

Abstract
Potent and specific inhibitors of protein•protein interactions have significant potential both as therapeutic compounds and biological tools, yet discovery of such molecules remains a significant challenge. Whereas small molecules typically bind proteins in small, well-defined, deep clefts, proteins generally recognize each other through formation of large, heterogeneous complementary surfaces. Our laboratory has recently described a general solution, called protein grafting, for the identification of highly functional miniature proteins by stabilization of α-helical binding epitopes on a protein scaffold. In protein grafting, those residues that comprise a functional epitope are grafted onto the solvent-exposed α-helical face of the small yet stable protein avian pancreatic polypeptide (aPP). In this study, we use protein grafting in combination with molecular evolution by phage display to identify phosphorylated peptide ligands that recognize the shallow surface of CBP KIX with high nanomolar to low micromolar affinity. Furthermore, we show that grafting of the CBP KIX-binding epitope of CREB KID onto the aPP scaffold yields molecules capable of high affinity recognition of CBP KIX even in the absence of phosphorylation. Importantly, both classes of designed ligands exhibit high specificity for the target CBP KIX domain over carbonic anhydrase and calmodulin, two unrelated proteins that bind hydrophobic or α-helical molecules that might be encountered in vivo.
Introduction
Inhibitors of protein•protein interactions are important both as therapeutics1-3 and chemical genetics tools,4 yet inhibitors that are both potent and selective remain largely elusive. Although efforts to design or identify non-natural small molecule inhibitors of protein•protein interactions have met with some success,5-7 and natural products with the ability to inhibit protein•protein interactions have been well studied,8 fundamental differences between the properties of small molecule•protein complexes and protein•protein complexes provide some insight into the difficulties associated with a small molecule approach to protein surface recognition.9 Whereas small molecules typically bind proteins in small, well-defined, deep clefts, proteins generally recognize each other through formation of large, heterogeneous complementary surfaces.10,11 Thus, small proteins with well-defined three-dimensional structures and finely tuned functional properties are perhaps ideally suited for protein surface recognition and disruption of protein•protein interfaces.
Our laboratory has recently described a general solution, called protein grafting, for the identification of highly functional miniature proteins by stabilization of α-helical binding epitopes on a protein scaffold12-16 (Figure 1A). In protein grafting, those residues that comprise a functional epitope are grafted onto the solvent-exposed α-helical face of the small yet stable protein avian pancreatic polypeptide (aPP).17 This procedure, often in combination with molecular evolution, identifies miniature protein ligands with high affinity and specificity for macromolecular targets. Initially developed in the context of DNA recognition,12-14 protein grafting has been applied recently to the identification of miniature proteins with nanomolar affinities for the related anti-apoptotic proteins Bcl-2 and Bcl-XL.5,15 Although interesting, the application of protein grafting in this case represented a rather conservative step, as Bcl-2 and Bcl-XL each contain a deep (approximately 7 Å at deepest point) hydrophobic groove which recognizes α-helical regions of protein partners such as Bak.18-22 Indeed, the nature of the interface formed between these anti-apoptotic proteins and their α-helical partners has recently been exploited in the discovery of a number of small molecule ligands for these proteins.5,23-29
Figure 1.

Protein grafting applied to the KIDP•KIX interaction. (A) Schematic representation of the protein grafting process. In the KIDP•KIX complex,30 the backbone of CREB KID helix B is in blue, the hydrophobic residues of helix B important for CBP KIX binding are in red, the PKA recognition site is in green, and the Ser phosphate moiety is in blue (phosphorous) and white (oxygen). In aPP,17 residues from the α-helix that form part of the hydrophobic core are in blue and residues from the polyproline helix are in orange. In PPKID Library 1, the Cα atoms at randomized positions are in orange. (B) Library design. The amino acid sequence of helix B of CREB KID is aligned with the sequence of the α-helix of aPP. The amino acid sequence of PPKID Library 1 is below. Residues important for aPP folding are in blue, the PKA recognition site is in green, and hydrophobic residues of helix B important for binding CBP KIX are in red. Randomized residues are represented by × in orange. (C) Comparison of the α-helix-binding surfaces of Bcl-XL (left) and CBP KIX (right). Bcl-XL contains a deep (~7 Å) hydrophobic cleft that recognizes the Bak BH3 α-helix.22 CBP KIX binds the CREB KID helix B in a shallow depression (< 5 Å at the deepest point) on its surface.30
The complex between the KIX domain of the transcriptional coactivator protein CBP and the kinase-inducible activation domain (KID) of the transcription factor CREB, though also mediated by an α-helix, is strikingly different from the complexes formed by Bcl-2 family members (Figure 1C). The KID-binding groove of the CBP KIX domain is quite shallow and more closely resembles the solvent-exposed protein surface than a typical α-helix-binding groove.30 In fact, only one hydrophobic residue of CREB KID is completely buried from solvent in the KID•KIX complex, and formation of a high affinity KID•KIX complex requires the enthalpic driving force provided by phosphorylation of CREB KID on Ser133.31,32 Thus, CBP KIX represents a difficult target for molecular recognition, and indeed, no small molecule ligands for CBP KIX have been reported. In this study, protein grafting and molecular evolution by phage display are used to identify phosphorylated peptide ligands that recognize the hydrophobic surface of CBP KIX with high nanomolar to low micromolar affinity and high specificity. Furthermore, grafting of the CBP KIX-binding epitope of CREB KID onto the aPP scaffold yields molecules capable of high affinity and specific recognition of CBP KIX even in the absence of phosphorylation.
Results
Library design and generation
The design of a CBP KIX-binding miniature protein (PPKID) library was based on the alignment of the α-helix of aPP and helix B of the CREB KID domain shown in Figure 1B. The otherwise unstructured phosphorylated CREB KID (KIDP) domain forms two α-helices, A and B, when bound to the CBP KIX domain; each helix contacts a different region of the CBP KIX surface.30 Mutagenesis studies have determined that most (though not all) of the residues that comprise the CBP KIX-binding epitope of CREB KIDP are located in helix B,30,33 and only residues from helix B were included in the miniature protein library. Four hydrophobic residues from CREB KID (Tyr134, Ile137, Leu138, Leu141) contribute significantly to the free energy of KIDP•KIX complex formation. The PPKID library contained three of these four residues (Ile137, Leu138, Leu141), and a conservative mutation of the fourth from Tyr to Phe, which in the context of CREB KIDP has no effect on CBP KIX binding.34 This mutation was included, along with the complete recognition site for protein kinase A (PKA; Arg130, Arg131, Ser133), to promote phosphorylation of the miniature protein library in vitro, if so desired. In the context of CREB KIDP, the Tyr to Phe mutation lowers five-fold the Km for phosphorylation by PKA.34 The structural scaffold of the α-helical portion of the library was provided by six of eight residues (Val14, Leu17, Phe20, Leu24, Tyr27, Leu28) from the aPP α-helix that contribute to the hydrophobic core.17 Based on our success using a similar approach to improve DNA-binding miniature proteins,14 the five residues from the polyproline helix of aPP known to participate in hydrophobic core formation (Pro2, Gln4, Pro5, Tyr7, Pro8) were varied to all 20 amino acids. Our expectation was that the CBP KIX-binding epitope on the α-helix would guide all library members to the CBP KIX surface, and the functional selection would identify those library members with increased CBP KIX affinity derived from packing of the polyproline helix against the otherwise exposed face of the bound α-helix. A 5 × 107-member library of miniature proteins (PPKID Library 1) based on this design was generated for use in phage display selection experiments.
Selection of phosphorylated miniature protein ligands for CBP KIX
Initially, eight rounds of selection were performed (selection 1). Each round included a PKA-catalyzed in vitro phosphorylation step designed to increase the CBP KIX-binding affinities of all library members. Phosphorylation of CREB KID is critical for high affinity recognition of CBP KIX; measurements of the contribution of the Ser133 phosphate moiety to the free energy of the KIDP•KIX complex range between 1.5 and 3.0 kcal•mol−1.31,32 In this selection, GST-KIX was immobilized on glutathione-coated microtiter plates, and stringency was increased over the course of the selection by increasing the binding and washing temperature, from 4 °C in round 1 to 25 °C by round 3, and by increasing the length and number of washes, from 10 × 1 min washes in round 1 to 20 × 5 min washes in round 8.35 Rounds 7 and 8 were performed in binding buffer containing 5 mM dithiothreitol (DTT), after sequencing of individual clones from rounds 4-6 indicated that a significant portion of the library members selected in these rounds contained single Cys residues. The Cys residues were evenly distributed over all five randomized positions, which suggested that library members were being selected based on their ability to form disulfide bonds with GST-KIX or glutathione, rather than based on high affinity, yet noncovalent, CBP KIX binding.
