

Abstract
A sensitive and rapid HTLC-ESI-MS/MS method with an advanced online sample preparation was developed for determination of the γ-secretase inhibitor MK-0752 in human plasma using an internal standard. Plasma samples (100 μL) were diluted and injected directly onto an online extraction column (Cohesive Cyclone MAX 0.5×50 mm, > 30 μm), the sample matrix was washed out with an aqueous solution, and retained analytes were eluted out and transferred directly to the analytical column (Phenomenex Gemini 3μ C18 110A, 50×2.0 mm at 50°C) for separation using a gradient mobile phase. The eluted analytes were then detected on an API-3000 LC–MS/MS System with ESI and a negative multiple reaction monitoring mode. The monitored ion transitions were m/z 441→175 for MK-0752 and 496→175 for the internal standard. Online extraction recoveries were 81%. The method was validated and was linear in the range of 0.05–50 μg/mL. Within-day and between-day precisions were < 8.6%, and accuracies were 0.7% and 7.1%. This method was applied to the measurement of plasma MK-0752 levels in a Phase I study of pediatric patients with recurrent or refractory brain tumors.
Keywords: MK-0752, online sample preparation, high turbulence liquid chromatography (HTLC), electrospray tandem mass spectrometry (ESI-MS/MS)
1. Introduction
MK-0752 (cis-3-[4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl] propanoic acid) is an inhibitor of γ-secretase which is being used to target the NOTCH pathway for treatment of pediatric and adult malignancies [1–3]. The NOTCH pathway consists of two families of ligands, four receptors (NOTCH1-4), and the CSL transcription factor [4]. Ligand activation of NOTCH1 triggers a series of proteolytic steps where the intracellular domain of NOTCH1 is cleaved, allowing it to translocate to the nucleus and activate CSL. Dysregulation of the NOTCH pathway has been observed in a variety of pediatric malignancies, including T-cell acute lymphoblastic leukemia (T-ALL), medulloblastoma, osteosarcoma, Ewing’s sarcoma, and neuroblastoma [2]. For example, 50% of T-ALL patients carried activating mutations in the NOTCH1 gene [5].
γ-secretase is responsible for the final cleavage step needed to activate NOTCH1 [4]. Therefore, although γ-secretase inhibitors were originally developed for the treatment of Alzheimer’s disease [6], this enzyme has now become a target for cancer therapies [1–3]. MK-0752 is a potent inhibitor of γ-secretase with adequate oral bioavailability.
An analytical method to measure MK-0752 in human plasma or CSF using an off-line solid phase extraction in an automated 96-well format was previous reported [7]. This method was developed to support pharmacokinetic studies in trials of adults with Alzheimer’s disease. However, pharmacokinetic studies in children with cancer presented unique challenges, which required the development of a new analytical method. Specifically, high sample throughput using an expensive automatic sample preparation system was not necessary due to the smaller number of patient samples, but a sensitive and relatively rapid assay using a small volume of blood was required.
Herein, we describe a sensitive and selective analytical method for the quantitation of MK-0752 in human plasma with online extraction and electrospray ionization tandem mass spectrometry (HTLC-ESI–MS/MS). This method is fully validated and application to a pharmacokinetic study in children with brain tumors is shown.
2. Experimental
2.1. Materials and Reagents
MK-0752 (>99% purity) and an analog, dimethyl MK-0752, used as the internal standard (ISTD) were obtained from Merck Research Laboratories (Rahway, NJ, USA). HPLC grade acetonitrile (ACN), methanol, and acetone were obtained from Burdick & Jackson (Mukegon, MI, USA), whereas formic acid (minimum 95%) and ammonium acetate (minimum 97.6%) were purchased from Sigma (St. Louis, MO, USA). Ammonium formate (HCOONH4 minimum 97.0%) was obtained from Fisher (Fairlawn, NJ, USA). Blank human plasma was obtained from Lifeblood (Memphis, TN, USA). All water (ddH2O) was distilled, deionized, and further purified via a Millipore Milli-Q UV plus and Ultra-Pure Water System (Tokyo, Japan) (resistance 18.2 MΩ). Other chemicals were purchased from standard sources and were of the highest quality available.
