

Abstract
The Drosophila brain and visual system are widely utilized model systems to study neuronal development, function and degeneration. Here we show three preparations of the brain and visual system that cover the range from the developing eye disc-brain complex in the developing pupae to individual eye and brain dissection from adult flies. All protocols are optimized for the live culture of the preparations. However, we also present the conditions for fixed tissue immunohistochemistry where applicable. Finally, we show live imaging conditions for these preparations using conventional and resonant 4D confocal live imaging in a perfusion chamber. Together, these protocols provide a basis for live imaging on different time scales ranging from functional intracellular assays on the scale of minutes to developmental or degenerative processes on the scale of many hours.
Keywords: Neuroscience, Issue 37, dissection technique, eye disc, brain culture, photoreceptor, confocal microscopy
Protocol
1. Introduction
In this video, we will demonstrate how to prepare Drosophila brains and retinae for live imaging and immunohistochemistry with a focus on photoreceptor cells. First, we will show how to prepare for brain dissections. Next, we will demonstrate two basic dissections commonly used for immunohistochemistry that form the basis for the live preparations. These two preparations are the adult brain and eye dissections. Next, we will demonstrate the dissections used for live imaging of the adult and developing brain with emphasis on their usage for live imaging of photoreceptors. Finally, we will describe how we image live tissue and show some examples of live imaging using resonant confocal microscopy.
2. Dissection preparation
For good dissections, you need sharp forceps. Using a sharpening block or very fine sand paper (>1500), gently pass the forceps back and forth on each side until the ends meet at a fine point. We use a standard sharpening stone. We will not cover the sharpening technique here, but note that sharp forceps are essential for live dissections. We sharpen forceps before every dissection.
For adult brain dissections, place the flies on a CO2 pad to anesthetize them and sort out the desired genotype. For pupal dissections, simply reach into the vial and carefully remove a pupa with your forceps, being careful to avoid breaking the pupal case.
Moisten a Kimwipe with water. You will use this during the dissection to remove debris from your forceps. Position a dissecting dish on the stage of a stereoscope and fill it with HL3 solution 1. All dissections will be conducted in HL3 to ensure that the tissue remains alive and healthy during the dissection procedure. Also, we use a dissection dish that has a bottom covering of Sylgard to protect forceps during the dissection.
Now, prepare fixing solution if you plan to perform immunohistochemistry. Place 180μL of HL3 into a 500μL microcentrifuge tube. Add 20μL of 37% formaldehyde to obtain a 3.7% formaldehyde solution.
Finally, proper hand position is important for good dissections. With good hand position, your forceps will be steady and capable of controlled, subtle movements. First, place the forceps on the side of your thumb. Then bring the index finger straight down such that the tip of the finger rests on top. Now rest the side of the forceps on the side of your middle finger and move to the dissection dish. Make physical contact with the dish by your thumb and middle finger. While resting your thumb and middle finger on the dish, plant your wrist on the microscope stage. This way, you have three points planted firmly on surfaces during the dissection and it is now possible to make very subtle manipulations of the forceps. This technique may feel a bit awkward at first, but with practice, you will soon be a master of brain dissections.
3. Adult brain
On the CO2 pad, orient an adult fly ventral side up with the head away from your hand. Grab the thorax just below the head. If done properly, the legs and proboscis will extend. While viewing under a stereoscope, grab the extended proboscis with your forceps to remove the head. Discard the body and submerge the head. Refocus onto the submerged head. The entire dissection will be performed under solution. It is very important to hold onto the head with forceps at all times. Otherwise, the head will float and is difficult to retrieve. If the head moves out of focus during the dissection, simply move it back into the focal plane without adjusting the microscope. This seemingly simple task may be difficult at first, but keeping your head in focus will become easier over time.
Begin the dissection by first tearing the connective tissue between the proboscis and the eye. Tear through the eye while holding firmly with at least one forceps at all times. Be certain that the toe of the forceps is only just beneath the retina or cuticle, to avoid damaging the brain underneath. Alternating left and right, use your forceps to tear away the retina, working toward the back of the head. Tearing away the retina will increase the chances that the lamina remains attached to the optic lobe throughout this dissection. Continue around the back of the head, tearing cuticle along the way. Tear through the other eye and begin pulling away cuticle. As long as you are grabbing cuticle, you know you are not crushing the brain! With this in mind, continue to pull cuticle and retina away until the brain is revealed.
