


Abstract
A novel template design for single-molecule sequencing is introduced, a structure we refer to as a SMRTbell™ template. This structure consists of a double-stranded portion, containing the insert of interest, and a single-stranded hairpin loop on either end, which provides a site for primer binding. Structurally, this format resembles a linear double-stranded molecule, and yet it is topologically circular. When placed into a single-molecule sequencing reaction, the SMRTbell template format enables a consensus sequence to be obtained from multiple passes on a single molecule. Furthermore, this consensus sequence is obtained from both the sense and antisense strands of the insert region. In this article, we present a universal method for constructing these templates, as well as an application of their use. We demonstrate the generation of high-quality consensus accuracy from single molecules, as well as the use of SMRTbell templates in the identification of rare sequence variants.
INTRODUCTION
Single-molecule real-time (SMRTTM) sequencing is a method for generating sequence data that harnesses the intrinsic speed, fidelity and processivity of polymerase molecules (1). The direct observation of polymerase molecules confined in zero-mode waveguides (ZMWs) makes this sequencing approach inherently flexible with respect to the size of the template that can be sequenced, as well as the topology of the template. This flexibility enables the production of sequencing data from short or long inserts. However, a novel application is created by combining circular templates with read lengths significantly longer than the insert size. In this application, a sequencing read produces multiple observations of each base, and these multiple observations can then be used to generate high-accuracy consensus sequence from single molecules. We refer to this use of SMRT sequencing as circular consensus sequencing.
Here, we describe a template format and method of production that, independent of insert size, allows for construction of molecules that are topologically circular. The resulting templates are called SMRTbell templates and consist of a double-stranded region flanked on either end by single-stranded loops (which are referred to as the insert and hairpins, respectively). In principle, the insert sequence can be of any length. In practice, we have created templates with inserts as short as 40 bp, and as large as 25 000 bp, and we currently see no evidence of an intrinsic limit in the size of template that can be created. The hairpins can be constructed to include a wide variety of sequences, including different lengths of sequence, limited only by the thermodynamics of loop formation and primer binding.
Due to the nature of the ligation reactions used to generate these templates, the products are covalently closed circles containing two complementary sequences. Observing the replication of these molecules in a SMRT sequencing system allows one to construct a consensus sequence from multiple reads of both a sense and an antisense strand, all from a single molecule.
In this work, we demonstrate the utility of this template format in a SMRT sequencing system by sequencing variants of a targeted region of the Staphylococcus aureus genome. This organism is a pathogen of increasing significance in hospital settings, particularly since the emergence of methicillin-resistant forms of the microorganism in the 1960s (2,3). More recently, there has been a dramatic increase in cases of non-hospital-acquired (or community-associated) S. aureus infections (2–5). These strains are genetically distinct from hospital-acquired strains (4,5).
Monitoring variation in housekeeping genes is a common approach to cataloging strains of microbes (6). Genetic fingerprints can be constructed by measuring the genotypes of these housekeeping genes. A number of markers were recently identified that efficiently divide clinical isolates of S. aureus into subpopulations (5,7). These markers were selected as a source of genetic diversity to test the ability of SMRTbellTM templates to identify variants when applied in a circular consensus sequencing mode.