The progress of the selection was monitored by measuring the retention of library phage in comparison to the retention of phage displaying aPP, which should not bind to GST-KIX, and by sequencing of individual clones after each round of selection. By round 8 of selection 1, the library phage were retained 13-fold over aPP phage. Furthermore, by round 7, three sequences (PPKID 1-3) had been identified in multiple independent clones (Table 1); two of these sequences (PPKID2, PPKID3) completely dominated the library by round 8. Surprisingly, the residues selected at each of the five randomized positions in PPKID2 and PPKID3 displayed no significant similarity.36
Table 1.
HisKIX-binding affinity of PPKID and control peptidesa
| Selection 1 | Kd PPKIDP (nM) | Kd PPKIDU (μM) | |
|---|---|---|---|
| PPKID1 | GASDMTYWGDDAPVRRLSFFYILLDLYLDAPGVC | 591 ± 59 | 24.1 ± 4.0 |
| PPKID2 | GMSRVTPGGDDAPVRRLSFFYILRDLYLDAPGVC | 729 ± 36 | 12.6 ± 1.4 |
| PPKID3 | GASPHTSSGDDAPVRRLSFFDILLDLYLDAPGVC | 1200 ± 100 | 6.7 ± 0.2 |
|
| |||
| Selections 2 & 4 | Kd PPKIDP (nM) | Kd PPKIDU (μM) | |
|
| |||
| PPKID4 | GPSQPTYPGDDAPVRRLSFFYILLDLYLDAPGVC | 515 ± 44 | 12.1 ± 2.4 |
| PPKID5 | GLSWPTYHGDDAPVRRLSFFYILLDLYLDAPGVC | 534 ± 31 | 6.6 ± 2.0 |
|
| |||
| Selections 3 & 4 | Kd PPKIDP (nM) | Kd PPKIDU (μM) | |
|
| |||
| PPKID6 | GISWPTFEGDDAPVRRLSFFYILLDLYLDAPGVC | 624 ± 49 | 1.5 ± 0.1 |
| PPKID6 S18E | GISWPTFEGDDAPVRRLEFFYILLDLYLDAPGVC | 10.9 ± 2.0 | |
| PPKID7 | GLSPYTEWGDDAPVRRLSFFYILLDLYLDAPGVC | 2.3 ± 0.2 | |
| PPKID8 | GLSWKTDPGDDAPVRRLSFFYILLDLYLDAPGVC | 3.1 ± 0.5 | |
|
| |||
| Control peptides | P: Kd | U: Kd (μM) | |
|
| |||
| KID-AB | TDSQKRREILSRRPSYRKILNDLSSDAPGVC | 562 ± 41 nM | >116 |
| KID-B | RRPSYRKILNDLSSDAPGVC | 51.6 ± 4.0 μM | >297 |
| Peptide C | RRLSFFYILLDLYLDAPGVC | 2.4 ± 0.2 μM | 21.5 ± 2.6 |
Each peptide was labeled on the C-terminal Cys residue with acetamidofluorescein for use in fluorescence polarization experiments. Kd values were determined by converting polarization data from three independent samples to fraction of fluorescently-labeled peptide bound values, which were fit to equilibrium binding equation (2). Residues selected at randomized positions are in red. Selected point mutations in PPKID2 and PPKID3 are underlined. P indicates a phosphopeptide. U indicates an unphosphorylated peptide. The phosphoserine residue in phosphopeptides is in bold.
CBP KIX-binding affinity
The PPKID peptides were synthesized as phosphopeptides (PPKIDP) and each was labeled with acetamidofluorescein on a C-terminal Cys residue. The affinity of each labeled peptide for a His-tagged CBP KIX domain (HisKIX) was measured by equilibrium fluorescence polarization. The HisKIX-binding affinities of three phosphorylated control peptides (KID-ABP, KID-BP and peptide CP) were also measured. Peptide KID-ABP comprises the full-length CREB KID domain (residues 119-148, A and B helices) and peptide KID-BP corresponds to the region of CREB KID whose residues were incorporated within the α-helix of aPP (residues 130-148, the PKA recognition site and helix B); these peptides allow direct comparison of our miniature proteins with natural CBP KIX-binding molecules. Peptide CP corresponds to the chimeric α-helical portion of the PPKID peptides (residues 15-33) and allows us to compare the contribution to CBP KIX-binding affinity of residues in the α-helix derived from aPP and residues in the randomized region of the PPKID library, which includes the putative polyproline helix37 and turn regions.
The results of the equilibrium fluorescence polarization experiments are shown in Figure 2A and Table 1. KID-ABP binds HisKIX with high affinity (Kd = 562 ± 41 nM) at 25 °C under the assay conditions used. This value is lower than previously reported Kds for similar KIDP•KIX complexes (3.1 μM to 9.7 μM)31,32,38 measured by a number of techniques (though not fluorescence polarization) and may result from slight differences in the buffers and the CREB KIDP and CBP KIX constructs used in each case. Peptides PPKIDP 1-3 bind HisKIX with affinities ranging from 591 nM to 1.2 μM, values that are comparable to the HisKIX-binding affinity of KID-ABP.
Figure 2.

HisKIX-binding affinity of PPKID and control peptides measured by fluorescence polarization. Serial dilutions of HisKIX were incubated with 25-50 nM of fluorescein-labeled peptide (peptideFlu) for 30 min at 25 °C. Each point represents an average of three independent samples; the error bars denote standard error. Observed polarization values were converted to fraction of peptideFlu bound using Pmin and Pmax values derived from the best fit of the polarization data to equation (1). Curves shown are the best fit of fraction of peptideFlu bound values to the equilibrium binding equation (2). Fraction of phosphorylated peptideFlu bound values are indicated with circular symbols, and fraction of unphosphorylated peptideFlu bound values are indicated with triangular symbols. (A) KID-ABP, KID-BP, peptide CP and PPKIDP 1-3. (B) KID-ABU, KID-BU, peptide CU and PPKIDU 1-3. (C) PPKIDP 4-5, PPKIDU 4-5 and PPKIDU 7-8. (D) PPKID6U, PPKID6P and PPKID6 S18E.
Remarkably, peptides PPKIDP 1-3 bind HisKIX with 43- to 87-fold higher affinity than does KID-BP (Kd = 51.6 ± 4.0 μM; this value is comparable to the Kd of 80 μM reported for the KID(129-149)P•KIX complex measured by isothermal titration calorimetry).32 Most of this increase in affinity can be attributed to the aPP-derived residues in the α-helical region of the miniature proteins; peptide CP (which comprises the α-helical region of PPKIDP 1-3) binds HisKIX with a Kd of 2.4 ± 0.2 μM, which represents a greater than 20-fold increase in affinity (ΔΔG = −1.8 kcal•mol−1) compared to the CBP KIX-binding affinity of KID-BP. The turn and polyproline helix regions (including selected residues) of the PPKIDP 1-3 peptides contribute a more modest −0.4 to −0.8 kcal•mol−1 to the free energy of complex formation with CBP KIX.
The HisKIX-binding affinities of unphosphorylated versions (denoted by a superscript U) of PPKID 1-3, KID-AB, KID-B and peptide C were also determined (Figure 2B and Table 1). As expected, the KID-ABU and KID-BU peptides possess very low affinities for HisKIX. Only a small change in polarization of the KID-ABU-Flu (61 mP) or KID-BU-Flu (76 mP) molecules was observed even at the highest HisKIX concentrations tested (150 μM and 325 μM, respectively). This experiment allows us to place a lower limit on the Kd of the complex formed between each of these peptides and HisKIX. If we estimate the change in polarization of KID-ABU-Flu to be 110 mP and the change in polarization of KID-BU-Flu to be 150 mP when fully bound by HisKIX (based on observed changes in polarization of 116 mP for fully HisKIX-bound KID-ABP and 161 mP for KID-BP), we can estimate that the Kd of the KID-ABU•HisKIX complex must be greater than 116 μM and the Kd of the KID-BU•HisKIX complex must be greater than 297 μM. Remarkably, the seven amino acid changes (including the conservative Tyr to Phe mutation) that convert KID-BU to peptide CU dramatically enhance CBP KIX-binding affinity (ΔΔG ≥ −1.5 kcal•mol−1). Peptide CU binds HisKIX with a Kd of 21.5 ± 2.6 μM. Addition of the turn and selected polyproline helix regions to yield peptides PPKIDU 1-3 slightly increases or even slightly decreases CBP KIX-binding affinity 1- to 3-fold (Kd = 6.7 to 24.1 μM; ΔΔG = −0.7 to +0.1 kcal•mol−1). As is true in the context of phosphorylated peptides, then, most of the free energy of complex formation with HisKIX is due to aPP-derived residues in the putative α-helical region of the PPKIDU peptides.
