignment that maximizes the SP score can be found using dynamic programming (9,10). However, this problem is NP-complete (11) and the dynamic programming approach becomes quickly infeasible once the number of sequences increases. For this reason, several heuristic techniques have been proposed as possible alternatives, which aim to find a good approximate solution at a reasonable computational cost (12–17). One of the most popular techniques that have been used to reduce the overall complexity is the progressive alignment scheme (16,17), which has been adopted by various alignment algorithms such as CLUSTALW (18), T-Coffee (19), ProbCons (20), MUSCLE (21), MAFFT (22–24), MUMMALS (25) and Pecan (26), just to name a few. The basic idea of the progressive scheme is to build a guide tree based on the similarities among sequences and to grow the MSA by repetitively aligning pairs of sequences or sequence profiles along the tree. Despite its usefulness, one significant weakness of the progressive alignment approach is that it tends to propagate the errors made in the early stages throughout the entire process, which may significantly degrade the quality of the final alignment.
A number of strategies have been proposed to overcome this problem, where the iterative refinement technique and the consistency-based approach have been shown to be especially useful. The ‘iterative refinement’ technique is carried out as a postprocessing step (20–22,25), during which it repetitively divides the aligned sequences into two random groups and realigns them. Unlike the iterative refinement approach, the ‘consistency-based’ approach tries to reduce the chance of early errors when constructing the alignment (19,20,25–28), instead of correcting existing errors via postprocessing. This is typically achieved by updating the pairwise sequence comparison scores based on other sequences in the alignment, to obtain pairwise alignments that are consistent with one another. T-Coffee (19) is one of the first methods that implemented this idea by using a consistency-based alignment measure based on a library of pairwise alignments. With a similar motivation, ProbCons (20) introduced the probabilistic consistency transformation. This algorithm transforms the pairwise residue alignment probabilities, computed using pair hidden Markov models (pair-HMMs), by probabilistically incorporating the information from other sequences. MUMMALS (25) adopts the same approach as ProbCons, but it computes the alignment probabilities using more complex HMMs that also consider local secondary structure similarities. ProbAlign (27) also uses the probabilistic consistency transformation scheme to construct MSAs, where a partition function-based methodology is used to estimate the residue alignment probabilities. In addition to the iterative refinement and consistency-based techniques, DIALIGN (29–31) proposed another solution to overcome the shortcomings of the progressive alignment approach, which constructs the global alignment by assembling the local segments with high similarity. Recently AMAP (32) introduced another non-progressive alignment technique, called sequence annealing, which incrementally constructs the multiple sequence alignment by merging single columns. This technique was adopted and further developed in FSA (33). It has been shown that such incremental approach can e.ectively reduce the number of incorrect residue alignments (32, 33).
As mentioned earlier, MSA algorithms generally aim to find the optimal alignment by optimizing an objective function. Optimality of the alignment, however, can be defined in different ways. For example, we may define the optimal alignment as the alignment with the highest probability or the one with the largest expected number of correctly aligned residues. According to the first definition, our goal would be to develop an algorithm that can find the MSA that has the maximum probability of being identical to the true (unknown) alignment among all possible alignments. In practice, it is not possible to pinpoint the correct alignment with certainty, in which case, it would be more useful to find an alignment that shares as many similarities as possible with the true alignment. In other words, it would be more beneficial to find the alignment that maximizes the expected accuracy, or the expected number of correctly aligned residue pairs. Do et al. (20) implemented this idea in ProbCons to construct the so-called ‘maximum expected accuracy’ (MEA) alignment, using consistency-transformed pairwise residue alignment probabilities. The MEA criterion has been also adopted in many other MSA algorithms, such as ProbAlign (27), Pecan (26), and NRAlign (34), which demonstrated that the MEA-based approach can improve the average accuracy of the predicted MSA. Note that all the aforementioned algorithms that try to construct MEA alignments are mainly based on the progressive technique (20, 26, 27, 34). The minimum expected distance criterion adopted by the sequence annealing technique is essentially identical to the MEA criterion, hence AMAP (32) and FSA (33) can be viewed as examples of non progressive algorithms for building MEA alignments.
In this article, we introduce PicXAA (probabilistic maximum accuracy alignment), an effective non-progressive algorithm that aims to find MEA alignment of multiple sequences. PicXAA greedily builds up the alignment from sequence regions with high local similarities, thereby yielding an accurate global alignment that effectively grasps local similarities among sequences. For a fast and accurate alignment, we adopt an efficient graph-based construction scheme, and also introduce a novel probabilistic consistency transformation and a robust refinement technique. To demonstrate the effectiveness of the proposed techniques, we conduct extensive experiments to compare PicXAA with other well-known alignment algorithms based on several alignment benchmarks. Experimental results confirm that PicXAA consistently yields accurate alignment results on various reference sets, with significant improvements in sequence sets with local similarities.
METHODS
The main goal of PicXAA is to find the MSA with the MEA, which maximizes the expected number of correctly aligned residue pairs. To effectively capture local similarities in the final alignment, while avoiding the propagation of early-stage errors that is often observed in progressive algorithms, we take a greedy approach to probabilistically build up the MSA, by starting from confidently alignable regions (with high similarities) and proceeding toward less confident regions (with lower similarities). The following subsections provide a brief overview of the proposed algorithm.
Improved probabilistic consistency transformation
To align m sequences in a set  , we first need to obtain the posterior pairwise alignment probability matrix
, we first need to obtain the posterior pairwise alignment probability matrix  for each sequence pair
for each sequence pair  for all
for all  .
.  is the probability that residues
is the probability that residues  and
and  are matched in the true (unknown) alignment
are matched in the true (unknown) alignment  . For simplicity, we hereafter denote the alignment probability as
. For simplicity, we hereafter denote the alignment probability as  instead of
instead of  . These posterior alignment probabilities can be estimated using various methods. Further discussion on several possible methods to compute these probabilities can be found in Supplementary Data.
. These posterior alignment probabilities can be estimated using various methods. Further discussion on several possible methods to compute these probabilities can be found in Supplementary Data.
Given the pairwise residue alignment probabilities, the probabilistic consistency transformation modifies the probabilities using the information from other sequences to make them suitable for constructing a more consistent and accurate MSA. One important observation in the consistency-based alignment approach is that all the pairwise alignments induced from a given MSA should be consistent with each other. This means that if residue  aligns with residue
aligns with residue  in the
in the  alignment, and if
alignment, and if  aligns with residue
aligns with residue  in the
in the  alignment, then
alignment, then  must align with
must align with  in the
in the  alignment. We can thus utilize the ‘intermediate’ sequence
alignment. We can thus utilize the ‘intermediate’ sequence  to improve the
to improve the  alignment by making it consistent with the alignments
alignment by making it consistent with the alignments  and
and  .
.
Based on this motivation, Do et al. (20) introduced the so called probabilistic consistency transformation that modifies the alignment probability for a residue pair  , by incorporating the alignment probability between
, by incorporating the alignment probability between  and
and  and that between
and that between  and
and  . This transformation can be written as:
. This transformation can be written as:
For multiple intermediate sequences, the overall probabilistic consistency transformation is defined as (20):
where  is the set of all sequences. This transformation assumes that every intermediate sequence
is the set of all sequences. This transformation assumes that every intermediate sequence  is homologous to both
is homologous to both  and
and  , hence it contains useful homology information that can be used to obtain an accurate and consistent
, hence it contains useful homology information that can be used to obtain an accurate and consistent  alignment. However, when
alignment. However, when  consists of distantly related sequences, this assumption does not necessarily hold, and the transformation can even degrade the quality of the resulting MSA. For instance, if sequences in two distantly related subfamilies are to be aligned, the alignment probability between sequences in the same family can be significantly degraded if we incorporate sequences in the other family. To address this problem, we propose a new probabilistic consistency transformation that explicitly considers the relative significance of each intermediate sequence
consists of distantly related sequences, this assumption does not necessarily hold, and the transformation can even degrade the quality of the resulting MSA. For instance, if sequences in two distantly related subfamilies are to be aligned, the alignment probability between sequences in the same family can be significantly degraded if we incorporate sequences in the other family. To address this problem, we propose a new probabilistic consistency transformation that explicitly considers the relative significance of each intermediate sequence  in improving the
in improving the  alignment.
alignment.