MATERIALS AND METHODS
Template construction
Genomic DNA for two strains of S. aureus was purchased from the American Type Culture Collection (ATCC): the methicillin-sensitive strain FDA 209 and the methicillin-resistant strain Mu50. A known variant in S. aureus strains was PCR amplified from these strains using Phusion DNA polymerase from New England Biolabs (NEB) with manufacturer recommended cycling conditions and the primers 5′-GTACGGGTCTCACCCGGTTAACTGCACCTGCATTAA-3′ and 5′-CCTAAGGTCTCGGAAGGAAATTATTTCGAAAAAAGA-3′. For demonstration of this approach on longer fragments, a 1 kb fragment of the ΦX174 genome (NEB) was PCR amplified with Phusion DNA polymerase using manufacturer recommended cycling parameters and the primers 5′-GTACGGGTCTCACCCGAGGCTCTAATGTTCCTAACC-3′ and 5′-CCTAAGGTCTCGGAAGATCTGCTTATGGAAGCCAAG-3′. In all cases, the primers contain a restriction site for the enzyme BsaI. The PCR products were purified using PCR purification columns (Qiagen) and digested with the restriction enzyme BsaI (NEB). The digested PCR products were then ligated to two hairpin-forming oligonucleotides. For the S. aureus products, the hairpin oligonucleotides were 5′-CTTCTCTCTCTCTTTTCCTCCTCCTCCGAAGAAGAAGCCGAGAGAGA-3′ and 5′-CGGGTTTGTTGCAAAGCCTAAACCAATATTGATACATTAGCAACAAA-3′. For the the ΦX174 PCR products, the hairpin oligonucleotides consisted of the sequences 5′-CGGGTCTCTCTCTTTTCCTCCTCCTCCGTTGTTGTTGTTGAGAGAGA-3′ and 5′-CTTCTCTCTCTCTTTTCCTCCTCCTCCGTTGTTGTTGTTGAGAGAGA-3′. The hairpin-forming oligonucleotides contained overhangs complementary to the BsaI product overhangs. Prior to ligation, the hairpins were annealed in stem–loop structures by diluting to 20 μM in the presence of 10 mM Tris (pH 7.5) and 100 mM NaCl. Annealed hairpins were added at molar excess relative to the insert and ligated using T4 DNA Ligase (NEB). Failed ligation products were removed through digestion in the presence of ExoIII and ExoVII exonucleases (NEB and USB, respectively).
Bulk extension reactions
To confirm the proper ligation of the inserts, extension reactions were performed in a similar method to that described previously (1). The sequencing primer (GGAGGAGGAGGA) was labeled with Cy5 on the 5′-end to facilitate detection. The template concentration in the extension reactions was 10 nM and the polymerase concentration was 100 nM. As described previously, a trap oligonucleotide was added to capture any polymerase that dissociated from the target during extension. Timepoints were taken at 2, 10, 30 and 60 min, quenched in the presence of 50 mM EDTA and run on a 1.5% agarose gel.
Sequencing reactions
Biotinylated DNA polymerases were incubated with 2–3 fold molar excess of primed DNA templates in loading buffer as described previously (1). The ternary complex was then kept at 4°C for the remainder of the sequencing experiment. For each chip, the ternary complex was immobilized onto the ZMW arrays at 22.5°C, and the array was prepared for sequencing by adding an enzymatic oxygen scavenging system, triplet state quencher and all four phospholinked dNTPs (at 0.5 μM final concentration of each) as described (1). Sequencing reactions were initiated by addition of Mn2+ to a final concentration of 0.5 mM.
Analysis
The consensus base calls from each single-molecule read are derived from a probabilistic sequence alignment method, modified from (8). The first step is to classify the different regions in the raw circular molecule reads into adapter and insert subreads. This is done by aligning a raw read sequence to the known adapter sequence and a putative reference sequence of the insert regions. Once all subreads are identified, we test at each base position whether the subreads are generated by the putative reference sequence of the template or some variation of that sequence (e.g. a single-nucleotide polymorphism or an indel).
To detect the potential single-nucleotide polymorphism at a given location of the template, we align all the insert subreads identified from a single-molecule raw read to four different sequences representing the four possible SNP candidates using a probabilistic sequence aligner (8). The sequences, denoted as 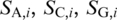 and
and 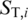 , are constructed by replacing the base in the original putative template sequence at position i by the four possible bases A, C, G and T, respectively. In contrast to a conventional maximum scoring alignment algorithm, e.g. a Smith–Waterman algorithm, the advantage of using a probabilistic alignment is that it naturally assigns the likelihood,
, are constructed by replacing the base in the original putative template sequence at position i by the four possible bases A, C, G and T, respectively. In contrast to a conventional maximum scoring alignment algorithm, e.g. a Smith–Waterman algorithm, the advantage of using a probabilistic alignment is that it naturally assigns the likelihood, 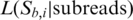 , for each of the candidates
, for each of the candidates 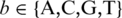 . We call the base
. We call the base 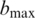 that gives the greatest likelihood
that gives the greatest likelihood 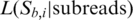 the consensus base call. Furthermore, the log-likelihood ratio,
the consensus base call. Furthermore, the log-likelihood ratio, 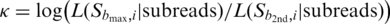 , between the best candidate
, between the best candidate 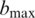 to the second best candidate
to the second best candidate 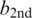 is used to assess the confidence or quality of the consensus call. If the likelihood ratio κ is zero, then the best call and the second best call are equally likely, and we cannot identify with confidence what template base at that position could have led to the observed insert subreads. In contrast, if κ is large, the alternative possibility that the second best call rather than the best one is the correct template base becomes proportionately less likely. Indeed, we find that κ is well correlated with the error rates of the consensus calls. Therefore, κ can be used to predict the quality of the consensus calls and filter out low-quality reads and base calls.