Once the brain is visible, you can begin to remove trachea attached to the brain (Fig. 1A). Continue to pull at trachea and cuticle until the brain is isolated. At this point, you should refocus to the bottom of your dish for the remaining steps. Remove the remaining trachea because your brain may otherwise float during overnight antibody washes. For imaging of photoreceptor terminals, it is desirable to retain the lamina, but you need to remove the retina, which contains the cell bodies and strongly autofluorescent pigment. To do this, carefully pull at the bushy and pigmented cellular remains without damaging the lamina. This is a finer skill and takes practice.
The adult brain can be kept alive for hours or even days using conditions that we will discuss in section 6. For immunohistochemistry, the adult brain is now ready to be fixed in formaldehyde.2 Use a P20 pipetteman set to 5μL to move the adult brain to the fixing solution you've already prepared. Continue dissecting adult brains for 20 minutes, which should yield 4-10 brains. Leave the brains in fix for an additional 40 minutes, however this duration may vary for optimal primary antibody staining. Remove the formaldehyde solution and wash the brains 3 times for 5 minutes each in 400μL of a solution containing PBS and 0.4% TritonX-100, hereafter referred to as PBS-Triton . After completely washing out the fix solution, you may add primary antibody solution.
4. Adult eye
Begin this dissection as described for the adult brain. After submerging the head and refocusing the microscope, start by tearing the connective tissue between the proboscis and the eye. Above the proboscis, separate the cuticle from the eye. Now, separate the cuticle from the underside of the eye, working toward the back of the head. With one forceps, grab the center of the back of the head. With the other, grab the eye and pull. Allow the eye to settle to the bottom of the dish (Fig. 1B). The lamina will probably still be attached to the eye and can be used for live imaging of the terminals of completely intact photoreceptor cells at this time. To continue with live imaging of photoreceptor cell bodies, go to section 4.5. The next three sections explain fixed eye immunohistochemistry.
For immunohistochemistry, move the eye to fixing solution using a P20 pipetteman set to 5μL. Continue dissecting adult eyes for a total of 10 minutes. Leave the eyes in fix for an additional 30 minutes. Remove the fix solution and conduct washes as described in the adult brain dissection.
If you ultimately wish to image the ommatidial structure of the eye the lamina must be removed now. Return the fixed eyes to your dissecting dish and remove any trachea or fat cells that may still be attached. Next, while holding the outside of the eye with one forceps, remove the lamina with the other. It should come off in one piece, exposing the cellbodies underneath (Fig. 1B). Be careful to grab the lamina only! Otherwise, you risk detaching the photoreceptors from the retina.
Using a P20 pipetteman, move the eyes to 400uL PBS-Triton in a 500uL tube. Gently rock them at 4 degrees overnight to remove the autofluorescent red pigment from the photoreceptors. The following day, you may discard the wash solution and add primary antibody solution.
For live imaging of adult eyes we only use pharate adult flies, i.e. late stage pupae shortly before eclosion. At this stage, the photoreceptor cell bodies separate from terminals in the lamina during the dissection.
Pharate adult pupae have well-defined wings, eyes, and cuticle visible through the pupal case. Select a pupa and remove the anterior portion of the pupal case to expose the head region. Carefully remove the thin inner membrane that additionally encloses the developing fly. Grab the base of the head with your forceps and pull. Discard the rest of the pupa while keeping the head submerged.
Proceed as demonstrated in the eye dissection for immunohistochemistry. At the pharate adult stage, however, the photoreceptor cell bodies will detach more easily from the lamina, allowing you to image the cell bodies nestled in the retina. Also, the lamina remains attached to the optic lobe, allowing you to image the distal lamina. If you wish to image the proximal lamina, where the photoreceptor terminal cartridges can be seen, then perform the eye dissection on an adult fly where the lamina is more strongly attached to the eye.