Let  be the set of sequences in
be the set of sequences in  that are related to both
that are related to both  and
and  , where
, where  means
means  and
and  are homologous. Using only the homologous sequences, in the set
are homologous. Using only the homologous sequences, in the set  , we define the probabilistic consistency transformation as:
, we define the probabilistic consistency transformation as:
This transformation can be also written as:
 |
(1) |
using the identity function 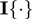 , where
, where  if
if  is homologous to both
is homologous to both  and
and  , and
, and  otherwise. In practice, we cannot judge with certainty whether two sequences are homologous or not. For this reason, it is useful to describe the relationship between the sequences probabilistically, instead of directly using the binary indicator function. Therefore, we use the expectation
otherwise. In practice, we cannot judge with certainty whether two sequences are homologous or not. For this reason, it is useful to describe the relationship between the sequences probabilistically, instead of directly using the binary indicator function. Therefore, we use the expectation  as a practical alternative, where
as a practical alternative, where  is the probability that
is the probability that  and
and  are homologous to each other. We estimate this probability
are homologous to each other. We estimate this probability  based on the similarity between the sequences
based on the similarity between the sequences  and
and  as:
as:
where  is the optimal pairwise alignment of
is the optimal pairwise alignment of  and
and  . Note that, when estimating
. Note that, when estimating  , we only consider the residue pairs that are aligned in the predicted optimal alignment
, we only consider the residue pairs that are aligned in the predicted optimal alignment  . By replacing the identity functions with their expected values in Equation (1) and by assuming the independence between
. By replacing the identity functions with their expected values in Equation (1) and by assuming the independence between  and
and  , we get:
, we get:
 |
Therefore, by using 
 , we obtain the following probabilistic consistency transformation:
, we obtain the following probabilistic consistency transformation:
 |
Conceptually, the new probabilistic consistency transformation improves the consistency of the alignment  with other pairwise alignments in the MSA, by transforming the residue alignment probabilities between
with other pairwise alignments in the MSA, by transforming the residue alignment probabilities between  and
and  using only the information from sequences that are homologous to both
using only the information from sequences that are homologous to both  and
and  . In this way, we can obtain more probabilistically consistent estimate of the posterior alignment probabilities, which ultimately helps enhance the quality of the final MSA. As in (20), we can efficiently implement this transformation based on sparse matrix multiplication, since most values in the matrices
. In this way, we can obtain more probabilistically consistent estimate of the posterior alignment probabilities, which ultimately helps enhance the quality of the final MSA. As in (20), we can efficiently implement this transformation based on sparse matrix multiplication, since most values in the matrices  and
and  will be close to zero. The transformation has a computational complexity of
will be close to zero. The transformation has a computational complexity of  , where
, where  is the average number of non-zero elements per row (typically
is the average number of non-zero elements per row (typically  in real examples),
in real examples),  is the number of sequences, and
is the number of sequences, and  is the length of each sequence.
is the length of each sequence.
More detailed derivation of the improved probabilistic consistency transformation and further discussion can be found in Supplementary Data.
Construction of the alignment graph
Given a set of sequences  , our ultimate goal is to find the MSA that maximizes the expected accuracy
, our ultimate goal is to find the MSA that maximizes the expected accuracy  (i.e. the expected number of correctly aligned residues) over all sequences in
(i.e. the expected number of correctly aligned residues) over all sequences in  . To this aim, we construct the alignment by successively adding the most confident pairwise residue alignments. This greedy alignment approach is conceptually similar to the one used in sequence annealing (32, 33). However, unlike sequence annealing, which greedily merges pairs of columns, we always add a single residue pair at a time, based on the consistency-transformed posterior alignment probabilities. During this process, we may encounter residue pairs that are incompatible with the current alignment. Verifying the compatibility of a new residue pair with the current alignment can be computationally expensive. For fast compatibility verification and efficient construction of the MSA, we adopt a graph-based framework described in the following. Note that a similar approach was also used in sequence annealing (32, 33).
. To this aim, we construct the alignment by successively adding the most confident pairwise residue alignments. This greedy alignment approach is conceptually similar to the one used in sequence annealing (32, 33). However, unlike sequence annealing, which greedily merges pairs of columns, we always add a single residue pair at a time, based on the consistency-transformed posterior alignment probabilities. During this process, we may encounter residue pairs that are incompatible with the current alignment. Verifying the compatibility of a new residue pair with the current alignment can be computationally expensive. For fast compatibility verification and efficient construction of the MSA, we adopt a graph-based framework described in the following. Note that a similar approach was also used in sequence annealing (32, 33).
Let us consider all possible residue pairs  for all
for all  and all
and all  . First, we sort these pairs according to their consistency-transformed alignment probability
. First, we sort these pairs according to their consistency-transformed alignment probability  to obtain an ordered set
to obtain an ordered set  , where
, where  corresponds to the most probable residue pair and
corresponds to the most probable residue pair and  corresponds to the least probable pair. To construct the MSA, we start by inserting the most probable residue pair
corresponds to the least probable pair. To construct the MSA, we start by inserting the most probable residue pair  in the alignment. Then we examine the next residue pair
in the alignment. Then we examine the next residue pair  and add it to the alignment only if it is compatible with the current alignment. Otherwise, we discard
and add it to the alignment only if it is compatible with the current alignment. Otherwise, we discard  and move on to the next most probable residue pair in
and move on to the next most probable residue pair in  . We continue this process for every residue pair
. We continue this process for every residue pair  . The multiple sequence alignment is modeled as a set
. The multiple sequence alignment is modeled as a set  of aligned ‘residue groups’. Each group
of aligned ‘residue groups’. Each group  consists of residues from different sequences that are aligned to each other, hence will be placed in the same column in the final MSA. We let
consists of residues from different sequences that are aligned to each other, hence will be placed in the same column in the final MSA. We let  , where
, where  is the
is the  residue of the sequence
residue of the sequence  . For convenience, we refer to each residue group
. For convenience, we refer to each residue group  as a column. MSA is represented as a directed acyclic graph
as a column. MSA is represented as a directed acyclic graph  , whose nodes correspond to the columns
, whose nodes correspond to the columns  in the current alignment. For convenience, we use the terms ‘node’ and ‘column’ interchangeably. The edges are defined such that
in the current alignment. For convenience, we use the terms ‘node’ and ‘column’ interchangeably. The edges are defined such that  implies that column
implies that column  precedes column
precedes column  in the given alignment. It can be easily shown that incompatibilities in the MSA introduce cycles in the alignment graph
in the given alignment. It can be easily shown that incompatibilities in the MSA introduce cycles in the alignment graph  . Therefore, we can maintain the compatibility of the MSA by adding only the residue pairs that keep the graph
. Therefore, we can maintain the compatibility of the MSA by adding only the residue pairs that keep the graph  acyclic.
acyclic.
Adding a new candidate residue pair  to the current alignment will lead to one of the following four possibilities:
to the current alignment will lead to one of the following four possibilities:
Adding a new column: If neither residue
 nor residue
nor residue  is included in the current alignment (
is included in the current alignment ( ), the residue pair
), the residue pair  should be added to the alignment as a new column, provided that it is compatible with the current MSA. Let us denote the new node for this column as
should be added to the alignment as a new column, provided that it is compatible with the current MSA. Let us denote the new node for this column as  and assume that residues
and assume that residues  and
and  belong to sequences
belong to sequences 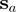 and
and  , respectively. To find the incoming and outgoing edges for this new node, we should identify the columns in the current alignment that contain residues in
, respectively. To find the incoming and outgoing edges for this new node, we should identify the columns in the current alignment that contain residues in  or
or  . This allows us to determine the relative position of the new column with respect to other existing columns. To determine the relative position of
. This allows us to determine the relative position of the new column with respect to other existing columns. To determine the relative position of  , we only need to identify the closest preceding and succeeding columns that contain residues in
, we only need to identify the closest preceding and succeeding columns that contain residues in  and
and  . Let
. Let  be the column that contains the closest residue in
be the column that contains the closest residue in 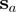 that precedes
that precedes  , and
, and  be the column that contains the closest residue in
be the column that contains the closest residue in  that comes after
that comes after  . Similarly, we define
. Similarly, we define  and
and  as the closest preceding and succeeding columns from the residue
as the closest preceding and succeeding columns from the residue  , respectively. Now we can figure out the relative position of the new column
, respectively. Now we can figure out the relative position of the new column 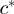 based on the four columns
based on the four columns  ,
,  ,
,  and
and  . Then we simultaneously consider the four new edges,
. Then we simultaneously consider the four new edges,  ,
,  ,
,  and
and  , to check whether these edges lead to any cycles. If the addition of the new column
, to check whether these edges lead to any cycles. If the addition of the new column  introduces a cycle in
introduces a cycle in  , we discard the residue pair
, we discard the residue pair  since it is not compatible with the current alignment, and we move on to the next residue pair in
since it is not compatible with the current alignment, and we move on to the next residue pair in  . Otherwise, the new node
. Otherwise, the new node  is retained in the graph. Figure 1A illustrates this process. In order to reduce the computational cost for verifying the compatibility, we prefer
is retained in the graph. Figure 1A illustrates this process. In order to reduce the computational cost for verifying the compatibility, we prefer  to have as few multipaths as possible between any two nodes in the graph. For this purpose, we prune the alignment graph after adding a new node (details described in Supplementary Data). During pruning, we try to remove redundant edges, which do not add any information about the relative position among the columns in the alignment graph
to have as few multipaths as possible between any two nodes in the graph. For this purpose, we prune the alignment graph after adding a new node (details described in Supplementary Data). During pruning, we try to remove redundant edges, which do not add any information about the relative position among the columns in the alignment graph  .