Minimizing fusion protein binding
Preliminary fluorescence polarization experiments using GST-KIX as a target indicated that two of the selected peptides (PPKID1 and PPKID3) possessed significantly higher (16- to 19-fold) affinity for GST-KIX than for HisKIX (data not shown). Therefore, we subjected the members of PPKID Library 1 to a second selection (selection 2) in which GST-KIX and HisKIX were alternated as the immobilized target protein to minimize selection of library members based on increased affinity for the GST-KIX or HisKIX fusion proteins relative to the isolated CBP KIX domain. Binding and washing conditions were similar to those used in selection 1,35 and each round included a PKA-catalyzed phosphorylation step. DTT (5 mM) was included in the binding buffer in all rounds where GST-KIX was used as a target (except for round 1) to minimize selection based on disulfide bond formation. After nine rounds of selection, the library phage were retained 44-fold over phage displaying aPP, although no consensus in miniature protein sequence was achieved. However, two sequences were identified in multiple independent clones from rounds 7-9 (PPKID 4-5). Interestingly, PPKID4 contains aPP-derived residues in all randomized positions. PPKID4 and PPKID5 contain identical residues at two of the randomized positions, 5 (Pro) and 7 (Tyr), but otherwise the selected residues are not conserved. Furthermore, none of the selected residues in either PPKID4 or PPKID5 are similar to the selected residues in PPKID 1-3. PPKID4 and PPKID5 exhibit high affinity for HisKIX (Figure 2C and Table 1), with Kds in both phosphorylated (515 ± 44 nM and 534 ± 31 nM, respectively) and unphosphorylated forms (12.1 ± 2.4 μM and 6.6 ± 2.0 μM, respectively) similar to those observed for PPKID 1-3.
Unphosphorylated selections
The significant CBP KIX-binding affinity displayed by peptide CU (as well as by short, unphosphorylated CBP KIX-binding peptides identified by Montminy and coworkers) encouraged us to perform selections with unphosphorylated PPKID Library 1. Unphosphorylated selections (selections 3 & 4) were performed in parallel with selections 1 & 2, with similar binding and washing conditions.35 After nine rounds of selection, the library phage in selection 3 were retained 32-fold over phage displaying aPP, and the library phage in selection 4 were retained 11-fold over phage displaying aPP. Although no consensus was reached in either selection, a number of sequences were identified in multiple independent clones. In selection 3, one sequence, PPKID6, was identified in rounds 6-9. Two of the sequences identified in selection 4, PPKID4 and PPKID5, were also identified in selection 2 (which included the phosphorylation step in each round) under the same conditions. Two additional sequences, PPKID7 and PPKID8, were identified in rounds 6-9 in selection 4.
Interestingly, four of five randomized positions (2, 4, 5, and 7) in peptides PPKID 4-9 approach consensus; Leu or Ile was selected at position 2, Trp at position 4, Pro at position 5, and aromatic or negatively charged residues at position 7. PPKID6U, PPKID7U and PPKID8U exhibit exceptionally high affinity for HisKIX, as measured by fluorescence polarization, with Kds ranging from 1.5 μM to 3.1 μM (Figures 2C and 2D and Table 1). These values correspond to at least 37- to 77-fold enhancements in HisKIX-binding affinity compared to KID-ABU and at least 96- to 198-fold enhancements relative to KID-BU. Furthermore, peptides PPKIDU 6-8 bind HisKIX with 7- to 14-fold enhancements in binding affinity compared to peptide CU. Thus, the selected polyproline helix and turn regions of the PPKIDU 6-8 peptides contribute −1.2 to −1.6 kcal•mol−1 to the free energy of complex formation with CBP KIX.
We investigated the HisKIX-binding affinities of two variants of PPKID6, each containing a simple modification of residue Ser18, phosphorylation and substitution of Ser by Glu. Phosphorylation of PPKID6 leads to only a two-fold enhancement in HisKIX-binding affinity (ΔΔG = −0.5 kcal•mol−1) (Figure 2D), a significantly smaller enhancement than is observed upon phosphorylation for the other PPKID peptides (6- to 41-fold; ΔΔG = −1.0 to −2.2 kcal•mol−1) and KID-AB (ΔΔG > 3.2 kcal•mol−1). Surprisingly, the Ser to Glu mutation actually decreases HisKIX-binding affinity 7-fold (Kd = 10.9 ± 2.0 μM; ΔΔG = +1.2 kcal•mol−1). A similar mutation in the context of the full length CREB KID domain leads to CBP KIX-binding affinity intermediate between that of unphosphorylated and phosphorylated CREB KID,39 presumably because the negative charge of Glu mimics the negatively charged phosphate moiety.
Binding modes of PPKID4P and PPKID6U
Two sets of experiments were performed to investigate the binding modes of PPKID4P and PPKID6U. First, competition fluorescence polarization experiments assessed the ability of PPKID4P and PPKID6U to compete with CREB KIDP for binding CBP KIX. In particular, the fraction of fluorescently tagged PPKID4P or PPKID6U bound to HisKIX at equilibrium was monitored as a function of the concentration of unlabeled KID-ABP. These experiments reveal that KID-ABP competes with both PPKID4P and PPKID6U for binding to CBP KIX (Figure 3). The concentration of KID-ABP needed to displace 50% of fluorescently tagged PPKID4P or PPKID6U from HisKIX (the IC50 value) is 3.2 μM or 2.4 μM, respectively. These values are, as expected given the conditions of the assay,40 slightly larger than the Kd of the KID-ABP•HisKIX complex determined by direct fluorescence polarization analysis (562 ± 41 nM). These results indicate that HisKIX cannot interact simultaneously with KID-ABP and either PPKID4P or PPKID6U, and are consistent with an interaction of both PPKID4P and PPKID6U within the CREB KIDP-binding cleft of CBP KIX.
Figure 3.
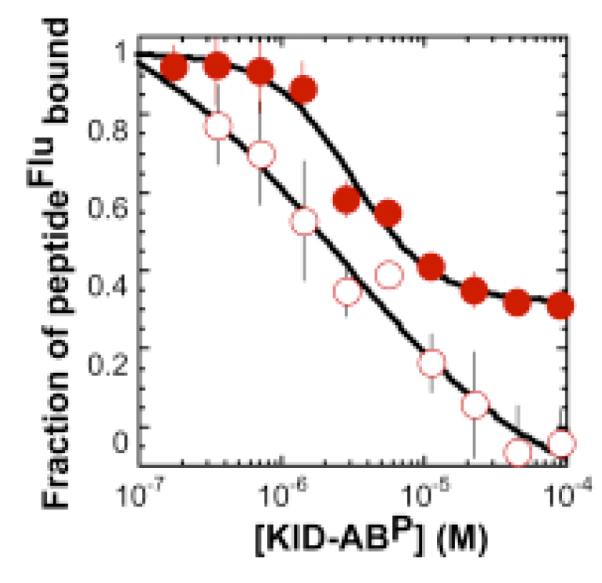
Competition between KID-ABP and PPKID4P (solid circle) or PPKID6U (open circle) for binding to HisKIX measured by fluorescence polarization. Serial dilutions of KID-ABP were incubated with 1.5 μM or 3.0 μM HisKIX and 25 nM fluorescein-labeled PPKID4P or PPKID6U (peptideFlu) for 60 min at 25 °C, respectively. Each point represents an average of three independent samples; the error bars denote standard error. Observed polarization values were converted to fraction of peptideFlu bound using experimentally determined Pmin and Pmax values corresponding to the polarization of samples containing 25 nM peptideFlu alone and peptideFlu with 1.5 μM or 3.0 μM HisKIX, respectively. Curves shown represent the best fit of fraction of peptideFlu bound values to equation (3). The close agreement between the Kd of the KID-ABP•HisKIX complex and the IC50 values determined here provides evidence that the fluorescein tag appended to KID-ABP contributes neither positively nor negatively to the stability of the KID-ABP•HisKIX complex.