5. Developing eye disc-brain complexes
The developing eye disc at 20% pupal development (P+20%) has become a favorite for live imaging in our lab because this single layer of relatively large developing cells is easily observable using confocal microscopy.3
To select a 20% pupa, look for the malpighian tubules, a green spot the appears beneath the pupal case, and avoid pupae that have a "dark body"4. After selecting a P+20% pupa, submerge it in HL3 in your dissecting dish and carefully remove an anterior portion of the pupal sack, being careful not to penetrate the thin inner membrane. Grasp the inner membrane with your forceps and pull apart.
Carefully search through the released contents to find the developing brain and eye discs which are quite prominent (Fig. 1C). The eye disc-brain complex is easily separated at this stage of metamorphosis. Isolate the brain at the bottom of the dissecting dish. Carefully separate the central brain from the optic lobe. The optic lobe/eye disk will be used in live imaging.
6. Live Imaging: At the microscope
If you wish to image for a short period of time in only one or two solutions, you may image your tissue on a glass slide. In this case, prepare the slide by first coating the surface with Sylgard using the manufacturer's instructions. Place 20μL HL3 in the middle of the slide. Transport your dissected tissue in 20 additional μL of HL3 to the slide. The tissue must NEVER be exposed to air. In addition, any stress or strain from pipette handling or solution surface tension must be avoided.
For live imaging, the tissue must be firmly secured with minimal handling and contact. We securely position the brain using the water-polymerizing surgical glue GluShield applied through a microelectrode, a technique routinely used for embryonic preparations for electrophysiology5. Obtain a glass electrode like those pulled for electrophysiological recordings. Break off the tip and fill the end with GluShield using negative air pressure generated by the mouth. Break off only enough of the electrode that glue may enter. Now use positive pressure to apply a small amount of glue to the Sylgard under a drop of HL3. Now, quickly set the tissue on the glue, maintaining the desired orientation for live imaging. A water-immersion lens can now be used for imaging. If you want to add a membrane permeable dye, you can add it to the bath while imaging.
We use a Resonant Scanning Confocal Microscope that allows fast imaging with reduced photobleaching.6 For imaging over long periods of time or to exchange solutions completely or multiple times, we use a perfusion chamber (Harvard IC30 confocal imaging chamber) which connects to a peristaltic pump that slowly perfuses culture solution over the live tissue (Fig. 2A). Movement of constant liquid exchange and minimal GluShield contact reduce the flattening of the tissue that has been observed over longer periods of time7. Note that for the imaging of developmental time periods, the eye disc-brain complex needs to remain intact and should have minimal contact with the glue.
Use a cover slip coated with a drop of Sylgard that is shaved to render it thin and flat. Dispense 20μL HL3 onto the center of the coverslip and glue the live tissue to the Sylgard. Generate a gasket that will allow bubbles to pass through the chamber without exposing the live tissue to air (Fig. 2B). The gasket should be thick enough to avoid crushing the tissue but thin enough to bring the tissue close to the confocal microscope lens; we use 250μm in our setup. Assemble the perfusion chamber, being careful to keep the tissue completely submerged at all times. Begin perfusion initially at a rate close to 100μL per minute using a peristaltic pump. This speed is rather fast depending on the gasket thickness that defines the chamber height. For imaging on the scale of hours, you may need to add 20-hydroxyecdysone and antibiotic, but not antifungal, and perform perfusion at much lower speeds around 10μl per minute. Finally, we bubble oxygen into the culture medium that supplies the perfusion chamber.
7. Representative Results:
At the end of our video protocol we show an example for live imaging in developing photoreceptor cells using the preparation presented in Section 5. This preparation takes advantage of Drosophila transgenesis to cell-specifically express fluorescent reporters. In the example shown, two fluorescent reporters are expressed under control of the photoreceptor-specific GMR-Gal4 driver8: the membrane-tagged myrRFP and the endosomal marker 2xFYVE-GFP9. Two-channel 3D datasets with a bounding box of 70x70x17.5μm and a voxel size of 137x137x700nm (512x512x26) were obtained every 1 min. The movie shows a selected plane over time in this 4D dataset that reveals dynamics of intracellular endosomal trafficking and vesicle fusion events. Representative images for fixed tissue preparations of the eye and lamina are shown in Fig. 3.