.Extending an existing column: suppose only one of the residues in
 appears in the current alignment (
appears in the current alignment ( or
or 
 ). Without loss of generality, we assume that only
). Without loss of generality, we assume that only  is included in the alignment. Let us denote the column that contains
is included in the alignment. Let us denote the column that contains  as
as  . In this case, the other residue
. In this case, the other residue  should be added to the same column as
should be added to the same column as  , hence we need to extend
, hence we need to extend  to include
to include  . Extension of the column may add new edges to the node
. Extension of the column may add new edges to the node  , and we have to examine whether this introduces any cycles in the graph
, and we have to examine whether this introduces any cycles in the graph  . Again, suppose
. Again, suppose  and
and  belong to sequences
belong to sequences 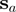 and
and  , respectively. As before, we look for the closest preceding and succeeding columns (with respect to
, respectively. As before, we look for the closest preceding and succeeding columns (with respect to  ) that contain residues in
) that contain residues in  . We denote these columns as
. We denote these columns as  and
and  . If the new edges
. If the new edges  and
and  do not introduce any cycle in the graph, we keep
do not introduce any cycle in the graph, we keep  in column
in column  . Otherwise, we discard the residue pair
. Otherwise, we discard the residue pair  . Figure 1B illustrates this process. After extending the column, we again prune the graph
. Figure 1B illustrates this process. After extending the column, we again prune the graph  as described in Supplementary Data.
as described in Supplementary Data.Merging two columns: suppose the residues
 and
and  are included in two distinct columns
are included in two distinct columns  and
and  , respectively (
, respectively ( ). In order to add the residue pair
). In order to add the residue pair  to the current alignment in a compatible manner, we have to merge the columns
to the current alignment in a compatible manner, we have to merge the columns  and
and  into a single column. This corresponds to merging the corresponding nodes
into a single column. This corresponds to merging the corresponding nodes  and
and  in the alignment graph
in the alignment graph  into a new node
into a new node  . In order to verify whether merging the columns leads to a legitimate MSA, we examine whether adding an edge between
. In order to verify whether merging the columns leads to a legitimate MSA, we examine whether adding an edge between  and
and  introduces any cycles. If the graph remains acyclic, we align all the residues in
introduces any cycles. If the graph remains acyclic, we align all the residues in  and
and  in the same column denoted as
in the same column denoted as 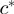 . Otherwise, we discard the residue pair
. Otherwise, we discard the residue pair  . Figure 1C illustrates the merging process. After merging the columns, the alignment graph is pruned as described in Supplementary Data.
. Figure 1C illustrates the merging process. After merging the columns, the alignment graph is pruned as described in Supplementary Data.Residue pair
 already included in the current alignment: no action is needed in this case, since the pair
already included in the current alignment: no action is needed in this case, since the pair  is already included in the current alignment (
is already included in the current alignment (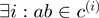 ).
).
Figure 1.

Graph Construction Process. (A) Adding a new column (node)  and the corresponding edges to the alignment graph. (B) Extending the column (node)
and the corresponding edges to the alignment graph. (B) Extending the column (node)  which may introduce new edges. (C) Merging the columns (nodes)
which may introduce new edges. (C) Merging the columns (nodes)  and
and  into a single column (node)
into a single column (node)  .
.
Mapping the alignment graph to an MSA
Once the graph construction process is complete, the resulting alignment graph  can serve as a skeleton of the final MSA. Each node in
can serve as a skeleton of the final MSA. Each node in  contains the residues that should be aligned in the same column in the final MSA. Suppose there is a sequence, none of whose residues is included in a given column. This implies that a gap symbol should be placed in the given column for this sequence. To obtain the final MSA from the graph
contains the residues that should be aligned in the same column in the final MSA. Suppose there is a sequence, none of whose residues is included in a given column. This implies that a gap symbol should be placed in the given column for this sequence. To obtain the final MSA from the graph  , we should arrange the nodes in a linear directed path
, we should arrange the nodes in a linear directed path  according to a legitimate topological ordering. More precisely, we should find a path
according to a legitimate topological ordering. More precisely, we should find a path 
 , so that there is no path from
, so that there is no path from  to
to  in
in  for any
for any  . This guarantees that the order of nodes in
. This guarantees that the order of nodes in  do not contradict their relative order in
do not contradict their relative order in  . We can easily find such
. We can easily find such  using the depth-first search algorithm starting from one of the root nodes in
using the depth-first search algorithm starting from one of the root nodes in  . Theoretically, we may have residues that are not included in any node due to their small alignment probability to other residues. These residues can be inserted in
. Theoretically, we may have residues that are not included in any node due to their small alignment probability to other residues. These residues can be inserted in  as single residue columns, according to their relative position to other residues. The resulting linear path
as single residue columns, according to their relative position to other residues. The resulting linear path  uniquely determines the final MSA. An illustrative example for the graph construction process using PicXAA can be found in the Supplementary Data (Supplementary Section S3 and Figure S4).
uniquely determines the final MSA. An illustrative example for the graph construction process using PicXAA can be found in the Supplementary Data (Supplementary Section S3 and Figure S4).
Improving the alignment quality in low confidence regions
The proposed greedy graph construction scheme is very effective in capturing local similarities among sequences and it faithfully preserves the confidently alignable residue pairs in the MSA. However, residue pairs with low alignment probabilities may have many other competing residue pairs, and the greedy selection of the most probable residue pair among these competing pairs may not necessarily yield an accurate alignment result. This can be a problem for sequence sets that consist of distant sequence families, since sequences that belong to different families will typically have low alignment probabilities. For such datasets, we can improve the alignment accuracy by first grouping similar sequences and performing a profile–profile alignment between distinct groups. This will allow us to improve the alignment quality in low confidence regions (between distantly related sequences that belong to different families), while preserving the confidently alignable residue pairs (between closely related sequences in the same family).
Based on this motivation, we propose the following discriminative refinement technique:
- For each sequence
 in the current multiple alignment, we find the set of similar sequences using the
in the current multiple alignment, we find the set of similar sequences using the  -means clustering. We measure the similarity between two sequences by computing the expected accuracy of their pairwise alignment, which is defined as:
-means clustering. We measure the similarity between two sequences by computing the expected accuracy of their pairwise alignment, which is defined as:
For each sequence
 , we try to partition the set
, we try to partition the set  into two clusters based on this similarity measure:
into two clusters based on this similarity measure:  , the set of sequences that are similar to
, the set of sequences that are similar to  , and
, and  (
( ), the set of remaining sequences in
), the set of remaining sequences in  that are not similar to
that are not similar to  .
. Then we repeat the following steps for all sequences
 :
:
Realign
 with the current alignment profile of
with the current alignment profile of  .
.Align the resulting profile of
 with the current alignment profile of
with the current alignment profile of  .
.Choose another sequence in
 and repeat the previous steps.
and repeat the previous steps.
The proposed refinement technique, based on a discriminative-split-and-realignment strategy, has a number of advantages over the conventional iterative refinement technique, based on a random-split-and-realignment strategy (20,27). First, it typically converges to an accurate alignment in a few (often in a single) iterations, since the overall process is not randomized. Second, the proposed technique takes advantage of both the intra-family similarity (by realigning  with
with  ) as well as the inter-family similarity (by aligning
) as well as the inter-family similarity (by aligning  with
with  ), thereby improving the alignment quality in low similarity regions without breaking the confidently aligned residues.
), thereby improving the alignment quality in low similarity regions without breaking the confidently aligned residues.