Although KID-ABP competes with both PPKID4P and PPKID6U for binding to HisKIX, small changes in KID-ABP concentration around the corresponding IC50 values have a larger effect on the change in thefraction of PPKID4P bound than on the change in the fraction of PPKID6U bound. This result suggests that there may exist differences in the orientation or geometry of PPKID4P and PPKID6U when bound to CBP KIX. To explore these differences in greater detail, we measured the affinities of KID-ABP, PPKID4P and PPKID6U for the Y650A variant of CBP KIX (GST-KIXY650A) using direct fluorescence polarization analysis (Figure 4). Tyr650 forms one side of the hydrophobic cleft within the CREB KIDP-binding groove of CBP KIX that accommodates Leu141 of helix B. As a result, CREB KIDP exhibits significantly lower affinity for the Y650A variant relative to wild type CBP KIX.32 These two factors make GST-KIXY650A an excellent surveyor of the CREB KIDP•CBP KIX interface.
Figure 4.
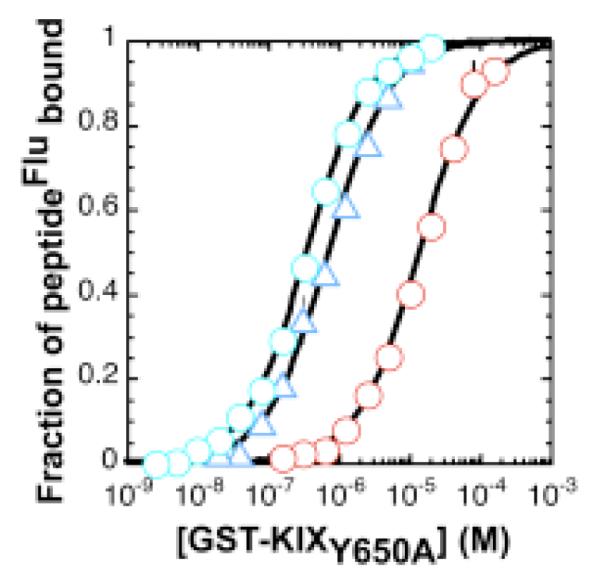
Affinity of PPKID4P (blue circle), PPKID6U (blue triangle) and KID-ABP (red circle) for GST-KIXY650A measured by fluorescence polarization. Serial dilutions of GST-KIXY650A were incubated with 25 nM of fluorescein-labeled peptide (peptideFlu) for 30 min at 25 °C. Each point represents an average of three independent samples; the error bars denote standard error. Observed polarization values were converted to fraction of peptideFlu bound using Pmin and Pmax values derived from the best fit of the polarization data to equation (1). Curves shown are the best fit of fraction of peptideFlu bound values to the equilibrium binding equation (2).
The GST-KIXY650A•KID-ABP complex is 15-fold less stable than the wild type GST-KIX•KID-ABP complex as measured by fluorescence polarization, a difference in stability similar to that observed previously in ITC experiments performed with the same GST-KIX constructs.32 Likewise, the PPKID4P• GST-KIXY650A complex is 24-fold less stable than the wild type GST-KIX•PPKID4P complex. The observation that mutation of Tyr650 to Ala has a similar effect on the binding of KID-ABP and PPKID4P, together with the equilibrium competition analysis, provides evidence that the two ligands interact with CBP KIX in a similar manner. Interestingly, despite the fact that PPKID6U and CREB KID-ABP compete for binding to CBP KIX, PPKID6U binds GST-KIXY650A with the same affinity (Kd = 712 ± 68 nM) as it binds wild type GST-KIX (Kd = 714 ± 128 nM). This observation suggests that PPKID6U interacts with CBP KIX in a manner that is somewhat different from the CBP KIX-binding mode of KID-ABP. Further work with an established panel of CBP KIX variants,41 currently in progress, will be necessary to characterize the binding mode of PPKID6U in detail.
PPKID specificity
Given the myriad protein surfaces present in the cell, the utility of molecules that recognize protein surfaces will depend on their ability to interact selectively with the desired protein. We investigated the specificity of our highest affinity phosphorylated (PPKID4P) and unphosphorylated (PPKID6U) CBP KIX ligands by measuring their affinity for two globular proteins, carbonic anhydrase II and calmodulin, known to recognize hydrophobic or helical molecules. To determine the effect of the region comprising the selected polyproline helix and turn on the specificity of PPKID4P and PPKID6U, we also examined the specificity of peptide C, in both its phosphorylated and unphosphorylated forms.
The PPKID peptides bind carbonic anhydrase with low affinity, with Kds of 106 ± 12 μM and 79 ± 13 μM for PPKID4P and PPKID6U, respectively (Figure 5A and Table 2). These values define specificity ratios (Krel = Kd (carbonic anhydrase) / Kd (HisKIX)) of 205 for PPKID4P and 53 for PPKID6U. The preference of PPKID4P for HisKIX over carbonic anhydrase (Krel = 205) is considerably higher than the preference of control peptide CP for HisKIX over carbonic anhydrase (Krel = 40), despite their approximately equal affinity for carbonic anhydrase (106 μM and 97 μM, respectively). Thus, the increased specificity of PPKID4P relative to peptide CP is due to enhanced affinity for HisKIX, and not a result of decreased affinity for carbonic anhydrase. Similar conclusions are drawn when comparing PPKID6U and peptide CU; although these two molecules display similar affinities for carbonic anhydrase, with Kd values of 79 μM and 66 μM, respectively, the specificity ratio for PPKID6U (Krel = 53) is significantly higher than the specificity ratio for peptide CU (Krel = 3).
Figure 5.

Specificity of protein surface recognition by PPKID and control peptides measured by fluorescence polarization. Binding reactions containing serially diluted target protein and 25-50 nM of fluorescein-labeled peptides (peptideFlu) were incubated for 30 min at 25 °C. Each point represents the average polarization of two to three independent samples; error bars denote standard error. Observed polarization values were converted to fraction of peptideFlu bound using Pmin and Pmax values derived from the best fit of the polarization data to equation (1). Curves shown are the best fit of fraction of peptideFlu bound values to the equilibrium binding equation (2). (A) Plot illustrating the polarization of fluorescently-labeled PPKID4P, PPKID6U, peptide CP and peptide CU molecules as a function of target protein (carbonic anhydrase II or HisKIX) concentration. Circular and triangular symbols indicate that HisKIX was used as the target protein; the symbols are colored as in Figure 2 with the exception of the points for peptide CU, which are in orange for clarity. Square symbols indicate that carbonic anhydrase was used as the target protein. (B) Plot illustrating the polarization of fluorescently-labeled PPKID4P, PPKID6U and peptide CP molecules as a function of target protein (calmodulin or HisKIX) concentration. Circular and triangular symbols indicate that HisKIX was used as the target protein, and the symbols are colored as in Figure 2. Square symbols indicate that calmodulin was used as the target protein.
Table 2.
Specificity of PPKID and control peptidesa
| Peptides | Kd HisKIX (μM) |
Kd CA (μM) (Krel) |
Kd CalM (μM) (Krel) |
|
|---|---|---|---|---|
| PPKID4P | GPSQPTYPGDDAPVRRLSFFYILLDLYLDAPGVC | 0.515 ± 0.044 | 106 ± 12 (205) |
52 ± 12 (100) |
| PPKID6U | GISWPTFEGDDAPVRRLSFFYILLDLYLDAPGVC | 1.5 ± 0.1 | 79 ± 13 (53) |
> 168 (> 112) |
| CP | RRLSFFYILLDLYLDAPGVC | 2.4 ± 0.2 | 97 ± 6 (40) |
178 ± 42 (74) |
| CU | RRLSFFYILLDLYLDAPGVC | 21.5 ± 2.6 | 66 ± 11 (3) |
N.D. |
Each peptide was labeled on the C-terminal Cys residue with acetamidofluorescein for use in fluorescence polarization experiments. Kd values were determined by converting polarization data from two to three independent samples to fraction of fluorescently-labeled peptide bound values, which were fit to equilibrium binding equation (2). Residues selected at randomized positions are in red. P indicates a phosphopeptide. U indicates an unphosphorylated peptide. The phosphoserine residue in phosphopeptides is in bold. CA indicates that carbonic anhydrase II was used as the target protein; CalM indicates calmodulin was used as the target protein. The specificity ratio Krel is defined as Krel = Kd (CA or CalM) / Kd (HisKIX). N.D. indicates that the value was not determined.