Discussion
Fluorescent Probes Used for Imaging
Here we describe the strengths and limitations of the Drosophila visual system preparation for a selection of fluorescent probes from three classes: (1) Probes that are added to a live tissue preparation exogenously; (2) fluorescent proteins that are genetically encoded for both live and fixed imaging; (3) fluorescent dyes that are added to fixed tissue for immunohistochemistry.
1. Exogenous probes:
Lysotracker Green DND-26 or Red DND-99 (Invitrogen, Inc.) These are commercially available fluorescent membrane-permeable compounds that accumulate in strongly acidified compartments in the cell, most prominently lysosomes, late endosomes, and autophagosomes. Lysotracker at nanomolar concentrations (we typically use 50 nM) penetrates the L3 and P+20% eye disc fully in less than one minute. Since Lysotracker continuously accumulates and exerts an alkalizing effect, we image in less than 5 minutes. In contrast to the eye discs, Lysotracker does not penetrate the brain without disruption of the tissue. However, careful tearing of the outer membrane allows penetration of a few cell diameters in the optic lobe.
Lysosensor DND-189 (Invitrogen, Inc.) In contrast to Lysotracker, Lysosensor does not accumulate in acidified compartments, but exhibits increased fluorescence at acidic pH. Lysosensor has penetration characteristics that are very similar to Lysotracker. We use Lysosensor at a concentration of 1μM.
2. Genetically encoded probes:
For many live imaging experiments, we express genes tagged with various fluorescent molecules like GFP, TagRFP10 or the pH-sensitive pHluorin11 under control of the binary Gal4/UAS expression system12. Gal4-lines that express in developing photoreceptors include GMR-Gal4 (all photoreceptors)8 and mDelta0.5-Gal4 (developing R4 and R7)13. Several subtype-specific Gal4-driver lines exist that use different Rhodopsin promoters14. Particularly useful fluorescent probes in live imaging are photoconvertible proteins. We have successfully used Dendra2, a monomeric green-to-red photoconvertable protein15. Note that Dendra2 is designed to be photoconvertible using a 488nm Argon laser line. However, we are unable to achieve conversion using visible blue light using our set-up in either conventional or resonant confocal scanning mode. In contrast, conversion with 405nm UV laser is efficient.
3. Immunohistochemistry:
For the fixed adult eye, we often choose either rhodamine phalloidin (Invitrogen, Inc.) or anti-Chaoptin (a photoreceptor-specific antibody16) to visualize rhabdomeric structure (Fig 1A). A good way to analyze lamina cartridge structure is to combine anti-chaoptin16, the glial marker anti-ebony17, and anti-sec618(Fig 1B). As shown in Fig. 3, this combination of antibodies distinguishes the major presynaptic (chaoptin) and postsynaptic (sec6) processes as well as epithelial glia (ebony) in the lamina19.
Assembling the Perfusion Chamber
For live imaging, we employ a perfusion chamber that allows slow exchange of solutions without disturbing the tissue preparation. A demonstration of perfusion chamber assembly is included in the video and schematically shown in Figure 2. Assembly of the perfusion chamber is illustrated in Fig. 2A. Begin by laying the gasket over the specimen first, and then continue with assembly as shown. The purpose of the gasket is to create a space between the base of the chamber and the lid, a space inside which the tissue is sealed and through which the solution will flow. The space inside the gasket must include the inlet (solution enters here), the tissue prep, and the outlet through which the solution leaves the chamber. Two features of the gasket are critical: First, the gasket must be thick enough for the brain yet thin enough to allow imaging with a high-resolution lens. The ideal gasket thickness depends on set-up-specific parameters. Second, a notch in the gasket provides a compartment that allows a bubble to pass through without contacting the specimen (Fig 2B).