RESULTS
Experimental results and comparison
We used six different benchmark datasets: BAliBASE 3.0 (35), IRMBASE 2.0 (31), SABmark 1.65 (36), PREFAB 4.0 (21), HOMSTRAD (37) and OXBench 1.3 (38) to assess the performance of PicXAA in comparison with other well-known MSA algorithms: ProbCons 1.12 (20), ProbAlign 1.1 (27), MAFFT 6.708 (24) with four different options (‘-linsi’, ‘-ginsi’, ‘-einsi’, and ‘-fftnsi’), MUMMALS 1.01 (25) with HMM_1_3_1 option, MUSCLE 3.7 (21), T-Coffee 6.00 (19), CLUSTALW 2.0.10 (18) and DIALIGN-TX (31). Note that all of these methods employ the progressive strategy except DIALIGN-TX. DIALIGN-TX, on the other hand, is a segment-based local alignment method that combines a greedy algorithm with the progressive alignment approach.
It is important to note that PicXAA does not depend on a specific method for computing the pairwise residue alignment probabilities, although it can certainly benefit from a more accurate estimation scheme. To demonstrate this point, in this work, we used three different methods for computing the alignment probabilities: (i) the pair-HMM approach implemented in ProbCons (20), (ii) the structural pair-HMM approach in MUMMALS (25), and (iii) the partition function-based scheme used in ProbAlign (27). We refer to these methods as PicXAA-PHMM, PicXAA-SPHMM and PicXAA-PF, respectively. Details on these posterior probability computation schemes can be found in Supplementary Data.
In the following, we compare these three implementations of PicXAA with the aforementioned MSA algorithms. In each table that summarizes the alignment results based on a specific database, the best result is shown in bold and the second best result is shown in underlined italic. For each test set, we chose the best performing version of PicXAA as the reference (marked by asterisk) and compared its performance with other techniques and estimated the statistical significance of the performance difference using the Wilcoxon signed-rank test. In each table, minus symbols are used to denote the statistically significant inferiority of the respective method compared with PicXAA, while plus symbols are used to denote statistically significant superiority of the method. Single plus or minus symbols denote that the p-value according to the Wilcoxon test is  , and double plus or minus symbols denote high statistical significance with
, and double plus or minus symbols denote high statistical significance with  . Finally, when there is no statistically significant difference between the methods (
. Finally, when there is no statistically significant difference between the methods ( ), it is denoted by the symbol 0.
), it is denoted by the symbol 0.
Results on BAliBASE 3.0
First, we evaluated the accuracy of PicXAA using the BAliBase 3.0 alignment benchmark. BAliBASE 3.0 is a widely used benchmark containing a total of 218 sequence alignments categorized into six reference sets. Two different criteria are used to score the alignment: the SP score, which is the percentage of the correctly aligned residue pairs in the alignment, and the column score (CS), which is the percentage of the correct columns in the alignment.
The SP and CS scores are given in Table 1. As shown in this table, PicXAA-PF outperforms other techniques both in terms of SP and CS scores on average. where this superiority is statistically significant in most cases. Moreover, we can see that, on average, all three versions of our algorithm (PicXAA-PF, PicXAA-PHMM and PicXAA-SPHMM) outperform their progressive counterparts (i.e., ProbAlign, ProbCons, and MUMMALS, respectively), which were used to estimate the alignment probabilities. This improvement is especially significant for the case of MUMMALS, where PicXAA-SPHMM improved the SP score by 1% and the CS score by 2.3%.
Table 1.
Performance evaluation on BAliBASE 3.0
| Method |
BALIBASE 3.0 |
||||||
|---|---|---|---|---|---|---|---|
| RV11 | RV12 | RV20 | RV30 | RV40 | RV50 | Overall | |
| SP/CS | SP/CS | SP/CS | SP/CS | SP/CS | SP/CS | SP/CS | |
| PicXAA-PF | 68.92 /46.16 /46.16
|
94.61 /86.20 /86.20
|
92.49 /41.51 /41.51
|
86.11 /57.80 /57.80
|
93.23 /63.27 /63.27
|
89.23 /53.00 /53.00
|
87.86 /59.32 /59.32
|
| PicXAA-PHMM | 66.26 /42.03 /42.03
|
94.23 /85.84 /85.84
|
91.69 /38.78 /38.78
|
84.95 /53.00 /53.00
|
90.90 /56.16 /56.16
|
90.16 /60.19 /60.19
|
86.55 /56.28 /56.28
|
| PicXAA-SPHMM |
69.46 /44.74 /44.74
|
94.79 /86.36 /86.36
|
91.65 /40.32 /40.32
|
84.06 /52.97 /52.97
|
89.14 /52.31 /52.31
|
89.83 /58.38 /58.38
|
86.67 /56.14 /56.14
|
|
| |||||||
| ProbAlign |
69.44 /44.53 /44.53
|
94.65 /86.27 /86.27
|
92.57 /43.93 /43.93
|
85.29 /56.57 /56.57
|
92.22 /60.35 /60.35
|
88.96 /54.94 /54.94
|
87.61 /58.82 /58.82
|
| ProbCons | 66.87 /41.61 /41.61
|
94.10 /85.48 /85.48
|
91.68 /40.63 /40.63
|
84.55 /54.37 /54.37
|
90.59 /54.63 /54.63
|
88.96 /55.88 /55.88
|
86.42 /56.01 /56.01
|
| MUMMALS | 66.95 /41.61 /41.61
|
94.30 /83.98 /83.98
|
91.04 /42.83 /42.83
|
84.79 /49.40 /49.40
|
87.15 /48.55 /48.55
|
87.91 /52.88 /52.88
|
85.53 /53.85 /53.85
|
| MAFFT-linsi | 66.19 /43.79 /43.79
|
93.46 /83.39 /83.39
|
92.70 /45.12 /45.12
|
86.80 /59.33 /59.33
|
92.61 /61.51 /61.51
|
90.25 /59.06 /59.06
|
87.22 /59.28 /59.28
|
| MAFFT-ginsi | 65.69 /42.66 /42.66
|
93.16 /83.41 /83.41
|
92.48 /41.85 /41.85
|
85.92 /55.23 /55.23
|
91.15 /56.24 /56.24
|
89.95 /59.25 /59.25
|
86.56 /56.73 /56.73
|
| MAFFT-einsi | 66.01 /43.74 /43.74
|
93.45 /83.39 /83.39
|
92.51 /44.32 /44.32
|
86.81 /59.43 /59.43
|
92.26 /60.53 /60.53
|
89.87 59.63 59.63
|
87.05 /58.95 /58.95
|
| MAFFT-fftnsi | 61.45 /39.45 /39.45
|
90.82 /79.57 /79.57
|
90.89 /38.24 /38.24
|
83.22 /49.00 /49.00
|
89.88 /54.67 /54.67
|
86.41 /53.38 /53.38
|
84.13 /53.08 /53.08
|
| T-Coffee | 65.98 /41.37 /41.37
|
94.07 /85.25 /85.25
|
91.49 /38.71 /38.71
|
83.71 /49.47 /49.47
|
89.69 /55.12 /55.12
|
89.41 /58.06 /58.06
|
85.94 /55.16 /55.16
|
| DIALIGN-TX | 51.52 /26.53 /26.53
|
89.18 /75.23 /75.23
|
87.87 /30.49 /30.49
|
76.18 /38.53 /38.53
|
83.64 /44.82 /44.82
|
82.28 /46.56 /46.56
|
78.83 /44.33 /44.33
|
| MUSCLE | 57.16 /31.79 /31.79
|
91.53 /80.39 /80.39
|
88.90 /35.00 /35.00
|
81.44 /40.87 /40.87
|
86.48 /45.02 /45.02
|
83.53 /45.94 /45.94
|
81.94 /47.46 /47.46
|
| CLUSTALW | 49.43 /23.95 /23.95
|
87.14 /71.86 /71.86
|
86.16 /23.46 /23.46
|
71.97 /26.90 /26.90
|
78.62 /40.00 /40.00
|
73.36 /30.38 /30.38
|
75.37 /38.01 /38.01
|
Further analysis of the alignment results indicates that the proposed alignment method yields the best (or close to the best) result for RV11, RV12, RV40 and RV50 reference sets, in terms of both SP and CS scores. Reference sets RV11 and RV12 contain equidistant families in which sequences that have large internal insertions ( residues) are excluded. The sequence identity in RV11 is <20%, while it is between 20% and 40% for RV12. As shown in the table, PicXAA has comparable performance to ProbAlign and ProbCons for these reference sets while it improves the performance of MUMMALS considerably. RV40 and RV50 contain sequences with large N/C-terminal extensions and large internal insertions, respectively. For these reference sets, alignment methods that can more effectively detect local similarities are expected to yield more accurate alignments. As we can see in Table 1, PicXAA yields the best performance on these reference sets, which shows that constructing the alignment from confidently alignable regions with high local similarities leads to more accurate results in such cases.
residues) are excluded. The sequence identity in RV11 is <20%, while it is between 20% and 40% for RV12. As shown in the table, PicXAA has comparable performance to ProbAlign and ProbCons for these reference sets while it improves the performance of MUMMALS considerably. RV40 and RV50 contain sequences with large N/C-terminal extensions and large internal insertions, respectively. For these reference sets, alignment methods that can more effectively detect local similarities are expected to yield more accurate alignments. As we can see in Table 1, PicXAA yields the best performance on these reference sets, which shows that constructing the alignment from confidently alignable regions with high local similarities leads to more accurate results in such cases.