is used to assess the confidence or quality of the consensus call. If the likelihood ratio κ is zero, then the best call and the second best call are equally likely, and we cannot identify with confidence what template base at that position could have led to the observed insert subreads. In contrast, if κ is large, the alternative possibility that the second best call rather than the best one is the correct template base becomes proportionately less likely. Indeed, we find that κ is well correlated with the error rates of the consensus calls. Therefore, κ can be used to predict the quality of the consensus calls and filter out low-quality reads and base calls.
RESULTS AND DISCUSSION
Production of SMRTbellTM templates
In deciding on a format for SMRT™ sequencing, a number of factors were considered, including ability to accommodate a range of insert sizes, suitability for circular consensus sequencing, simplicity and speed of construction, uniformity of structure and compatibility with ZMW geometry (9). The SMRTbell template format meets all of these criteria. As depicted in Figure 1A, a SMRTbell template structurally resembles a linear double-stranded DNA fragment. At either end, the double strand is capped with a hairpin sequence, such that there are no free 5′- or 3′-ends. These hairpins contain a sequence complementary to a primer. When incubated in the presence of a DNA polymerase, the enzyme can bind to the primer/template complex, leading to a sequencing-productive complex. As the SMRTbell template is constructed starting from a double strand, it possesses complementary strand information. Therefore, in a circular consensus application, sequence information can be obtained from both the sense and antisense strands of a template, which have different sequence contexts (Figure 1B). It is expected that the performance of a polymerase will vary with sequence context. The ability to read both strands on a single DNA molecule therefore enables correction for sequence context-dependent variation.
Figure 1.

Schematic of a SMRTbell™ template. (A) A SMRTbell template consists of a double-stranded region (the insert) flanked by two hairpin loops. The hairpin loops present a single-stranded region to which a sequencing primer can bind (orange). (B) As a strand-displacing polymerase (gray) extends a primer from one of the hairpin loops, it uses one strand as the template strand and displaces the other. When the polymerase returns to the 5′-end of the primer, it begins strand displacement of the primer and continues to synthesize DNA (moving in the direction of the blue arrow). Therefore, the length of sequence obtained from these templates is not limited by the insert length. Furthermore, the resulting sequence is derived from both sense- and anti-sense strands.
Methods for producing these structures have been described previously for short hairpin loop sequences (10–13). For templates prepared from PCR fragments, we follow a similar approach, but with modifications to the hairpin design. In this case, PCR fragments are digested with a restriction enzyme and then ligated to hairpin-forming oligonucleotides containing a complementary overhang (Figure 2A). We find that one of the by-products of ligation, dimeric and higher-order multimers of the insert, form during this ligation. These by-products are eliminated through the use of Type IIS restriction enzymes. As adapter ligation is a bimolecular process, the efficiency of ligation is relatively independent of insert size, enabling the generation of templates across a wide range of insert sizes.
Figure 2.

Method of construction of SMRTbell templates. (A) Method for template generation. The boxes on the left depict the process for SMRTbell generation from a PCR fragment. The boxes on the right illustrate the process for randomly generated fragments of DNA. Whereas PCR products are produced in a defined length and, if digested with a restriction enzyme, contain defined overhangs, genomic DNA must be sheared down to an appropriate size, end polished to generate blunt ends and then extended by 1 nt to generate a single A overhang. This is represented schematically with a generic ‘N’ overhang. Hairpin loops with an overhang complementary to the overhang on the DNA fragments are ligated to the ends of the insert in the final step. (B) The single-nucleotide polymorphism-containing constructs used in this work. The two templates contain an insert of ∼140 bp, with either a T/A or a G/C base pair at the site of the polymorphism (indicated in bold).