Imaging at the Microscope
We use a 63x (aperture 1.3) glycerine immersion lens for the perfusion chamber or a 63x water immersion lens directly into the culture medium on a slide. Glycerine lenses provide more working distance than oil lenses. We use a resonant scanning confocal microscope which scans at much faster rates than a conventional scanner (8000 Hz versus 1400 Hz, respectively). Resonant scanning allows 3-dimensional recordings over time at faster frame rates. In addition, faster scan speeds significantly reduce laser phototoxicity6 which is a major concern for repeated scans of live tissue. A balance of laser intensity and PMT voltage (gain) must be achieved to maximize signal while minimizing noise and photobleaching. An important further consideration is that the amount of laser impact on a given region is determined by resolution and zoom. For example, a region of choice is scanned at the same resolution at 256x256, zoom 2x as at 512x512, zoom 1x, yet the maximally possible live imaging speed is 4 times faster at 256x256, zoom 2x. To record the activity of quickly moving intracellular compartments, we have successfully recorded with the following settings: 5 frames per z-stack at a rate of one z-stack per 10 seconds with a scan speed of 8000 Hz and 2x line averaging. For long imaging sessions on the scale of hours, we often reduce the frame rate to one per several minutes and choose larger areas as the preparation is more likely to shift.
Drosophila Visual System Anatomy
Figure 3 shows the anatomy of the Drosophila adult eye (Fig. 3A), the adult brain (Fig. 3B), and the 20% -25% pupal eye disc/brain complex (Fig. 3C). Figure 3A illustrates the adult eye both with and without the lamina and fat cells attached. During the adult eye dissection, the lamina, which is in the center of the eye nestled in a bowl formed by the photoreceptor cell bodies (left), must be removed without disrupting the cell bodies underneath (right). For the adult brain dissection, one should be able to distinguish the photoreceptor cell bodies, the lamina, the optic lobe, the central brain, and trachea (Fig. 3B). The photoreceptors and trachea need to be removed for this dissection while leaving the lamina attached to the optic lobe. To aid in the identification of the 20-25% pupal eye disc/brain complex, a live image is presented in Figure 3C.
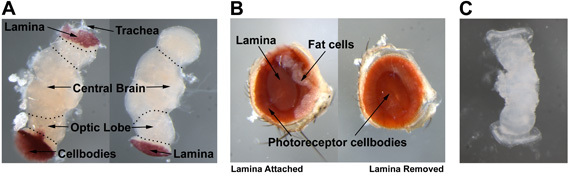 Figure 1. (A) adult brain. (B) adult eye. (C) 20%-25% pupal eye-disc/brain complex.
Figure 1. (A) adult brain. (B) adult eye. (C) 20%-25% pupal eye-disc/brain complex.
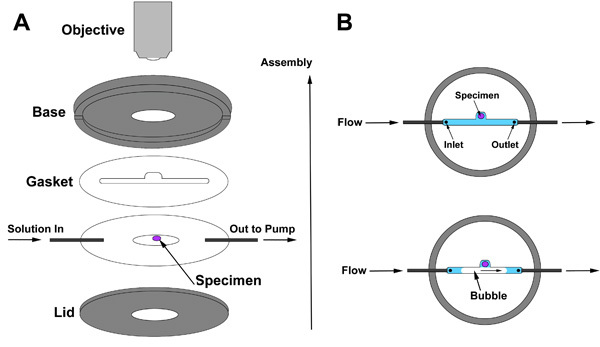 Figure 2. (A) Perfusion chamber assembly and (B) gasket positioning relative to the specimen.
Figure 2. (A) Perfusion chamber assembly and (B) gasket positioning relative to the specimen.
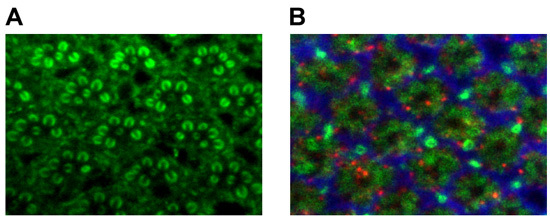 Figure 3. (A) Adult rhabdomere staining using anti-chaoptin. (B) Adult lamina staining using anti-chaoptin (green, photoreceptors), anti-ebony (red, glia), and sec6 (blue, interneurons).