In RV20, each alignment consists of a family with  similarity and an orphan sequence that shares
similarity and an orphan sequence that shares  identity to other sequences. RV30 contains subfamilies with
identity to other sequences. RV30 contains subfamilies with  identity within each subfamily while having
identity within each subfamily while having  similarity between the sequences of different subfamilies. Since the alignments in RV20 and RV30 consist of sequences that belong to distantly related subfamilies, we would expect the progressive approach to perform better, since it first aligns the homologous sequences in each subfamily and then align the sequence profiles of the respective families. In fact, MAFFT-(le)insi shows the best performance on these reference sets, and the high CS scores of various progressive algorithms demonstrate their advantage on such sets. However, Wilcoxon test shows that the superiority of the progressive techniques over PicXAA on these reference sets is mostly not statistically significant.
similarity between the sequences of different subfamilies. Since the alignments in RV20 and RV30 consist of sequences that belong to distantly related subfamilies, we would expect the progressive approach to perform better, since it first aligns the homologous sequences in each subfamily and then align the sequence profiles of the respective families. In fact, MAFFT-(le)insi shows the best performance on these reference sets, and the high CS scores of various progressive algorithms demonstrate their advantage on such sets. However, Wilcoxon test shows that the superiority of the progressive techniques over PicXAA on these reference sets is mostly not statistically significant.
Results on IRMBASE 2.0
To assess the effectiveness of PicXAA in capturing local similarities among sequences, we carried out another experiment based on the IRMBASE 2.0 alignment benchmark. IRMBASE 2.0 has been constructed by inserting highly conserved motifs generated by ROSE (39) in long random sequences. IRMBASE 2.0 consists of four reference sets with a total of 192 alignments. Similar to BAliBASE 3.0, we use the SP and CS scores to evaluate the alignment quality.
The average SP and CS scores of the tested methods are given in the first column of Table 2. The SP and CS scores for different reference sets of IRMBASE 2.0 can be found in Supplementary Table S1. As we can see in these tables, DIALIGN-TX shows the best overall performance on IRMBASE 2.0. However, it must be noted that IRMBASE 2.0 was originally developed to evaluate the performance of DIALIGN-TX to align sequences with local similarities, hence DIALIGN-TX may have been especially fine-tuned to perform well on this data set. In general, DIALIGN-TX does not perform well on other alignment benchmarks (Tables 1 and 2). Besides, we can see that even on IRMBASE 2.0, the superiority of DIALIGN-TX over PicXAA is not statistically significant for the SP score. Except for DIALIGN-TX, we can see that PicXAA yields the best overall performance among all other alignment methods with statistically significant superiority in most cases.
Table 2.
Performance evaluation on IRMBASE 2.0, SABmark 1.65, PREFAB 4.0, HOMSTRAD, and OXBench 1.3
| Method | IRMBASE | SABmark | PREFAB | HOMSTRAD | OXBench | |||
|---|---|---|---|---|---|---|---|---|
| 2.0 |
1.65 |
4.0 |
1.3 |
|||||
| Overall | Twilight | Superfamily | master | full | extended | |||
| SP/CS |
 / /
|
 / /
|
Q | SP/CS | SP/CS | SP/CS | SP/CS | |
| PicXAA-PF | 89.00 /50.08 /50.08
|
16.75 /15.37 /15.37
|
49.66 /41.41 /41.41
|
71.34
|
82.36 /7813 /7813
|
89.72 /84.72 /84.72
|
84.15 /76.46 /76.46
|
92.41 /88.08 /88.08
|
| PicXAA-PHMM | 90.76 /54.48 /54.48
|
17.12 /14.65 /14.65
|
50.37 /41.13 /41.13
|
71.16
|
82.08 /77.74 /77.74
|
89.28 /84.05 /84.05
|
83.23 /75.16 /75.16
|
92.10 /87.65 /87.65
|
| PicXAA-SPHMM | 72.75 /33.02 /33.02
|
20.99 /17.12 /17.12
|
53.53 /42.77 /42.77
|
72.38
|
84.04 /80.04 /80.04
|
90.55 /85.68 /85.68
|
83.78 /75.96 /75.96
|
92.99 /88.86 /88.86
|
|
| ||||||||
| ProbAlign | 81.68 /36.69 /36.69
|
15.86 /13.05 /13.05
|
48.66 /39.82 /39.82
|
71.90
|
82.39 /78.15 /78.15
|
89.79 /84.93 /84.93
|
84.07 /76.42 /76.42
|
92.55 /88.41 /88.41
|
| ProbCons | 85.30 /42.51 /42.51
|
16.64 /13.55 /13.55
|
48.56 /39.51 /39.51
|
71.64
|
82.04 /77.74 /77.74
|
89.29 /84.10 /84.10
|
83.25 /75.18 /75.18
|
92.43 /88.15 /88.15
|
| MUMMALS | 68.44 /24.62 /24.62
|
19.99 /18.23 /18.23
|
52.08 /42.74 /42.74
|
72.73
|
83.79 /79.66 /79.66
|
90.20 /85.21 /85.21
|
82.81 /75.05 /75.05
|
92.52 /87.79 /87.79
|
| MAFFT-linsi | 89.44 /46.02 /46.02
|
17.42 /13.16 /13.16
|
50.47 /40.01 /40.01
|
72.16
|
80.31 /75.78 /75.78
|
88.30 /82.79 /82.79
|
82.83 /74.74 /74.74
|
92.95 /88.92 /88.92
|
| MAFFT-ginsi | 84.53 /40.24 /40.24
|
17.40 /13.13 /13.13
|
50.23 /39.76 /39.76
|
71.65
|
80.20 /75.60 /75.60
|
88.71 /83.27 /83.27
|
81.91 /73.50 /73.50
|
93.19 /89.14 /89.14
|
| MAFFT-einsi |
91.77 /48.21 /48.21
|
17.77 /13.07 /13.07
|
49.94 /39.23 /39.23
|
72.04
|
80.32 /75.80 /75.80
|
88.32 /82.83 /82.83
|
82.98 /74.87 /74.87
|
92.99 /88.97 /88.97
|
| MAFFT-fftnsi | 82.76 /38.51 /38.51
|
11.54 /9.04 /9.04
|
42.43 /34.89 /34.89
|
68.78
|
78.31 /73.51 /73.51
|
87.69 /82.02 /82.02
|
81.50 /73.07 /73.07
|
91.35 /86.83 /86.83
|
| T-Coffee | 87.75 /46.33 /46.33
|
15.18 /11.88 /11.88
|
42.47 /34.20 /34.20
|
70.75
|
76.79 /71.97 /71.97
|
81.78 /75.04 /75.04
|
73.38 /63.75 /63.75
|
91.28 /86.64 /86.64
|
| DIALIGN-TX |
92.92 /71.02 /71.02
|
11.01 /11.59 /11.59
|
39.30 /35.28 /35.28
|
62.47
|
76.85 /71.53 /71.53
|
85.96 /79.65 /79.65
|
80.76 /72.26 /72.26
|
88.79 /82.89 /82.89
|
| MUSCLE | 43.73 /11.12 /11.12
|
12.91 /8.94 /8.94
|
43.07 /33.56 /33.56
|
68.26
|
80.91 /76.36 /76.36
|
89.25 /84.07 /84.07
|
82.46 /74.20 /74.20
|
91.61 /87.01 /87.01
|
| CLUSTALW | 26.34 /2.44 /2.44
|
12.87 /8.72 /8.72
|
38.62 /30.27 /30.27
|
61.75
|
80.14 /75.42 /75.42
|
89.29 /83.79 /83.79
|
81.56 /72.66 /72.66
|
89.40 /83.86 /83.86
|
Comparing the three implementations of PicXAA with their progressive counterparts (ProbAlign, ProbCons and MUMMALS) shows that the proposed alignment method leads to significant improvement (with P ) in the overall accuracy for all reference sets. On average, the SP score is improved by 5–8% and the CS score is improved by 8–13%. These results clearly demonstrate the strength of PicXAA in picking up local similarities among the aligned sequences that are often missed by progressive methods. Table 2 also shows that MUMMALS, T-Coffee, MUSCLE, and CLUSTALW do not perform well on IRMBASE 2.0. In terms of the SP score, MAFFT-einsi performs well on IRMBASE 2.0 (with no statistically significant superiority over PicXAA-PHMM), where PicXAA-PHMM yields comparable results. However, PicXAA-PHMM results in significantly higher CS score in all the reference sets (with P
) in the overall accuracy for all reference sets. On average, the SP score is improved by 5–8% and the CS score is improved by 8–13%. These results clearly demonstrate the strength of PicXAA in picking up local similarities among the aligned sequences that are often missed by progressive methods. Table 2 also shows that MUMMALS, T-Coffee, MUSCLE, and CLUSTALW do not perform well on IRMBASE 2.0. In terms of the SP score, MAFFT-einsi performs well on IRMBASE 2.0 (with no statistically significant superiority over PicXAA-PHMM), where PicXAA-PHMM yields comparable results. However, PicXAA-PHMM results in significantly higher CS score in all the reference sets (with P
 ), which is on average 6.2% higher than that of the runner-up (i.e. MAFFT-einsi).