We have extended this strategy to make it amenable to libraries of randomly generated fragments. Whereas the PCR-based strategy is useful for targeting specific, known template sequences, randomly generated fragments are useful for sequencing much larger target regions. Fragments can be readily generated through any of the common approaches that are used in the production of libraries, including sonication (14), mechanical shearing (15), restriction enzyme digestion (16) and other enzymatic digestions (17). Randomly generated fragments must be cleaned up in an end-polishing reaction, generating blunt ends. Simultaneously, the 5′-ends of the blunt fragments are phosphorylated. We prevent the formation of chimeric fragments by utilizing a tailing reaction to incorporate a single adenine at the 3′-end of every fragment. Finally, hairpin adapters are ligated to the resulting single-nucleotide overhang.
The circular topology of the SMRTbell format enables a circular consensus sequencing application, where observations of polymerase activity can be made repeatedly from the same molecule from both strands of the insert region. Single-molecule consensus data can be used to identify with high confidence the different alleles that may be present within an individual template molecule. To demonstrate the suitability of this format for application to a circular consensus sequencing application, a targeted, PCR-based strategy was used to generate templates. Primers were designed to target a region of the housekeeping gene aroE132 of S. aureus. The primers were designed to include a specific recognition sequence such that digestion with a restriction enzyme would result in a unique 4-nt overhang on each end of the PCR product.
Housekeeping genes have been sequenced from a large number of S. aureus strains (5,7) and have a number of positions of variation mapped. These variations can be used as markers to distinguish different strain isolates of S. aureus. One of these markers is contained within the aroE132 gene. Two strains of S. aureus (the FDA 209 and Mu50 strains) with a single-nucleotide difference within the aroE132 gene were selected for this study. Two amplicons were produced, one from each strain, with a single-nucleotide difference between the two templates (Figure 2B). Each end of the products was then ligated to a unique hairpin-forming sequence. One of these hairpins contained a sequence complementary to a primer sequence and could therefore be used as a priming site for DNA synthesis.
Generation of a long SMRTbellTM template for sequencing
The circular consensus sequencing application requires the ability to generate long reads from each molecule. We assessed the ability of these templates to support the generation of long products such as would be seen in rolling-circle replication in a bulk extension assay. A fluorescently labeled primer was annealed to one of the two hairpin loops on the template. The primed template was then incubated in the presence of polymerase and allowed to extend for up to 1 h (Figure 3).
Figure 3.

Bulk extension products of two SMRTbell templates. Lanes 1–4 contain the product from a template derived from strain FDA 209 (the ‘T’ allele) at timepoints of 2, 10, 30 and 60 min. Lanes 5–8 contain the product from a template derived from strain Mu50 (the ‘C’ allele) at timepoints of 2, 10, 30 and 60 min. Approximate sizes of products were determined relative to a 1 kb molecular weight ladder (indicated on the left). The position of the sequencing primer is indicated with an arrow.
In the full time course of these extension reactions, the product is converted into material that is nearly 3000 bp in length. Including both strands of the insert and both hairpins of these templates, the template length is 336 nt. Therefore, these products represent nearly 10 complete passes around the template, indicating that this format will indeed support the circular consensus sequencing application.
The circular molecules were then applied to SMRTTM sequencing, described in (1). In brief, this system utilizes a number of recently developed technologies to enable multiplex single-molecule observation. Nucleotide analogs, each containing a fluorophore linked to its terminal phosphate, are incorporated into a nascent chain. These fluorophores are cleaved during incorporation, leaving a native product and a free fluorophore. Polymerization is confined to the bottom of nanostructures known as ZMWs through a streptavidin/polymerase complex bound to a biotinylated surface. The ZMWs allow for a zeptoliter scale illumination and detection volume (9), such that free-nucleotide analogs and fluorophore products diffuse through the illumination volume on a microsecond timescale and are consequently not detected. In contrast, bound analogs are retained by the polymerase on a timescale governed by the rate of catalysis (on the order of 10 s of ms). Therefore, they remain in the illumination volume long enough to provide the signal to noise required for detection of a single event, even in the presence of micromolar analog concentrations. The fluorescence signal for each of the analogs is observed by a CCD camera. Consequently, a time series of observed pulses reveals the template sequence (1).