Figure 3. (A) Adult rhabdomere staining using anti-chaoptin. (B) Adult lamina staining using anti-chaoptin (green, photoreceptors), anti-ebony (red, glia), and sec6 (blue, interneurons).
Acknowledgments
This work was supported by grants from the Welch Foundation (I-1657) and the NIH (RO1EY18884). P.R.H. is a Eugene McDermott Scholar in Biomedical Research.
References
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A. 1994;175(2):179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- Walther RF, Pichaud F. Immunofluorescent staining and imaging of the pupal and adult Drosophila visual system. Nat Protoc. 2006;1:2635–2642. doi: 10.1038/nprot.2006.379. [DOI] [PubMed] [Google Scholar]
- Fischbach KF, Hiesinger PR. Optic lobe development. Adv Exp Med Biol. 2008;628:115–136. doi: 10.1007/978-0-387-78261-4_8. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Golic KG, Hawley RS. Cold Spring Harbor, New York: Cold Spring Harbor Press; 2005. Drosophila - A Laboratory Handbook. [Google Scholar]
- Featherstone DE, Chen K, Broadie K. Harvesting and preparing Drosophila embryos for electrophysiological recording and other procedures. J Vis Exp. 2009. [DOI] [PMC free article] [PubMed]
- Borlinghaus RT. MRT letter: high speed scanning has the potential to increase fluorescence yield and to reduce photobleaching. Microsc Res Tech. 2006;69:689–692. doi: 10.1002/jemt.20363. [DOI] [PubMed] [Google Scholar]
- Ayaz D. Axonal injury and regeneration in the adult brain of Drosophila. J Neurosci. 2008;28(23):6010–6021. doi: 10.1523/JNEUROSCI.0101-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M. Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell. 1996;87(4):651–660. doi: 10.1016/s0092-8674(00)81385-9. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig T, Wilsch-Brauninger M, Gonzalez-Gaitan M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J Cell Biol. 2003;161:609–624. doi: 10.1083/jcb.200211087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merzlyak EM. monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods. 2007;4(7):555–557. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- Ng M. Transmission of olfactory information between three populations of neurons in the antennal lobe of the fly. Neuron. 2002;36(3):463–474. doi: 10.1016/s0896-6273(02)00975-3. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Cooper MT, Bray SJ. Frizzled regulation of Notch signalling polarizes cell fate in the Drosophila eye. Nature. 1999;397(6719):526–530. doi: 10.1038/17395. [DOI] [PubMed] [Google Scholar]
- Cook T, Desplan C. Photoreceptor subtype specification: from flies to humans. Semin Cell Dev Biol. 2001;12(6):509–518. doi: 10.1006/scdb.2001.0275. [DOI] [PubMed] [Google Scholar]
- Gurskaya NG. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24(4):461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- Reinke R, Krantz DE, Yen D, Zipursky SL. Chaoptin, a cell surface glycoprotein required for Drosophila photoreceptor cell morphogenesis, contains a repeat motif found in yeast and human. Cell. 1988;52(2):291–301. doi: 10.1016/0092-8674(88)90518-1. [DOI] [PubMed] [Google Scholar]
- Richardt A, Rybak J, Stortkuhl KF, Meinertzhagen IA, Hovemann BT. Ebony protein in the Drosophila nervous system: optic neuropile expression in glial cells. J Comp Neurol. 2002;452(1):93–102. doi: 10.1002/cne.10360. [DOI] [PubMed] [Google Scholar]
- Beronja S. Essential function of Drosophila Sec6 in apical exocytosis of epithelial photoreceptor cells. J Cell Biol. 2005;169(4):635–646. doi: 10.1083/jcb.200410081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SQ. Mutations in Drosophila sec15 reveal a function in neuronal targeting for a subset of exocyst components. Neuron. 2005;46(2):219–232. doi: 10.1016/j.neuron.2005.02.029. [DOI] [PubMed] [Google Scholar]