2.2. Apparatus and Chromatographic Conditions
2.2.1. Online Plasma Sample Extraction and Chromatographic Conditions
The turboflow TLX-1 online extraction system (ThermoFisher Scientific, Franklin, MA), which contains both loading and eluting pumps with a proprietary valve-switching module (Agilent Technologies, 1200 series), was interfaced to a computer workstation running Aria® OS software. After an online injection of prepared sample, a Cyclone MAX micro-bore large particle size column (50×0.5 mm, ≥30μm, ThermoFisher Scientific, Franklin, MA) was used to extract/cleanup the analytes from human plasma. Extracted sample was transferred onto an analytical column (Gemini C18 column, 3μ, 50×2.0 mm, Torrance, CA, USA) preceded by a guard column with the same material (4×2.0 mm, 5μ). Separation was performed at 50°C. The loading pump was coupled with a Shimadzu FCV-10ALvp and carried three loading solutions for the online extraction with Cyclone MAX column: Loading Solution A (5mM HCOONH4, pH 10), Loading Solution B (ACN with 1.0% formic acid), Loading Solution C (0.5% formic acid in ddH2O). The transferring loop was previously filled with 75% ACN in 0.5% formic acid water. The flow rate of loading pump was 2.0 mL/min for online sample extraction except transferring step at 0.10mL/min. The eluting pump delivered two eluting solutions for chromatographic separation on the Gemini C18 column with a gradient profile of (B% from 5 to 95): Eluting Solution A (the mixture of 5mM HCOONH4/ACN = 98/2, v/v) and Eluting Solution B (100% ACN). Prime flow rate of eluting pump was 0.70mL/min, which was split by 50:50 at the inlet of ion source. A flow diagram is published [8] and the settings for the HTLC method is shown in Table 1.
Table 1.
LC method for the focus mode HTLC online extraction method
| Loading Pump | Eluting Pump | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Step | Start | Sec | Flow (mL/min) | Grad | A% | B% | C% | D% | Tee | Loop | Flow (mL/min) | Grad | A% | B% |
| 1 | 0.00 | 60 | 2.00 | Step | 100.0 | - | - | - | ==== | out | 0.70 | Step | 100.0 | - |
| 2 | 1.00 | 120 | 0.10 | Step | 100.0 | - | - | - | T | in | 0.60 | Step | 100.0 | - |
| 3 | 3.00 | 90 | 2.00 | Step | - | 100.0 | - | - | ==== | in | 0.70 | Ramp | 5.0 | 95.0 |
| 4 | 4.50 | 90 | 2.00 | Step | - | 75.0 | 25.0 | - | ==== | in | 0.70 | Step | 5.0 | 95.0 |
| 5 | 6.00 | 160 | 2.00 | Step | 100.0 | - | - | - | ==== | out | 0.70 | Step | 100.0 | - |
10μL of supernatant was directly injected onto micro-bore online extraction column. MK-0752 and its ISTD were retained on the extraction column while the plasma matrix components were rapidly washed out with 100% aqueous Loading Solution A for 60 s at a flow rate of 2.0mL/min. The analytes were eluted from the extraction column by back-flushing with acidified 100% organic Loading Solution B and then transferred onto the analytical column at 0.10mL/min, where the analytes were separated on the analytical column from all other endogenous compounds. The analytes were then eluted to the mass spectrometer using a one-step gradient organic Eluting Solution B from 5% to 95% at flow rate 0.70mL/min for 90 s. The extraction column was washed with a combination of Loading Solution B and C for 90 s at 2.0 mL/min, and then each column was equilibrated with its aqueous Solution A for 160 s prior to the next injection at 2.0 mL/min and 0.7 mL/min, respectively. We observed back pressure values of approximately 20 and 130 bars on the extraction and analytical column, respectively. Total run time for each sample was about 6.5 minutes per cycle, which included extraction, separation, and MS detection.