Figure 4A is the result of such a sequencing reaction. This plot shows a time series of total fluorescence signal observation. The different regions of the observed trace are colored by its corresponding region of the molecule. This trace shows an alternating pattern of sense strand, first hairpin, antisense strand and second hairpin alignments, as expected for a long sequencing reaction from a short template. We call the set of called bases corresponding to the insert region a subread. For example, Figure 4A shows four sense subreads (shown in blue) and four antisense subreads (shown in orange). The sequence from these subreads are used for building single-molecule consensus.
Figure 4.

Demonstration of single-molecule traces that include both sense and antisense strands of a single molecule. The pulses within the sequencing traces are colored according to location within the template, with blue corresponding to the sense strand, orange corresponding to the antisense strand, and light blue and green corresponding to the hairpin adapters. (A) A representative trace from an aroE132 template. In this example, incorporations are observed on four complete passes of the template, generating four-fold coverage of the sense strand and four-fold coverage of the antisense strand. (B) A representative trace from a 1000-bp PCR product derived from the PhiX174 genome. In this case, there are two sub-reads that correspond to the sense strand and one to the antisense strand.
A similar demonstration is shown in Figure 4B for a 1000-bp template. Due to the longer size of the insert, there are fewer subreads observed in the trace. However, this trace also shows two subreads corresponding to the sense strand and one subread corresponding to the antisense strand, with pulses corresponding to the hairpins separating each subread (shown in light blue and green).
Single-molecule consensus from SMRTbellTM templates
The subreads resulting from sequencing the two aroE132 templates were identified and used to generate consensus base calls for reads from all ZMWs as described in the ‘Materials and Methods’ section. To asses the quality of the consensus base calls, κ (defined in ‘Materials and Methods’ section) is calculated and tested for its power to predict the consensus base call quality. A data set of reads was randomly split into two equal size sets, a training set and a test set. We calculated the κ of all reads at non-SNP positions in the training set. The data is binned and the number of errors of the consensus base calls in each bin is tallied. For each bin, the phred-style quality value (QV) was calculated as QV = −10 log10 (number of base call errors/total number of base calls) to derive the quality value of the consensus call as a function of κ. In the test set, κ is calculated for each position and the predicted consensus quality value is calculated by 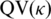 . Figure 5 shows that the prediction agrees with the measured empirical quality values. For each read, we also define a read-level quality value (RQV) as the average of per base quality values over the template sequence. The RQVs are used for filtering low-quality reads.
. Figure 5 shows that the prediction agrees with the measured empirical quality values. For each read, we also define a read-level quality value (RQV) as the average of per base quality values over the template sequence. The RQVs are used for filtering low-quality reads.
Figure 5.

Comparison of measured empirical quality values to predicted consensus quality values. In the test set, data are binned around the predicted consensus QVs and the numbers of errors are tallied for calculation of the empirical consensus base call QV. We normalize the number of total base calls of each bin to 10 000. We repeat this sampling procedure 10 times for each predicted QV to derive the error bar to represent sampling errors.
Detection of variants using circular consensus sequencing
The minimum criterion for detecting variants from individual molecules is the ability to obtain a high-accuracy sequencing result from a single molecule. However, single-molecule detection should also enable high sensitivity of detection. To test the sensitivity of SNP detection using this circular consensus sequencing approach, we mixed the two variant aroE132 templates across a wide range of mixing ratios. The two templates to be sequenced were mixed at percentages of 0:100, 2.5:97.5, 5:95, 10:90, 25:75, 50:50 and 100:0 [T allele (%):C allele (%) at position 79]. Sequence data were generated in the system described above. A conventional threshold RQV value of 20 was used as a quality control to filter the data.