The selected PPKID molecules also display a dramatic preference for binding CBP KIX over calmodulin (Figure 5B and Table 2). PPKID4P binds calmodulin with a Kd of 51 ± 12 μM, which corresponds to a Krel value of 100. Peptide CP displays slightly lower specificity (Krel = 74) than PPKID4P for CBP KIX over calmodulin, a result of 5-fold lower affinity for HisKIX and 4-fold lower affinity for calmodulin (Kd = 178 ± 42 μM). The Kd for the PPKID6U•calmodulin complex could not be determined definitively, but we could place a lower limit of 168 μM on the Kd value by defining the minimum change in polarization between the fully calmodulin-bound and fully unbound states of fluorescently labeled PPKID6U as 100 mP (the observed change in the presence of 185 μM calmodulin was 66 mP). Thus, PPKID6u, like PPKID4P, exhibits a significant preference for CBP KIX over calmodulin, with a specificity ratio of at least 112.
Discussion
Protein grafting has proven to be a versatile solution to the problem of macromolecular recognition, having been used previously to identify miniature protein ligands for both the DNA major groove12-14,16 and deep protein clefts.15 The complex formed between the α-helical phosphorylated KID domain of CREB and the KIX domain of CBP30 provided us with a unique context in which to address two questions about the generality and utility of protein grafting in the design of ligands for protein surfaces. First, we asked whether protein grafting could provide access to ligands for a shallow protein surface, namely that of the CBP KIX domain. Second, we asked whether a post-translational modification step, phosphorylation, could be incorporated into the molecular evolution protocol used in protein grafting.
Can a miniature protein recognize the shallow, extended cleft on the surface of CBP KIX?
Structural and energetic information about the KIDP•KIX complex guided the design of a library of miniature proteins based on the CREB KID domain. Five peptides, PPKID 1-5, were identified in phosphorylated phage display selections 1 and 2. Despite their divergent sequences in the randomized region, these peptides bind to the CBP KIX domain with similar affinity when phosphorylated, with Kds ranging from 515 nM to 1.2 μM. PPKIDP 1-5 display CBP KIX-binding affinities comparable to that of the full length phosphorylated CREB KID domain (KID-ABP) and 50- to 100-fold greater than that of phosphorylated helix B of CREB KID (KID-BP), which corresponds to the portion of CREB KID that was grafted onto the aPP scaffold. Thus, in the context of phosphorylated peptides, the aPP scaffold and selected residues enhance the CBP KIX-binding affinity of the CREB KID helix B functional epitope by an amount comparable to the 92-fold enhancement provided by the addition of helix A. The increases in affinity seen in this work (2.2 to 2.7 kcal•mol−1) compare well with increases observed in previous applications of protein grafting in which the miniature protein target contains a much deeper binding pocket.15
Two lines of evidence suggest that PPKID4P, the highest affinity phosphorylated CBP KIX ligand identified here, interacts with the CREB KIDP-binding groove on the CBP KIX surface in a manner similar to the natural ligand CREB KIDP. First, PPKID4P and the full-length CREB KID domain (KID-ABP) compete for binding CBP KIX in an equilibrium fluorescence polarization assay. The concentration of KID-ABP needed to displace 50% of fluorescently tagged PPKID4P is consistent with the Kd of the KID-ABP•CBP KIX complex determined by direct fluorescence polarization analysis. Second, the stabilities of the CBP KIX complexes of PPKID4P and KID-ABP decrease by a comparable amount (24- and 15-fold, respectively) when an energetically significant residue within the binding cleft (Tyr650) is altered to alanine. These results confirm that protein grafting can produce ligands capable of high affinity recognition of a shallow hydrophobic groove such as found in CBP KIX.
Distribution of labor between the polyproline helix and phosphorylated α-helix in CBP KIX recognition by PPKIDs
Interestingly, peptide CP, which contains the residues comprising the putative α-helical portion of the PPKID peptides, has significantly higher affinity for CBP KIX (Kd = 2.4 μM) than does KID-BP (Kd = 51.6 μM). Peptides CP and KID-BP differ at seven of twenty residues, including the conservative Tyr to Phe mutation known to have no effect on binding in the context of the full-length CREB KID domain.34 The enhanced CBP KIX-binding affinity exhibited by peptide CP relative to KID-BP may result from direct contacts between the aPP-derived residues in peptide CP and the CBP KIX surface or by virtue of increased helical propensity of aPP-derived residues in peptide CP compared to the corresponding residues in KID-BP, or by a combination of these two factors. The 1.8 kcal•mol−1 increase in stability of the peptide CP•KIX complex compared to the KID-BP•KIX complex is significantly larger than the more modest 0.4-0.9 kcal•mol−1 increases in stability of the PPKIDP•KIX complexes compared to the peptide CP•KIX complex. Thus, in the context of phosphorylated peptides, changes within the α-helical region contribute more to CBP KIX-binding affinity than residues (even selected residues) within the polyproline helix region. Despite the lack of conservation among the residues of the putative polyproline helix regions of PPKIDP 1-5, it seems likely that these molecules bind to CBP KIX in a similar manner, as most of their CBP KIX-binding affinity is derived from their common α-helical portion.
Minimizing the functional epitope as well as the globular fold
Although the PPKID library members contained all residues from CREB KID helix B known to be important for KIDP•KIX complex formation, no residues from helix A of CREB KID were included in the library. However, at least one residue from helix A, Leu128, makes energetically significant contacts to CBP KIX in the KIDP•KIX complex; when Leu128 is mutated to alanine, CBP KIX binding is abolished in a GST pull-down assay.33 The identification of the PPKID peptides, which have high affinity for CBP KIX despite the absence of residues from helix A, thus provides a second example of a successful protein grafting application (and the first in the context of protein recognition) using a thermodynamically incomplete set of binding residues, akin to the recent example of a miniature engrailed homeodomain which binds DNA with high affinity despite the absence of residues known to be critical for formation of the natural engrailed•DNA complex.16
Ramifications of phosphorylation on CBP KIX binding affinity
Though the CBP KIX-binding affinities of the phosphorylated PPKID peptides are similar, the importance of phosphorylation for CBP KIX binding varies more substantially among PPKID 1-5. The Kds for PPKIDU 1-5 vary between 6.6 μM and 24.1 μM, between 6-fold (for PPKID3) and 41-fold (PPKID1) lower than the values measured for the analogous phosphorylated peptides (ΔΔG = −0.5 to −1.9 kcal•mol−1). Taken together, the similarity in the CBP-KIX binding affinities of PPKIDP 1-5 and the more substantial variation in the CBP-KIX binding affinities of PPKIDU 1-5 suggest that the desired phosphorylation step was successfully incorporated into the molecular evolution step of the protein grafting protocol.
In contrast to peptides PPKID 1-5, the KID-AB peptide requires phosphorylation to achieve high affinity for CBP KIX, exhibiting a greater than 206-fold enhancement (ΔΔG > 3.2 kcal•mol−1) in CBP KIX-binding affinity upon phosphorylation. Peptide C, like the PPKID peptides, exhibits less dependence on phosphorylation for high affinity CBP KIX binding than does the KID-AB peptide; upon phosphorylation, the affinity of peptide C for CBP KIX is enhanced 9-fold (ΔΔG = −1.3 kcal•mol−1). The significant CBP KIX-binding affinity exhibited by peptide CU suggests that the relatively high affinity of the unphosphorylated PPKID peptides is derived from their common α-helical region. As is true in the context of phosphorylated peptides, peptide CU exhibits significantly increased affinity for CBP KIX when compared to KID-BU, with a Kd at least 14-fold better (ΔΔG > −1.6 kcal•mol−1). However, peptide CU also exhibits significantly higher affinity (> 5-fold) for CBP KIX than the unphosphorylated full length CREB KID domain (KID-ABU) (ΔΔG > −1.0 kcal•mol−1). Thus, the seven amino acid changes which convert KID-BU to peptide CU provide a significant increase (> 14-fold) in CBP KIX-binding affinity, whereas the addition of helix A to KID-BU does not result in high affinity CBP KIX binding (Kd > 116 μM). In contrast, in the context of phosphorylated peptides, the seven residue changes which convert KID-BP to peptide CP provide an enhancement in CBP KIX-binding affinity comparable to the addition of helix A to KID-BP.