2.2.2. Mass Spectrometric Conditions
Detection was performed with an API 3000 LC–MS/MS System (Toronto, Canada) equipped with a Turbo IonSpray® source (thermally and pneumatically assisted electrospray), which was run at the unit resolution of Q1 and the low resolution of Q3 in negative mode with multiple reaction monitoring (MRM). The optimized conditions of MS/MS with electrospray and HTLC conditions were as follows: ion spray source temperature at 400 °C, nebulizer (NEB) gas at 13, curtain (CUR) gas at 13, turbo gas flow at 6 L/min, ionspray voltage (IS) at −4500 V, and collision-activated dissociation (CAD) at 8.0 units; declustering potential (DP) at −61 V, focusing potential (FP) at −209 V, entrance potential (EP) at −10.5 V, collision energy (CE) at −34.0 V, and collision exit potential (CXP) at −13.0 V. The mass spectrometer was interfaced to a computer workstation running both Aria® OS software and Analyst software (Version 1.4.1 Applied Biosystems, Foster City, CA) for data acquisition and processing.
2.3. Sample Preparation
2.3.1. Stock Solutions
The stock solutions were prepared by dissolving MK-0752 and ISTD in 80% Methanol water to yield a concentration of 1.0 mg/mL, respectively. The internal standard stock solution needed to be sonicated to dissolve well from the chemical powder. Stock solutions were stored at −80 °C, and less than 5% of their nominal values were lost over 6 months. The working solutions were prepared by diluting the 1.0mg/mL stock solution with 80% Methanol in water to the concentrations of 100, 10, and 1 μg/mL for MK-0752 and 60 μg/mL for ISTD.
2.3.2. Calibration Curve and Quality Controls
Calibrators were made by adding MK-0752 working stock solutions to blank human plasma in correct proportions to give final MK-0752 concentrations of 50, 500, 1500, 3500, 5000, 15000, 35000, and 50000 ng/mL, with 3000 ng/mL of ISTD. The quality controls of three different concentrations were prepared in the same manner at 60, 6000, and 45,000 ng/mL.
2.3.3. Plasma Sample Preparation
A 100 μL aliquot of patient plasma or blank human plasma spiked with MK-0752 was transferred to a 1.5 mL centrifuge tube with 5 μL of 60 μg/mL ISTD working solution, and vortexed for 15 s. Each sample was then diluted with 200 μL of ACN with 0.1% formic acid. The tubes were then vortexed well again and centrifuged at 14,000 rpm for five min at 4°C. 150 μL of the supernatant were transferred into autosampler vials and 10 μL was injected onto the HTLC-ESI-MS/MS system.
2.3.4. Patient Sample Collection and storage
Whole blood samples were collected in sodium heparin tubes and immediately centrifuged at 10,000 rpm for 2 min to separate the plasma. Plasma was immediately frozen and stored at −80°C until analysis.
2.4. Assay Validation
2.4.1 Accuracy, precision, and linearity
MK-0752 quality control samples were used for validation of this analytical method. The within-day and between-day precision and accuracy for the method were determined over five days. Ten injections of low and high concentrations of MK-0752 control samples were made on days one and two to assess within-day variability and again on days three, four, and five to evaluate between-day variability. Two calibration curves were prepared and the linear regression of the ratio of MK-0752/ISTD peak areas was weighted by 1/x2. The coefficient of determination (R2) was used to evaluate the linearity of the calibration curve. The lower limit of quantitation (LLOQ) was defined as the lowest plasma concentration at which the signal/noise ratio was ≥ 10 and the within-day accuracy and precision (%CV) were within 80%~120% and ≤ 15%, respectively. The limit of detection (LOD, S/N≥3) was also determined by triplicate analysis of an extensive calibration curve in the low concentration range (1.0–50 ng/mL).
2.4.2 Selectivity and carryover
Selectivity of the assay was assessed by monitoring all selected ion transitions of MK-0752 and the ISTD after injection of blank human plasma from six different sources. Carryover was assessed by injecting MK-0752 at 50 μg/mL and monitoring selected ion transitions of MK-0752 and the ISTD through four blank plasma samples.