Figure 6 demonstrates the ability of single-molecule sequencing to quantitate allele frequency. The x-axis is the expected frequency of observing the SNP position called as a ‘T’ in the sample according to the mixing ratio. The y-axis is the measured frequencies of all four possible base calls at the SNP position. At each titration level, one to two thousand single-molecule reads pass the quality filter. This allows us to determine the range of allele frequencies in the sample to within 2–3%.
Figure 6.

Comparison of the expected SNP frequency to the measured SNP frequency. The SMRTbell templates derived from each of the alleles were mixed in ratios of 0:100, 2.5:97.5, 5:95, 10:90, 25:75, 50:50 and 100:0 (listed as T:C). The frequency of calls for all four possible bases are shown.
CONCLUSIONS
We have developed a DNA format and a universal methodology for generating templates in that format for SMRTTM sequencing, a format we call a SMRTbell template. This format structurally resembles a linear DNA fragment. Topologically however, the resulting DNA is circular. Therefore, this strategy provides a means for construction of circular molecules across a wide range of insert sizes, from <100 bp to at least 25 000 bp. As SMRT sequencing allows for a wide range of insert sizes, this method of template generation provides a universal protocol for the upfront sample preparation. Furthermore, this protocol for template preparation requires a minimal number of steps and does not depend on amplification.
We have illustrated the application of this DNA format by sequencing a polymorphic region of the MRSA genome and in doing so demonstrated reads as high as QV40 at the single-molecule level, by generating a consensus sequence from multiple reads of the same molecule. In a mixture of two alleles, we were able to accurately call the frequency of the polymorphism, even when it was present at only 2.5% of the population. In contrast with other systems, where quality values are a fundamental limit of the chemistry used, the strategy of applying circular consensus sequencing to rare variant detection allows yield and read length to be exchanged for higher QV.
To illustrate this trade-off, consider a system with an average read length of 1000 nt. With a template of approximately 300 bp, an average of three reads can be obtained from the template. With a template size of approximately 250 bp, four reads can be obtained and with a template size of approximately 200 bp, five reads can be obtained. In this manner, the choice of template size determines the number of reads that can be obtained from that template and therefore the final empirical QV (EQV) of the consensus data.
This trade-off is illustrated in Figure 7. Here, we have plotted the unfiltered distributions of EQV obtained from different numbers of reads from individual templates. This plot demonstrates that the majority of the bases are called with consensus EQV >30 by the time we have reached four subreads. In this manner, improvement in rare variant detection does not depend on improvements in the raw accuracy. As previously described (18), the identification of sequence polymorphisms requires that allelic variation can be separated from sequencing error. In other words, the identification of a true variant requires that the quality values of the base calls are high enough that the observed variant could not occur by chance. The circular consensus sequencing approach described here allows one to tune the quality values of consensus base calls to the level demanded by the expected rate of variation.
Figure 7.

The unfiltered distributions of EQV as a function of single-molecule coverage (each full synthesis of a SMRTbell corresponds to 2× single-molecule coverage representing the forward and reverse strands). The upper and lower error bars represent the 75th and 25th quartile, respectively.
FUNDING
National Institutes of Health (5R01HG003710-02 to D.R.R., J.S.E. and S.T.). Funding for open access charge: Pacific Biosciences, a privately held corporation.
Conflict of interest statement. All the authors are employees of Pacific Biosciences.
ACKNOWLEDGEMENTS
The authors would like to acknowledge the entire staff at Pacific Biosciences, in particular Primo Baybayan, Benson Chau, Paul Peluso, Eric Olivares and Susana Wang for help with running sequencing experiments.