), which is on average 6.2% higher than that of the runner-up (i.e. MAFFT-einsi).
Results on SABmark 1.65
We also evaluated the performance of PicXAA on SABmark 1.65. This dataset includes two sets of reference alignments derived from the SCOP classification (40): ‘Twilight Zone’ and ‘Superfamilies’. For each alignment, the collection of pairwise reference structural alignments is provided. The accuracy of the alignment is measured using the  score, an equivalent of the SP score. An additional
score, an equivalent of the SP score. An additional  score is also computed in SABmark 1.65 to measure the specificity of the alignment.
score is also computed in SABmark 1.65 to measure the specificity of the alignment.
As we can see in Table 2, PicXAA-SPHMM yields the highest  score with statistically significant superiority on both the Twilight and Superfamily sets. In terms of specificity (
score with statistically significant superiority on both the Twilight and Superfamily sets. In terms of specificity ( score), PicXAA-SPHMM performs best on the Superfamily set, while MUMMALS outperforms other methods on the Twilight reference set. However, the superiority of MUMMALS over PicXAA-SPHMM on the Twilight set (in terms of
score), PicXAA-SPHMM performs best on the Superfamily set, while MUMMALS outperforms other methods on the Twilight reference set. However, the superiority of MUMMALS over PicXAA-SPHMM on the Twilight set (in terms of  score) is not statistically significant, while the superiority of PicXAA-SPHMM (in terms of
score) is not statistically significant, while the superiority of PicXAA-SPHMM (in terms of  score) is statistically significant (
score) is statistically significant ( ). We can observe that the use of structural pair-HMM leads to more accurate alignments results for PicXAA-SPHMM and MUMMALS on this benchmark, owing to more accurate estimation of the pairwise residue alignment probabilities. PicXAA-PHMM and PicXAA-PF, which do not incorporate structural information in estimating the probabilities, yield lower
). We can observe that the use of structural pair-HMM leads to more accurate alignments results for PicXAA-SPHMM and MUMMALS on this benchmark, owing to more accurate estimation of the pairwise residue alignment probabilities. PicXAA-PHMM and PicXAA-PF, which do not incorporate structural information in estimating the probabilities, yield lower  and
and  scores compared PicXAA-SPHMM. However, both PicXAA-PHMM and PicXAA-PF significantly outperform their progressive counterparts (i.e. ProbCons and ProbAlign), and we obtain 1–2% improvement in
scores compared PicXAA-SPHMM. However, both PicXAA-PHMM and PicXAA-PF significantly outperform their progressive counterparts (i.e. ProbCons and ProbAlign), and we obtain 1–2% improvement in  and
and  scores.
scores.
Results on PREFAB 4.0
Here, we evaluated PicXAA on the PREFAB 4.0 alignment benchmark. PREFAB 4.0 (21) consists of 1682 alignments. Each alignment contains one pair of sequences with known 3D structure and additional sequences that are obtained from high scoring hits of these two sequences in a database search.
Table 2 shows the average Q-score over the 1682 alignments in PREFAB 4.0 for every alignment method. For each alignment, the Q-score is computed according to the 3D structural alignment of the core sequence pair contained in the alignment. Therefore, it would be natural to expect that MUMMALS, which employs structural information for computing the alignment probabilities, will perform well on PREFAB 4.0 as on SABmark 1.65. This can be observed in Table 2, where MUMMALS shows best performance among all the compared methods. However, the performance difference between MUMMALS and PicXAA-SPHMM is not statistically significant. For all other methods, PicXAA-SPHMM shows significantly better performance (with  ) in terms of the Q-score.
) in terms of the Q-score.
Table 2 also shows that PicXAA-PF, PicXAA-PHMM and PicXAA-SPHMM yield slightly lower average Q-scores compared with the corresponding progressive methods, ProbAlign, ProbCons and MUMMALS. One possible explanation for this observation can be found from the benchmark generation process. As mentioned earlier, each alignment in PREFAB 4.0 consists of a pair of protein sequences with known 3D structure and other homologous sequences obtained from database search. For this reason, every alignment in the PREFAB 4.0 basically consists of two groups of similar sequences, or two subfamilies. When using the progressive alignment technique, the guide tree will separate the sequences in the two subfamilies, hence the algorithm will first align the sequences in respective families and align the two subfamilies at the end. As noted in (41), recruiting homologous sequences via database search and performing a profile–profile alignment is known to improve the alignment accuracy compared with pairwise alignment of individual sequences, which explains why the progressive techniques slightly outperform PicXAA on PREFAB 4.0.
Results on HOMSTRAD
We performed a comparison based on HOMSTRAD (37) alignment benchmark (dated 1 January 2010), containing 1031 homologous protein families aligned based on their 3D structures. Table 2 shows the average SP and CS scores. We can see that PicXAA-SPHMM performed best among all the compared methods, with high statistical significance. In fact, PicXAA-SPHMM outperformed the runner-up (i.e. MUMMALS) with a P
 .
.
As shown in Supplementary Table S2, the performance of PicXAA on HOMSTARD stands out even more when we consider only the alignments with  sequences (totally 233 alignments). In this case, PicXAA-SPHMM outperforms MUMMALS by 0.65% in the SP and 1.12% in the CS score.
sequences (totally 233 alignments). In this case, PicXAA-SPHMM outperforms MUMMALS by 0.65% in the SP and 1.12% in the CS score.
Results on OXBench 1.3
Finally, we used OXBench 1.3 (38) benchmark, a set of structure-based alignments of protein domains. OXBench consists of three reference sets: ‘master’, a set of 672 reference alignments of protein structural domains; ‘full’, a set of 605 full-length sequences of the domains in the master dataset; and ‘extended’, the master set of domains augmented by sequences of unknown structure. We excluded seven large alignments in the extended reference set due to running time consideration. Last three columns of Table 2 report the SP and CS scores.
We can see that in both scores, PicXAA-SPHMM and PicXAA-PF outperform all other techniques for the master and full reference sets, respectively. We can also see that, in most cases, this superiority is statistically significant with P
 . For the extended set, MAFFT-[lge]insi shows the best performance, while PicXAA-SPHMM yields the next best result. However, this inferiority is not statistically significant, and PicXAA-SPHMM significantly outperforms most of the other methods.
. For the extended set, MAFFT-[lge]insi shows the best performance, while PicXAA-SPHMM yields the next best result. However, this inferiority is not statistically significant, and PicXAA-SPHMM significantly outperforms most of the other methods.
Expected number of correctly aligned residues
The primary goal of PicXAA is to find the MSA that maximizes the expected accuracy, or the expected number of correctly aligned residues. To assess the effectiveness of PicXAA in achieving this goal, we introduce the ‘sum-of-pairwise probabilities’ (SPP) score. The SPP score computes the total expected number of correct pairwise alignments, and it can serve as a good indicator of the expected accuracy of a predicted alignment. The SPP score is computed as
by summing up the posterior pairwise alignment probabilities of all aligned residue pairs in the final alignment  . It is important to remember that the original (non-transformed) probabilities should be used to compute the SPP score, since the transformed probabilities do not directly reflect the pairwise residue alignment probabilities. In fact, the consistency transformation mainly aims to adjust the original pairwise alignment probabilities to obtain a consistent, hence more accurate, MSA.