2.4.3 Ion suppression
Matrix effects were evaluated by post-column infusion of the analyte [9]. Briefly, a solution of neat MK-0752 (5 ng/ml) and ISTD (500 ng/ml) was infused, post-analytical column, through a Valco zero dead volume tee using a Harvard Apparatus syringe pump 11 (Harvard Apparatus, Holliston, MA, USA) at a constant flow rate of 5.0 μL/min into the LC effluent (300 μL/min) prior to entering the MS. Mobile phase solution and blank human plasma from 6 different sources were then injected onto the online extraction column for sample cleanup and then transferred onto the analytical column. Effluent from the HTLC combined with the infused compound, entered the electrospray interface and was analyzed under the operating conditions to evaluate the matrix effect.
2.4.4 Stability
Stability of MK-0752 at two concentrations (60 and 45000 ng/mL) was evaluated in triplicate in both plasma and supernatant at 4°C and 25°C for up to 24 h, and at −80°C in plasma for 90 days. The freeze-thaw stability of MK-0752 at −80°C was evaluated over three freeze-thaw cycles. For each freeze-thaw cycle, plasma samples spiked with MK-752 (60 and 45000 ng/mL) were stored at −80 °C overnight. The next day samples were thawed at room temperature for 15–20 min, an aliquot from each of the samples was analyzed in duplicate, and the remaining unprocessed sample was returned to −80 °C storage. Internal standard was added to samples prior to the sample preparation and analysis. Data presented are the mean percent of the initial concentration measured immediately (<1 h) after preparation.
2.5. Application of Method to Patient Samples
Serial samples for pharmacokinetic studies of MK-0752 were obtained as part of a clinical study of pediatric patients with relapsed or refractory brain tumors. Whole blood was collected at the following times: pre-dose, and 1, 2, 4, 8 (±2), and 24 (±2) hours after oral MK-752 administration. Plasma was obtained, stored, prepared, and analyzed as per the described method.
Results and discussion
3.1. Online sample extraction and chromatography
The focus mode of the TLX instrument is ideal for online extraction of biological samples as it provides quick sample cleanup, rapid elution, sharp peaks, and low noise level. As we have done previously [10], we used a TurboFlow column (0.5 mm i.d. × 50 mm length) rather than the conventional 1.0mm i.d. column, in order to reduce the flow rate (2.0 mL/min) without loss of extraction efficiency. While the analyte was on the TurboFlow column, a wash with 100% aqueous loading solution A (5.0 mM HCOONH4, pH 10), and transfer with 100% organic loading solution B (ACN containing 1% formic acid) minimized the transfer time between the extraction and the analytical columns, which allowed for an optimized flow rate of 0.10 mL/min. In addition, a 75% organic mobile phase (75% of the loading solution B with 25% of 0.5% formic acid water) was optimized to fill the transfer loop and reduce the organic solvent strength via the tee while permitting a low organic eluting mobile phase containing 5% acetonitrile to focus the analytes prior to the analytical column. The analytes were then eluted by a gradient of acetonitrile from 5%–95% at a flow rate of 0.70 mL/min.
A C18 column with a smaller particle size and internal dimension (3μm, 2.0mm i.d. × 100 mm) was chosen for separation of MK-0752 based upon its chemical structure and the column characteristics. The HPLC conditions were optimized to increase mass signal strength for both compounds, an optimal gradient to improve signal response and peak retention time was achieved by a gradient of ACN from 5% to 95 within 90 seconds at the flow rate of 0.70 mL/min. Column temperatures of 25°C, 40°C, and 50°C were evaluated and a column temperature of 50°C resulted in the best signal response, retention time, and peak shape within 5 min and a proper pump pressure. Under these conditions, the typical retention time was 2.2 min for both MK-0752 and its ISTD (Figure 1).
Figure 1.

Chromatogram of human plasma spiked with MK-0752 at 3.5 μg/mL and ISTD at 3.0 μg/mL (Note: MK-0752 and ISTD eluted at 2.17 and 2.22 min, respectively)
3.2 Mass spectrometry
The full-scan MS1 negative-ion mass spectra of the analyte and ITSD in mobile phase showed the parent (M-H)− ions for MK-0752 and ISTD at m/z 441.1 and m/z 469.3, respectively (Figure 2A and C). These were used as precursor ions for collision-induced dissociation. The product ion scans showed a single abundant product ion for both MK-0752 and ISTD (m/z 174.8), which was used for multiple reaction monitoring (Figure 2B and D).