REFERENCES
- 1.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 2.Fridkin SK, Hageman JC, Morrison M, Sanza LT, Como-Sabetti K, Jernigan JA, Harriman K, Harrison LH, Lynfield R, Farley MM. Methicillin-resistant Staphylococcus aureus disease in three communities. N. Engl. J. Med. 2005;352:1436–1444. doi: 10.1056/NEJMoa043252. [DOI] [PubMed] [Google Scholar]
- 3.Woodford N, Livermore DM. Infections caused by Gram-positive bacteria: a review of the global challenge. J. Infect. 2009;59(Suppl. 1):S4–S16. doi: 10.1016/S0163-4453(09)60003-7. [DOI] [PubMed] [Google Scholar]
- 4.Orscheln RC, Hunstad DA, Fritz SA, Loughman JA, Mitchell K, Storch EK, Gaudreault M, Sellenriek PL, Armstrong JR, Mardis ER, et al. Contribution of genetically restricted, methicillin-susceptible strains to the ongoing epidemic of community-acquired Staphylococcus aureus infections. Clin. Infect. Dis. 2009;49:536–542. doi: 10.1086/600881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephens AJ, Huygens F, Inman-Bamber J, Price EP, Nimmo GR, Schooneveldt J, Munckhof W, Giffard PM. Methicillin-resistant Staphylococcus aureus genotyping using a small set of polymorphisms. J. Med. Microbiol. 2006;55:43–51. doi: 10.1099/jmm.0.46157-0. [DOI] [PubMed] [Google Scholar]
- 6.Maiden MC. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 2006;60:561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- 7.Robertson GA, Thiruvenkataswamy V, Shilling H, Price EP, Huygens F, Henskens FA, Giffard PM. Identification and interrogation of highly informative single nucleotide polymorphism sets defined by bacterial multilocus sequence typing databases. J. Med. Microbiol. 2004;53:35–45. doi: 10.1099/jmm.0.05365-0. [DOI] [PubMed] [Google Scholar]
- 8.Yu YK, Hwa T. Statistical significance of probabilistic sequence alignment and related local hidden Markov models. J. Comput. Biol. 2001;8:249–282. doi: 10.1089/10665270152530845. [DOI] [PubMed] [Google Scholar]
- 9.Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. Zero-mode waveguides for single-molecule analysis at high concentrations. Science. 2003;299:682–686. doi: 10.1126/science.1079700. [DOI] [PubMed] [Google Scholar]
- 10.Kuhn H, Frank-Kamenetskii MD, Demidov VV. High-purity preparation of a large DNA dumbbell. Antisense Nucleic Acid Drug Dev. 2001;11:149–153. doi: 10.1089/108729001300338672. [DOI] [PubMed] [Google Scholar]
- 11.Schakowski F, Gorschluter M, Junghans C, Schroff M, Buttgereit P, Ziske C, Schottker B, Konig-Merediz SA, Sauerbruch T, Wittig B, et al. A novel minimal-size vector (MIDGE) improves transgene expression in colon carcinoma cells and avoids transfection of undesired DNA. Mol. Ther. 2001;3:793–800. doi: 10.1006/mthe.2001.0322. [DOI] [PubMed] [Google Scholar]
- 12.Taki M, Kato Y, Miyagishi M, Takagi Y, Sano M, Taira K. A direct and efficient synthesis method for dumbell-shaped linear DNA using PCR in vitro. Nucleic Acids Res. Suppl. 2003;3:191–192. doi: 10.1093/nass/3.1.191. [DOI] [PubMed] [Google Scholar]
- 13.Zanta MA, Belguise-Valladier P, Behr JP. Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc. Natl Acad. Sci. USA. 1999;96:91–96. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deininger PL. Random subcloning of sonicated DNA: application to shotgun DNA sequence analysis. Anal. Biochem. 1983;129:216–223. doi: 10.1016/0003-2697(83)90072-6. [DOI] [PubMed] [Google Scholar]
- 15.Oefner PJ, Hunicke-Smith SP, Chiang L, Dietrich F, Mulligan J, Davis RW. Efficient random subcloning of DNA sheared in a recirculating point-sink flow system. Nucleic Acids Res. 1996;24:3879–3886. doi: 10.1093/nar/24.20.3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanger F, Coulson AR, Barrell BG, Smith AJ, Roe BA. Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing. J. Mol. Biol. 1980;143:161–178. doi: 10.1016/0022-2836(80)90196-5. [DOI] [PubMed] [Google Scholar]
- 17.Anderson S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981;9:3015–3027. doi: 10.1093/nar/9.13.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marth GT, Korf I, Yandell MD, Yeh RT, Gu Z, Zakeri H, Stitziel NO, Hillier L, Kwok PY, Gish WR. A general approach to single-nucleotide polymorphism discovery. Nat. Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]