Can a functional selection identify improved unphosphorylated miniature protein ligands for the shallow cleft of CBP KIX?
The high CBP KIX-binding affinity of peptide CU (and PPKIDU 1-5) led us to investigate the possibility that PPKID Library 1 might contain unphosphorylated miniature protein ligands with significantly enhanced affinity (relative to peptide CU) for CBP KIX. The five PPKID peptides identified in unphosphorylated selections 3 and 4 (PPKIDU 4-8) display a wider range of affinities for CBP KIX than is observed among the PPKIDP peptides, with Kds from 1.5 μM to 12.1 μM. At four of the five randomized positions, the selected residues approach consensus in PPKID 4-8, whereas they are essentially non-conserved in the same positions among PPKID 1-5. Notably, the three peptides identified solely in the selections lacking a phosphorylation step (PPKIDU 6-8) exhibit the highest affinity for CBP KIX, with Kds from 1.5 μM to 3.1 μM. This result provides further evidence that the library members in selections 1 and 2 were successfully phosphorylated by PKA, as the PPKID molecules identified in those selections (PPKID 1-5) display significantly lower CBP KIX-binding affinity when unphosphorylated than PPKIDU 6-8.
The best unphosphorylated peptide, PPKID6U, binds CBP KIX with extremely high affinity (Kd = 1.5 μM), only two-fold worse than the full length phosphorylated CREB KID domain, and at least 200-fold better than unphosphorylated helix B of CREB KID (Kd > 297 μM). The aPP scaffold contributes at least 3.1 kcal•mol−1 stability to the PPKID6U•KIX complex, of which 1.5 kcal•mol−1 is due to the putative polyproline helix and turn region (including the selected residues). This contribution is significantly larger than is observed for the polyproline regions of PPKIDP 1-5 (ΔΔG = −0.7 to +0.1 kcal•mol−1). In contrast, the dependence of the CBP KIX-binding affinity of PPKID6 on phosphorylation (ΔΔG = −0.5 kcal•mol−1) was significantly smaller than is observed for PPKID 1-5 (ΔΔG = −1.0 to −2.2 kcal•mol−1). Notably, PPKID6U binds CBP KIX with 8-fold greater affinity than PPKID4U, which contains all aPP-derived residues at the randomized positions. In the unphosphorylated case, then, the simple graft of the functional epitope of CREB KID helix B onto the aPP scaffold did not produce the optimal CBP KIX-binding molecule and a functional selection successfully identified a molecule with significantly higher affinity.
Two results suggest that the CBP KIX-binding mode of PPKID6U may differ somewhat from that of CREB KIDP. First, mutation of PPKID6U residue Ser18 to Glu results in a 7-fold decrease in affinity for CBP KIX, whereas a similar mutation in the context of CREB KID leads to an affinity intermediate between the higher affinity phosphorylated and lower affinity unphosphorylated forms.39 Second, mutation of CBP KIX residue Tyr650 to Ala has no effect on the stability of the PPKID6U•CBP KIX complex, whereas it reduces the stability of the CREB KIDp•CBP KIX complex by 15-fold. Interestingly, despite these observed differences in the binding modes of PPKID6U and CREB KIDP, PPKID6U and full-length CREB KIDP cannot bind simultaneously to CBP KIX, as KID-ABP and PPKID6U compete for binding HisKIX in an equilibrium fluorescence polarization assay. These results are consistent with overlapping, but not identical, binding sites of PPKID6U and CREB KIDP on CBP KIX. Further experiments, with an established panel of CBP KIX variants,41 will be necessary to clarify the binding mode of PPKID6U and are underway.
The quest for specificity
A unique advantage of molecules capable of binding other proteins in the nanomolar concentration range is their ability to discriminate effectively among the numerous protein surfaces inside the cell. To evaluate the specificity of the interaction between the selected PPKID molecules and CBP KIX, we investigated the affinity of PPKID4P and PPKID6U for two proteins, carbonic anhydrase and calmodulin, that bind hydrophobic α-helical peptides or small molecules. PPKID4P and PPKID6U bind both calmodulin and carbonic anhydrase with low affinity, exhibiting 53- to 205-fold preferences for recognition of CBP KIX over calmodulin and carbonic anhydrase. Interestingly, control peptides CP and CU exhibit lower specificity ratios (ranging from 3 to 74), suggesting that the selected polyproline helix residues in PPKID4P and PPKID6U have a significant effect on the ability of these peptides to selectively interact with CBP KIX over unrelated proteins. The specificity ratios observed for PPKID4P and PPKID6U are lower than the specificity ratios observed for a miniature protein (PPBH3-1) that recognizes the anti-apoptotic protein Bcl-2 (Krel values from 577 to >1900).15 Both the PPKID peptides and PPBH3-1 bind proteins unrelated to their targets with affinities in the high micromolar concentration range; the lower specificity ratios observed for the PPKID peptides are a function of their high nanomolar to low micromolar affinities for their protein target, CBP KIX, whereas PPBH3-1 recognizes its protein target, Bcl-2, with approximately 10-fold lower affinity. Nevertheless, the PPKID peptides clearly exhibit a dramatic preference for recognition of CBP KIX over unrelated proteins that might be encountered inside the cell.
The relationship between affinity and inherent α-helicity
Surprisingly, given their high affinity for CBP KIX, the PPKID peptides display only nascent α-helicity in the absence of CBP KIX as determined using circular dichroism, although at least one PPKID peptide (PPKID6U) exhibits TFE-inducible α-helicity 35. Phosphorylation of the PPKID peptides had no effect on their α-helical content, suggesting that the increased affinity of the phosphorylated PPKID peptides is not due to phosphorylation-dependent changes in their structures. The CREB KID domain is likewise mostly disordered in the absence of CBP KIX, with less than 10% helicity observed in helix B.38 Furthermore, CD and NMR experiments have shown that phosphorylation of Ser133 has little or no effect on the (lack of) structure of helix B,42,43 despite the dramatic effect of phosphorylation on CBP KIX-binding affinity. Complex formation between CREB KID and CBP KIX is primarily driven by a favorable enthalpy change41 and is a consequence of intermolecular contacts between the phosphoserine moiety of CREB KID and two residues on the CBP KIX surface.30 Based on mutagenesis of helix B of CREB KID, it has been proposed that the stability of CBP KIX-bound helix B (inferred from measurements of peptide helicity in the presence of TFE) rather than the stability of unbound helix B is the major determinant of CBP KIX-binding affinity.32 Interestingly, residues Arg135 and Lys136 of CREB KID have been suggested to contain a negative determinant of helicity.32,41 Two of the seven amino acid differences between peptide C and helix B correspond to these residues, and this may, in part, explain the enhanced affinity of peptide C for CBP KIX, and further, the high affinity of the unphosphorylated PPKID peptides for CBP KIX.
The lack of helical content in PPKID4, which corresponds to the simple graft of the CBP KIX-binding epitope onto the aPP scaffold, indicates that the changes made in the putative α-helical region are sufficient to disrupt the hydrophobic core of aPP, presumably due to the absence of Val30 and Val31. We chose to randomize five residues from the polyproline helix which form part of the hydrophobic core of aPP, based on our success using a similar strategy to evolve DNA-binding miniature proteins.14 We hoped to identify those members of the PPKID Library 1 that exhibited enhanced affinity due to stabilization of the CBP KIX-bound α-helical region by the randomized polyproline helix. In retrospect, this strategy may not have been the best one for this system, given the structure of the complex formed between full-length CREB KIDP and CBP KIX.30 Although most of the energetically important contacts between CREB KIDP and CBP KIX are mediated by helix B, which binds into a shallow groove on the surface of CBP KIX, helix A contacts another region of the CBP KIX surface, and thereby contributes to CBP KIX-binding affinity. The conditions used in our functional selections would not distinguish between those library members which have enhanced affinity for CBP KIX by virtue of stabilization of the CBP KIX-bound α-helix and those which have enhanced affinity due to the gain of contacts between residues in the randomized region and the CBP KIX surface. One explanation, then, for the high affinity of the miniature proteins for CBP KIX despite their lack of well-defined structure might be that the randomized region is acting akin to helix A, and contacting the CBP KIX surface outside of the helix B-binding groove. It might contact the same surface which binds helix A, or another region of the CBP KIX surface, as at least two other proteins (c-Jun44 and MLL 45) bind to CBP KIX in ternary complexes with CREB KID, contacting distinct regions of the CBP KIX surface.