Figure 2.

Negative ion scan spectra for (A) MK-0752 and (C) ISTD (dimethyl MK-0752), and collision induced dissociation spectra for (B) MK-0752 and (D) ISTD.
3.3 Assay validation
3.3.1 Accuracy, precision, and linearity
Accuracy (as % error) and precision (as % RSD) are reported in Table 2. Within- and between-day accuracy ranged from −0.7 to 7.1% and precision was < 8.6%. The LOD and LLOQ of MK-0752 with this method were 1.5 ng/mL and 50 ng/mL, respectively. The plasma calibration curves were linear from 50 to 50,000 ng/mL with correlation coefficients (R2) greater than 0.9975. The average recoveries of plasma MK-0752 were 77% (60 ng/mL), 85% (6000 ng/mL), and 81% (45000 ng/mL).
Table 2.
Validation parameters of MK-752 in human plasma
| MK-0752 Control (ng/mL) | Within-day (n=10) | Between-day (n=5) | ||
|---|---|---|---|---|
| %RSD | %Error | %RSD | %Error | |
| 60 | 3.39 | 7.05 | 4.33 | 3.68 |
| 45,000 | 8.55 | 4.98 | 2.85 | −0.7 |
3.3.2 Selectivity and carry-over
When monitoring all selected ion transitions after injection of blank human plasma from six different sources, no co-eluting peaks for MK-0752 or ISTD were observed. After injection of MK-0752, no carryover was seen through four sequential blank plasma samples.
3.3.3 Ion Suppression
Ion suppression is an important factor affecting the quantitative performance of a mass detector, especially when using an electrospray interface [11]. Figure 3 depicts the response obtained from post-column infusion of neat MK-0752 and ISTD through injection of blank plasma from 6 different sources. Ion suppression at the retention time of MK-0752 and ISTD (2.2 min) was about 50% for both MK-0752 and ISTD and was similar in all plasma sources, indicating that the ion suppression effect is not a significant concern for this method.
Figure 3.

Ion suppression of MK-0752 and ISTD. Chromatogram resulting from a neat mixture of (A) MK-752 (5 ng/mL) and (B) ISTD (500 ng/mL) infused post-column at a flow rate of 5.0 μL/min into the LC effluent while blank human plasma from 6 sources was injected.
3.3.4 Stability
MK-0752 in spiked plasma samples stored at 4 °C and 25 °C degraded by less than 6.4% and MK-0752 in the extract degraded by less than 5.7% over 24 h (Table 3). When stored at −80°C, there was never more than 6.9% change at any time point up to 95 days (Table 3). Over three freeze-thaw cycles, MK-0752 samples did not show more than 7.6% change compared to the initial concentration (Table 3).
Table 3a.
Validation of MK-0752 stability under assay conditions.
| Condition | Sample | % difference from control |
|
|---|---|---|---|
| 60 ng/mL | 45,000 ng/mL | ||
| 24 h at 25°C | Plasma | 6.4 | −4.2 |
| Extract | 0.7 | 1.1 | |
| 24 h at 4°C | Plasma | 6.4 | −4.5 |
| Extract | 1.7 | 5.7 | |
| 95 days at −80°C | Plasma | −1.6 | −2.4 |
| 3 freeze-thaw cycles | Plasma | −5.3 | 2 |
3.4 Application of assay to patient samples
Plasma samples from pediatric patient with refractory or recurrent brain tumors receiving MK-0752 were analyzed to understand the disposition of MK-0752 in this patient population. Serial samples were collected, processed, and analyzed according to the methods described in this report. A representative plasma concentration-time profile for MK-0752 after a single oral dose of MK-0752 (260 mg/m2) is depicted in Figure 4.
Figure 4.