. It is important to remember that the original (non-transformed) probabilities should be used to compute the SPP score, since the transformed probabilities do not directly reflect the pairwise residue alignment probabilities. In fact, the consistency transformation mainly aims to adjust the original pairwise alignment probabilities to obtain a consistent, hence more accurate, MSA.
In this experiment, we computed the sum of SPP scores for all alignments in BAliBASE 3.0 and IRMBASE 2.0 for two versions of the proposed alignment method, PicXAA-PF and PicXAA-PHMM, and compared the results with their progressive counterparts, i.e. ProbAlign and ProbCons, respectively. We did not compare PicXAA-SPHMM and MUMMALS, since MUMMALS does not compute all pairwise residue alignment probabilities, in general. As shown in Supplementary Table S3, the PicXAA slightly improves the SPP score compared with other progressive methods, when evaluated on BAliBASE 3.0. This improvement is especially significant for the RV30 reference set, which consists of distantly related subfamilies. Evaluation on IRMBASE 2.0 shows that PicXAA remarkably improves the SPP score over the progressive methods, where we improve the score by 9.5% compared with ProbAlign and 3.2% compared with ProbCons. These results demonstrate the effectiveness of the probabilistic greedy construction scheme, adopted by PicXAA, in enhancing the expected accuracy of the MSA, especially for sequences with only local similarities.
Computational complexity
As shown in Figure 2, PicXAA-PF and PicXAA-PHMM has comparable speed with their progressive counterparts, ProbAlign and ProbCons. This implies that we can obtain more accurate MSAs, which effectively capture the local similarities among sequences, by using the proposed probabilistic greedy alignment approach without any substantial increase in the overall computational complexity compared with the conventional progressive approach. Further discussion on the computational complexity of PicXAA can be found in Supplementary Data.
Figure 2.

Total CPU time for aligning the sequences in BAliBASE 3.0 (shown in seconds). For different implementations of PicXAA, the total CPU time is divided into two parts: the time that takes for probability estimation and the time needed in the remaining steps for constructing the alignment.
DISCUSSION
Overall performance of PicXAA
To find the MEA alignment for multiple sequences, we developed PicXAA, a probabilistic non-progressive algorithm that greedily builds up the alignment by successively adding the most probable residue pair to the MSA. For fast construction of the multiple sequence alignment, we took a graph-based approach as in (32, 33), in which the overall compatibility of the alignment is verified and maintained using efficient graph-based techniques. We also introduced a novel consistency transformation and a discriminative refinement technique, which can altogether improve the accuracy of the final MSA when combined with the probabilistic greedy alignment technique.
As demonstrated in our results, PicXAA consistently shows excellent performance on various reference sets with different characteristics, from reference sets containing sequences that belong to closely related families, to reference sets containing sequences that belong to distant families, hence share only local similarities. In other words, PicXAA is not fine-tuned nor overtrained to perform well on specific types of sequence sets, and it can yield accurate alignment results under various circumstances. However, its advantage may be most clearly seen on datasets with only local similarities, where progressive techniques fail to capture those similarities, while PicXAA effectively grasps such similarities through the probabilistic greedy alignment approach.
Experimental results on IRMBASE 2.0 clearly show PicXAA's strength in grasping local similarities among distantly related sequences. In fact, the proposed method exhibits a significant improvement over various progressive alignment techniques, and it is outperformed only by DIALIGN-TX (not statistically significant in terms of the SP score) on this dataset, which does not generally perform well on reference sets that contain sequences with global similarities. As shown in Tables 1 and 2, PicXAA also yields accurate results on other benchmarks such as BAliBASE 3.0, SABmark 1.65, PREFAB 4.0, HOMSTRAD and OXBench 1.3. PicXAA has both the highest average SP and CS scores on BAliBASE 3.0, HOMSTRAD and OXBENCH 1.3, and it results in the best overall performance, in terms of average  (sensitivity) and
(sensitivity) and  (specificity) scores, on SABmark 1.65.
(specificity) scores, on SABmark 1.65.
The statistical significance test also demonstrates that PicXAA consistently outperforms other alignment techniques on various benchmark datasets. When PicXAA yields a higher average score compared with another algorithm, this superiority is statistically significant in most cases (usually with very low P-values). However, when another technique outperforms PicXAA, this performance difference is often not statistically significant (except for the CS score of DIALIGN-TX on IRMBASE 2.0).
Furthermore, Supplementary Table S3 shows that PicXAA can effectively increase the number of correctly aligned residues, hence capable of finding more accurate MSAs according to the MEA criterion, especially for sequence sets with local similarities.
Proposed consistency transformation improves the alignment accuracy
The new probabilistic consistency transformation updates the residue alignment probabilities in a given sequence pair, by using the information from other sequences in the alignment according to their relation to the given pair. When we have divergent sequences with only local similarities or a set of sequences that belong to distantly related subfamilies, this technique can improve the overall quality of the final MSA, by incorporating only the information from related sequences in the transformation. In fact, the conventional consistency transformation tends to water down local similarities that are shared only by a subset of sequences, thereby breaking up related residues into different columns.
To assess the effectiveness of the proposed transformation, we conducted the following experiment. In this test, we examined the performance improvement that can be achieved by incorporating the new consistency transformation in two popular probabilistic consistency-based alignment algorithms, ProbAlign (27) and ProbCons (20). Figure 3A and B summarize the average SP and CS scores on IRMBASE 2.0 and BAliBASE 3.0 benchmarks. As we can observe in these figures, the new consistency transformation significantly improves the SP and CS scores of both algorithms on the IRMBASE 2.0 dataset. The improvement is near 1.5% in the SP score and 3.3% in the CS score for ProbAlign, and 1.6% in the SP score and 4.2% in the CS score for ProbCons. This clearly shows the advantage of the proposed probabilistic consistency transformation over the conventional transformation on reference sets with local similarities. For BAliBASE 3.0, the proposed transformation does not result in a significant change in average the SP score, but it leads to about 0.3% improvement in the average CS score. The improvement is especially significant for the reference set RV30, which consists of sequences that belong to diverse subfamilies, where we have 1.6% (0.1%) improvement in the SP score and 3.6% (1.7%) improvement in the CS score for ProbAlign (ProbCons).
Figure 3.

Effectiveness of the proposed techniques. (A and B) Novel consistency transformation; (C and D) Discriminative refinement strategy; (E and F) Greedy graph-based alignment.
Discriminative refinement improves the alignment quality in low confidence regions
Residues with low alignment probability are difficult to correctly align. In low similarity regions, the profile–profile alignment approach can help improve the overall alignment accuracy. In this work, we proposed a discriminative refinement technique, which divides the alignment into two sequence groups based on sequence similarity and realigns them in an iterative manner (see ‘Methods’ section). For a given MSA, this technique can improve the alignment quality in low confidence regions, while preserving residues pairs that have been confidently aligned.
To investigate the effectiveness of the proposed refinement strategy, we replaced the conventional refinement scheme used in ProbAlign and ProbCons by the proposed scheme, and analyzed the performance changes. The average SP and CS scores are shown in Figure 3C and D. Experimental results on IRMBASE 2.0 demonstrate that the new refinement strategy leads to considerable improvement over the conventional refinement strategy. This improvement is about 5% in the SP score and 7.7% in the CS score for ProbAlign, and 4% in the SP score and 4.6% in the CS score for ProbCons. Similar experiments on BAliBASE 3.0 did not result in significant changes, as we can see in Figure 3C and D. These results show that the ‘discriminative’-split-and-realignment technique proposed in this article is especially effective for improving the alignment accuracy in sequence sets with local similarities. In fact, we could observe that the conventional refinement strategy tends to break many confidently aligned residue pairs in such datasets, due to the ‘random’-split-and-realignment process.
Greedy alignment construction leads to more accurate alignments
The proposed probabilistic greedy alignment technique, which adds a single residue pair with the highest alignment probability at each step, has a crucial positive impact on the overall alignment accuracy. In fact, remarkable performance gain can be obtained by considering all possible residue pairs between all sequence pairs simultaneously, and constructing the alignment by adding the most probable residue pairs one after another. By building up the MSA from the confidently alignable regions, PicXAA significantly reduces the risk of propagating the alignment errors made at the early stage to the final alignment, which is a commonly observed problem in many progressive algorithms. Furthermore, the proposed alignment scheme is also effective in maximizing the number of correctly aligned residues, as can be observed in Supplementary Table S3.