The minimal unphosphorylated activation domain of the proto-oncoprotein c-Myb (residues 291-315) binds constitutively to the KID-binding groove of the CBP KIX domain with a Kd of 15 μM.41 The c-Myb activation domain displays significant helical content (30%) in aqueous buffer and is 90% helical in the presence of 40% TFE.32 In contrast to CREB KID, binding of c-Myb to CBP KIX is driven both by entropy and enthalpy.41 Furthermore, α-helix-destabilizing mutations of c-Myb significantly compromise CBP KIX binding. Taken together, these studies strongly suggest that the significant helicity of c-Myb is crucial for its high (at least as compared to unphosphorylated CREB KID) affinity recognition of CBP KIX. In light of this, it is quite surprising that the unphosphorylated PPKID peptides display affinities for CBP KIX comparable to or up to 10-fold greater than the CBP KIX-binding affinity of c-Myb, despite their lack of helical structure in the absence of CBP KIX. The similarity between c-Myb and the PPKID miniature proteins (or CREB KID, upon which they are based) is limited to the conservation in spacing of three hydrophobic residues known to be important for CBP KIX binding by both c-Myb and CREB. Thus, it is likely that the molecular details and energetic components of the PPKIDU•KIX and c-Myb•KIX complexes differ significantly, but further experiments will be required to confirm this.
The PPKID peptides represent only the second example of synthetic CBP KIX-binding molecules. A recent study identified short, unphosphorylated KIX-binding peptides (KBPs) by phage display screening of a degenerate peptide library.46 An 11mer peptide containing the selected eight amino acid KBP consensus sequence binds CBP KIX with a Kd of 16 μM. The KBPs, like the PPKID peptides, appear to bind overlapping, but distinct regions of CBP KIX compared to CREB KID. Homology between the KBP consensus sequence and the PPKID peptides is limited, as was the case with c-Myb, to the spacing of the three energetically important CBP KIX-contacting hydrophobic residues. The selected KBPs can serve as transcriptional activation domains in 293 cells when fused to a DNA-binding domain. Interestingly, the in vitro CBP KIX-binding affinity of the (albeit only) two KBPs examined correlated with their potency as activation domains. It will be of interest to determine if the PPKID molecules can act as transcriptional activation domains. If the correlation between CBP KIX-binding affinity and transcriptional potency holds, PPKID6U, which binds CBP KIX 10-fold more tightly than the best KBP, could potentially act as an extremely potent constitutive activation domain.
Conclusions
The work described here extends the utility of the protein grafting and molecular evolution procedure to the significant problem of high affinity and specific recognition of shallow protein surfaces. Taken together with previous applications, the protein grafting strategy has now proven to be extremely general in scope, enabling the discovery of highly functional miniature proteins capable of molecular recognition of diverse nucleic acid and protein targets. In addition, a posttranslational modification step, phosphorylation, was introduced here for the first time into the molecular evolution protocol used in protein grafting. Phosphorylated peptide ligands based on the functional epitope of the CREB KID domain were discovered which possess high nanomolar to low micromolar affinity and high specificity for the shallow surface groove of the CBP KIX domain. Furthermore, presentation of the CREB KID domain functional epitope on the aPP scaffold protein yielded peptide ligands for CBP KIX which bypass the need for phosphorylation to achieve high affinity CBP KIX recognition and have potential for use as extremely potent transcriptional activation domains.
Experimental Section
HisKIX expression vector cloning
The CBP KIX-coding region (residues 586 to 672) of pGEX-KT KIX 10-67241 (a gift from Marc Montminy) was amplified by PCR using 5′ and 3′ primers containing NdeI and BamHI restriction sites, respectively. Primers KIX5P and KIX3P had the following sequences: KIX5P: 5′-GCCGCGCGGCAGCCATATGGGTGTTCGAAAAGCCTGGC-3′; KIX3P: 5′-CCAGGCCGCTGCGGATCCTCATCATAAACGTGACCTCCGC-3′. The CBP KIX-coding duplex DNA insert was digested with NdeI and BamHI and ligated into NdeI- and BamHI-digested pET15b (Novagen) using T4 DNA ligase (New England Biolabs). The resulting plasmid, pHisKIX, codes for the CBP KIX domain in-frame with an amino-terminal hexahistidine tag under control of a T7 promoter. Plasmid identity was confirmed by DNA sequencing of the CBP KIX-coding region of pHisKIX.
Overexpression and purification of GST-KIX and HisKIX
pGST-ΔKIX(588-683)47 (a gift from Jennifer Nyborg) or pHisKIX was transformed into BL21(DE3) pArg E. coli cells by electroporation. A single colony was used to inoculate a 1 L culture of LB media containing 0.2 mg/mL ampicillin and 0.05 mg/mL kanamycin. The culture was incubated at 37 °C with shaking at 250 rpm until the solution reached an optical density of 0.6 absorbance units at 600 nm. Isopropyl β-D-thiogalactoside (IPTG) was added to a final concentration of 1 mM and incubation continued for 3 h at 37 °C. Cells were harvested by centrifugation for 20 min at 10,800 g and resuspended in 15-20 mL of buffer (GST-KIX: 50 mM potassium phosphate (pH 7.2), 150 mM NaCl, 1 mM DTT; HisKIX: 50 mM sodium phosphate (pH 8.0), 300 mM NaCl, 10 mM imidazole). Cells were lysed by sonication, insoluble material was pelleted by centrifugation for 30 min at 37,000 g, and the supernatant was retained. GST-KIX and HisKIX proteins were purified by glutathione and nickel-nitrolotriacetic acid (Ni-NTA) affinity chromatography, respectively. Fractions containing the desired protein were combined, desalted on a NAP 10 (GST-KIX) or NAP 25 (HisKIX) column (Amersham) and stored in buffer containing 50 mM Tris (pH 8.0), 100 mM KCl, 12.5 mM MgCl2, 1 mM ethylenediaminetetraacetic acid (EDTA) and 0.05% Tween-20 (GST-KIX storage buffer also contained 1 mM DTT) at −70 °C. Protein identity and concentration were confirmed by amino acid analysis.
Phage library construction
PPKID Library 1 was created by cassette mutagenesis of the phagemid vector pJC2013 using the synthetic oligonucleotides Align1 and PPLib. These oligonucleotides possessed the following sequences (N indicates an equimolar mixture of G, C, A and T, and S indicates an equimolar mixture of G and C): Align1: 5′- TGTTCCTTTCTATGCACCGGTTCGTCTCTGTCCTTCTTCTACATCCTGCTGGACCTGTACCTGG ACGCACCGGCGGCCGCAGGTGCGCCGGGCC-3′; PPLib: 5′-TGTTCCTTTCTATGCGGCCCAGCCGGCCGTNNSTCCNNSNNSACCNNSNNSGGTGACGACGC ACCGGTAGGTGCGCCGGTGCC-3′. Double stranded Align1 and PPLib inserts were generated by primer extension of appropriate primers using Sequenase version 2.0 T7 DNA polymerase (US Biochemicals). The duplex Align1 insert was digested with AgeI and NotI, and purified from a preparative agarose gel using the QIAquick gel extraction kit (Qiagen) and ethanol precipitation. Purified Align1 insert was ligated into AgeI- and NotI-digested pJC20 using the Ligation Express Kit (Clontech) to yield the phagemid vector pAlign1. Double stranded PPLib insert was digested with AgeI and SfiI and purified as per Align1. PPLib insert was ligated into AgeI- and SfiI-digested pAlign1 using the Ligation Express Kit (Clontech) to generate PPKID Library 1. The ligated PPKID Library 1 phagemid vector was transformed into XL1 Blue E. coli cells by electroporation and amplified by overnight growth at 37 °C in 2X YT-AG media (2X YT media containing 2% (w/v) glucose and 0.1 mg/mL ampicillin). Glycerol stocks of this culture were used as the initial pool for selection experiments. PPKID Library 1 contained 5 × 107 independent transformants, which covered the theoretical diversity of the library (325 = 3.36 × 107) with 77% confidence. Sequencing of twenty individual clones from the initial pool verified the quality of the library; none of the sequenced clones contained mutations, deletions or insertions in the PPKID-coding region.