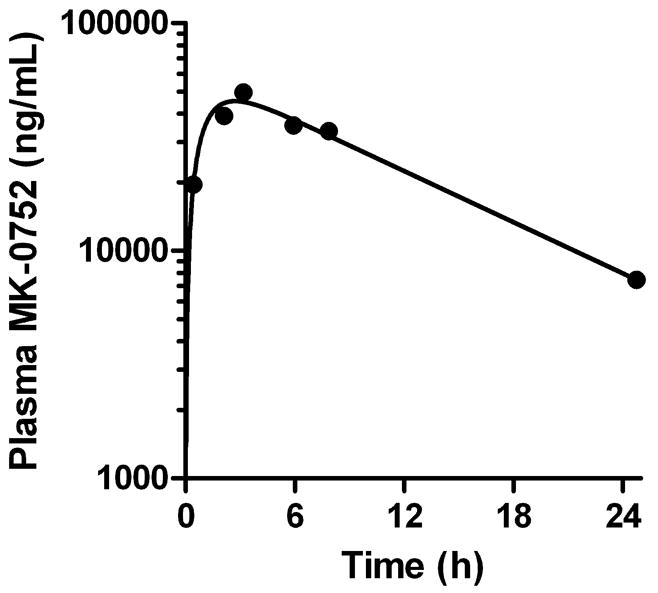
Example MK-0752 plasma concentration-time profile in a pediatric patient after a single oral dose of MK-0752 (260 mg/m2).
3. Conclusion
We report here an online extraction HTLC–ESI-MS/MS method for the rapid and precise quantitation of MK-0752 in human plasma samples. This method requires only 100 μL of plasma instead of 1000 uL [7], making it suitable for studies of MK-0752 in children. The use of turbulence flow online sample extraction decreases sample preparation time, saves labor, and reduces consumable expense. The use of a narrow-bore short column and ESI ion source instead of APCI [7] with a low flow rate reduces solvent costs and minimizes the use of organic solvents. This method was developed to facilitate the quantitation of MK-0752 in human plasma samples and performs with sufficient precision (CV ≤ 8.6%) and accuracy (−0.7 ≤ %Error ≤ 7.1) for application in clinical studies of MK-0752 pharmacokinetics in both pediatric and adult populations. The method is linear in the range of expected plasma concentrations. Finally, we have successfully applied this HTLC–ESI-MS/MS method by measuring MK-0752 in human plasma from a clinical pharmacokinetic study of children treated with MK-0752.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Nat Rev Drug Discov. 2009;8:806. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 2.Zweidler-McKay PA. Curr Oncol Rep. 2008;10:459. doi: 10.1007/s11912-008-0071-2. [DOI] [PubMed] [Google Scholar]
- 3.Macy ME, Sawczyn KK, Garrington TP, Graham DK, Gore L. Curr Oncol Rep. 2008;10:477. doi: 10.1007/s11912-008-0073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aster JC, Pear WS, Blacklow SC. Annu Rev Pathol. 2008;3:587. doi: 10.1146/annurev.pathmechdis.3.121806.154300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Science. 2004;306:269. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 6.Imbimbo BP. Curr Top Med Chem. 2008;8:54. doi: 10.2174/156802608783334015. [DOI] [PubMed] [Google Scholar]
- 7.Matthews CZ, Woolf EJ. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;863:36. doi: 10.1016/j.jchromb.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 8.Xu RN, Fan L, Rieser MJ, El-Shourbagy TA. J Pharm Biomed Anal. 2007;44:342. doi: 10.1016/j.jpba.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Annesley TM. Clinical Chemistry. 2003;49:1041. doi: 10.1373/49.7.1041. [DOI] [PubMed] [Google Scholar]
- 10.Bai F, Fraga CH, Tagen M, Schaiquevich P, Hagedorn N, Stewart CF. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1709. doi: 10.1016/j.jchromb.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai F, Minkin P, Fraga CH, O’Shaughnessy MA, Gururangan S, Stewart CF, Chromatogr J. B Analyt Technol Biomed Life Sci. 2007 doi: 10.1016/j.jchromb.2007.02.062. [DOI] [PubMed] [Google Scholar]