To analyze the effect of the proposed probabilistic greedy alignment approach on the alignment quality, we examined the performance change of ProbAlign and ProbCons after replacing the progressive alignment scheme with the proposed greedy alignment scheme. Figure 3E and F show the average SP and CS scores of the different alignment methods. For IRMBASE 2.0, we can see a remarkable enhancement in both the SP and CS scores that results from replacing the progressive scheme with the proposed greedy scheme, while retaining the conventional consistency transformation and refinement technique. The resulting enhancement is about 6.8% in the SP score and 14.2% in the CS score for ProbAlign, and 6.1% in the SP score and 11.7% in the CS score for ProbCons. Experiments based on BAliBASE 3.0 show that the greedy alignment scheme alone, without employing the proposed consistency transformation and the discriminative refinement technique, does not necessarily improve the overall accuracy of the alignment. This is particularly evident when we look at the CS score, where we see 2–3% reduction. This degradation is mainly due to the lower performance on the reference sets RV20 and RV30, where each alignment consists of divergent subfamilies. (In the case of RV20, each alignment contains one orphan sequence that bears low similarity with the rest of the sequences.) As discussed earlier, the conventional progressive alignment technique is expected to work well on such reference sets. However, combination of the greedy alignment scheme with the new consistency transformation and refinement strategy effectively overcomes these problems by considering both the intra- and inter-family similarities. We can see in Figure 3E and F that the greedy approach integrated with the proposed consistency transformation and the new refinement technique improves the alignment accuracy. Such a phenomenon is not observed in experiments based on IRMBASE 2.0, since IRMBASE 2.0 does not contain reference sets with different subfamilies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Texas A&M faculty start up fund.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the anonymous reviewers for their helpful remarks.
REFERENCES
- 1.Phillips A, Janies D, Wheeler W. Multiple sequence alignment in phylogenetic analysis. Mol. Phylogenet. Evol. 2000;16:317–330. doi: 10.1006/mpev.2000.0785. [DOI] [PubMed] [Google Scholar]
- 2.Wong KM, Suchard MA, Huelsenbeck JP. Alignment uncertainty and genomic analysis. Science. 2008;319:473–476. doi: 10.1126/science.1151532. [DOI] [PubMed] [Google Scholar]
- 3.Cuff JA, Barton GJ. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 2000;40:502–511. doi: 10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 4.Kemena C, Notredame C. Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics. 2009;25:2455–2465. doi: 10.1093/bioinformatics/btp452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edgar RC, Batzoglou S. Multiple sequence alignment. Curr. Opin. Struct. Biol. 2006;16:368–373. doi: 10.1016/j.sbi.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Pei J. Multiple protein sequence alignment. Curr. Opin. Struct. Biol. 2008;18:382–386. doi: 10.1016/j.sbi.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, Filipski A. Multiple sequence alignment: in pursuit of homologous DNA positions. Genome Res. 2007;17:127–135. doi: 10.1101/gr.5232407. [DOI] [PubMed] [Google Scholar]
- 8.Notredame C. Recent progress in multiple sequence alignment: a survey. Pharmacogenomics. 2002;3:131–144. doi: 10.1517/14622416.3.1.131. [DOI] [PubMed] [Google Scholar]
- 9.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 10.Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Jiang T. On the complexity of multiple sequence alignment. J. Comput. Biol. 1994;1:337–348. doi: 10.1089/cmb.1994.1.337. [DOI] [PubMed] [Google Scholar]
- 12.Gondro C, Kinghorn BP. A simple genetic algorithm for multiple sequence alignment. Genet. Mol. Res. 2007;6:964–982. [PubMed] [Google Scholar]
- 13.Riaz T, Yi W, Li KB. A tabu search algorithm for post-processing multiple sequence alignment. J. Bioinform. Comput. Biol. 2005;3:145–156. doi: 10.1142/s0219720005000928. [DOI] [PubMed] [Google Scholar]
- 14.Lenhof H-P, Reinert K, Vingron M. Proceedings of the Second Annual International Conference on Computational Molecular Biology (RECOMB-98) New York, NY, USA: ACM Press; 1998. A polyhedral approach to RNA sequence alignment; pp. 153–162. [DOI] [PubMed] [Google Scholar]
- 15.Eddy SR. Multiple alignment using hidden Markov models. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995;3:114–120. [PubMed] [Google Scholar]
- 16.Hogeweg P, Hesper B. The alignment of sets of sequences and the construction of phyletic trees: an integrated method. J. Mol. Evol. 1984;20:175–186. doi: 10.1007/BF02257378. [DOI] [PubMed] [Google Scholar]
- 17.Feng DF, Doolittle RF. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol. 1987;25:351–360. doi: 10.1007/BF02603120. [DOI] [PubMed] [Google Scholar]
- 18.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 20.Do CB, Mahabhashyam MS, Brudno M, Batzoglou S. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 25.Pei J, Grishin NV. MUMMALS: multiple sequence alignment improved by using hidden Markov models with local structural information. Nucleic Acids Res. 2006;34:4364–4374. doi: 10.1093/nar/gkl514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paten B, Herrero J, Beal K, Birney E. Sequence progressive alignment, a framework for practical large-scale probabilistic consistency alignment. Bioinformatics. 2009;25:295–301. doi: 10.1093/bioinformatics/btn630. [DOI] [PubMed] [Google Scholar]
- 27.Roshan U, Livesay DR. Probalign: multiple sequence alignment using partition function posterior probabilities. Bioinformatics. 2006;22:2715–2721. doi: 10.1093/bioinformatics/btl472. [DOI] [PubMed] [Google Scholar]
- 28.Rausch T, Emde AK, Weese D, Dring A, Notredame C, Reinert K. Segment-based multiple sequence alignment. Bioinformatics. 2008;24:i187–i192. doi: 10.1093/bioinformatics/btn281. [DOI] [PubMed] [Google Scholar]
- 29.Morgenstern B, Frech K, Dress A, Werner T. DIALIGN: finding local similarities by multiple sequence alignment. Bioinformatics. 1998;14:290–294. doi: 10.1093/bioinformatics/14.3.290. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian AR, Weyer-Menkhoff J, Kaufmann M, Morgenstern B. DIALIGN-T: an improved algorithm for segment-based multiple sequence alignment. BMC Bioinformatics. 2005;6:66. doi: 10.1186/1471-2105-6-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian AR, Kaufmann M, Morgenstern B. DIALIGN-TX: greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol. Biol. 2008;3:6. doi: 10.1186/1748-7188-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwartz AS, Pachter L. Multiple alignment by sequence annealing. Bioinformatics. 2007;23:e24–e29. doi: 10.1093/bioinformatics/btl311. [DOI] [PubMed] [Google Scholar]
- 33.Bradley RK, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, Holmes I, Pachter L. Fast statistical alignment. PLoS Comput. Biol. 2009;5:e1000392. doi: 10.1371/journal.pcbi.1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, Sze SH. Improving accuracy of multiple sequence alignment algorithms based on alignment of neighboring residues. Nucleic Acids Res. 2009;37:463–472. doi: 10.1093/nar/gkn945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson JD, Koehl P, Ripp R, Poch O. BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins. 2005;61:127–136. doi: 10.1002/prot.20527. [DOI] [PubMed] [Google Scholar]
- 36.Van Walle I, Lasters I, Wyns L. SABmark—a benchmark for sequence alignment that covers the entire known fold space. Bioinformatics. 2005;21:1267–1268. doi: 10.1093/bioinformatics/bth493. [DOI] [PubMed] [Google Scholar]
- 37.Mizuguchi K, Deane CM, Blundell TL, Overington JP. HOMSTRAD: a database of protein structure alignments for homologous families. Protein Sci. 1998;7:2469–2471. doi: 10.1002/pro.5560071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raghava GP, Searle SM, Audley PC, Barber JD, Barton GJ. OXBench: a benchmark for evaluation of protein multiple sequence alignment accuracy. BMC Bioinformatics. 2003;4:47. doi: 10.1186/1471-2105-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoye J, Evers D, Meyer F. Rose: generating sequence families. Bioinformatics. 1998;14:157–163. doi: 10.1093/bioinformatics/14.2.157. [DOI] [PubMed] [Google Scholar]
- 40.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 41.Altschul SF, Madden TL, Schffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.