Phage display procedure
A glycerol stock of the initial pool (round 1) or output from the previous round (rounds 2-9) was used to inoculate 10 mL 2X YT-AG media. The culture was incubated at 37 °C until it reached an optical density of 0.6 absorbance units at 600 nm. The culture was then infected with 4 × 1011 pfu M13K07 helper phage and incubated at 37 °C for 1 h. Cells were pelleted by centrifugation, resuspended in 10 mL 2X YT-AK (2X YT media containing 0.1 mg/mL ampicillin and 0.05 mg/mL kanamycin) and incubated for 12-13 h at 37 °C. Cells were then pelleted by centrifugation and the retained supernatant was filtered through a 0.45 μm syringe filter. Phage were precipitated with 1/5 volume PEG/NaCl (20% (w/v) PEG-8000, 2.5 M NaCl) on ice for 45 min, and then pelleted by centrifugation for 35 min at 24,000 g. For phosphorylated selections, the precipitated phage were resuspended in water and approximately 1010 phage were phosphorylated in vitro with 2500 U PKA (Promega) in 100 μM ATP, 40 mM Tris (pH 8), 20 mM magnesium acetate for 2 h at 30 °C. Phosphorylated phage were precipitated on ice for 45 min with PEG/NaCl and then pelleted by centrifugation at maximum speed in a microcentrifuge for 30 min. Mock phosphorylation reactions were performed in parallel without PKA, and purified in the same manner. Precipitated phage (+/− PKA treatment) were resuspended in binding buffer for use in selections. HisKIX binding buffer contained 50 mM potassium phosphate (pH 7.2), 150 mM NaCl, 0.05% Tween-20 and GST-KIX binding buffer contained 20 mM Tris (pH 8.0), 150 mM NaCl, 0.1% Tween-20.
Selections against HisKIX were performed in Ni-NTA HisSorb microtiter 8-well strips (Qiagen) and selections against GST-KIX were performed in glutathione-coated 96-well microtiter plates (Pierce). 200 μL target protein was added to each well (final concentration of 30 nM for GST-KIX and 100 nM for HisKIX) and incubated overnight with shaking at 4 °C. Wells were washed three times with HisKIX or GST-KIX binding buffer to remove unbound protein. For blocking, binding buffer containing 6% milk was added to each well and incubated at 4 °C for 3 h. After blocking, wells were washed three times with binding buffer. Phage purified as described were added to each well and incubated for 3 h at 4 °C or 25 °C. Nonbinding or weakly binding phage were removed by repeated washing (10 to 20 times, 1 min to 5 min in length, according to round)35 with binding buffer. Bound phage were eluted by incubation with 0.1 M glycine (pH 2.2) for 20 min. After neutralization of the eluted phage solution with 2 M Tris (pH 9.2), XL1 Blue E. coli cells in log phase were infected with input and output phage and incubated at 37 °C for 1 h. Serial dilutions of infected cells were plated on SOB agar plates containing 2% (w/v) glucose and 0.1 mg/mL ampicillin. Cells infected with output phage were used to make glycerol stocks and stored at −70 °C.
Peptide synthesis and modification
Peptides were synthesized on a 25 μmol scale at the HHMI Biopolymer/Keck Foundation Biotechnology Resource Laboratory at the Yale University School of Medicine, New Haven, CT. All peptides contained free N-terminal amines and amidated C-termini. Phosphoserine residues were introduced using an Fmoc-protected O-benzyl-phosphoserine derivative with standard coupling conditions. Crude peptides were purified by reverse-phase HPLC on a Vydac semi-preparative C18 column (300 Å, 5 μm, 10 mm × 150 mm). Matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) mass spectrometry was used to confirm peptide identity before further modification. Fluorescein-conjugated derivatives were generated by reaction of purified peptides containing single C-terminal cysteine residues with a 10-fold molar excess of 5-iodoacetamidofluorescein (Molecular Probes) in a 3:2 mixture of dimethylformamide:phosphate-buffered saline (DMF:PBS). Labeling reactions were incubated with rotation for 3-16 h at room temperature. Fluorescein-labeled peptides were purified by reverse-phase HPLC as described above, and characterized by MALDI-TOF mass spectrometry and amino acid analysis
Fluorescence polarization
Fluorescence polarization experiments were performed with a Photon Technology International QuantaMaster C-60 spectrofluorimeter at 25 °C in a 1 cm pathlength Hellma cuvette. Serial dilutions of HisKIX were made in buffer containing 50 mM Tris (pH 8.0), 100 mM KCl, 12.5 mM MgCl2, 1 mM EDTA, 0.1% Tween-20. Briefly, an aliquot of fluorescently labeled peptide was added to a final concentration of 25-50 nM and the binding reaction was incubated for 30 min at 25 °C. Thirty minutes was a sufficient length of time for the binding reaction to reach equilibrium, as judged by an absence of change in the observed polarization value of the sample with the highest HisKIX concentration over 1 h. For competition experiments, serial dilutions of KID-ABP were incubated with 1.5 μM or 3 μM HisKIX and 25 nM fluorescein-labeled PPKID4P or PPKID6U (peptideFlu) for 60 min at 25 °C, respectively. For specificity measurements, carbonic anhydrase II (Sigma) or calmodulin (Sigma) was used as the target protein in place of HisKIX, and fluorescently labeled peptide was used at a final concentration of 50 nM. Carbonic anhydrase was serially diluted in binding buffer as described for HisKIX. Calmodulin was serially diluted in calmodulin folding buffer containing 20 mM Hepes (pH 7.5), 130 mM KCl, 1 mM CaCl2, 0.05% Tween-20.
Polarization was measured by excitation with vertically polarized light at a wavelength of 492 nm (10 nm slit width) and subsequent measurement of the fluorescence emission at a wavelength of 515 nm (10 nm slit width) for 10 s in the vertical and horizontal directions. The polarization data were fit using Kaleidagraph v3.51 software to equilibrium binding equation (1), derived from first principles.
| (1) |
In this equation, Pobs is the observed polarization at any target protein (HisKIX, carbonic anhydrase or calmodulin) concentration, Pmax is the maximum polarization value, Pmin is the minimum observed polarization value, and Kd is the equilibrium dissociation constant. Measurements from two to three independent sets of samples were averaged for each dissociation constant determination. For plots of fraction of fluorescently labeled peptide (peptideFlu) bound as a function of target protein concentration, polarization values were converted to fraction of peptideFlu bound using the Pmin and Pmax values derived from equation (1), and the fraction of peptideFlu bound data were fit to equilibrium binding equation (2) using Kaleidagraph v3.51 software.
| (2) |
In this equation, θobs is the observed fraction of peptideFlu bound at any target protein concentration and Kd is the equilibrium dissociation constant.
For competition experiments, observed polarization values were converted to fraction of peptideFlu bound using experimentally determined Pmin and Pmax values corresponding to the polarization of samples containing 25 nM peptideFlu alone and peptideFlu with 1.5 μM or 3.0 μM HisKIX, respectively. The fraction of peptideFlu bound data were fit to equation (3) using Kaleidagraph v3.51 software to determine the IC50 value.
| (3) |
In this equation, θobs is the observed fraction of peptideFlu bound at any competitor peptide concentration, slope is defined as the slope at the inflection point and IC50 is the concentration of competitor that reduces binding of peptideFlu by 50%.
Supplementary Material
Acknowledgment
This work was supported by the NIH (GM 65453) and in part by a grant to Yale University, in support of AS, from the Howard Hughes Medical Institute. S.E.R. thanks the Medicinal Chemistry Division of the American Chemical Society for a predoctoral fellowship. We are grateful to Marc Montminy and Jennifer Nyborg for expression vectors. Oligonucleotide and peptide synthesis, DNA sequencing, and amino acid analysis were performed by the HHMI Biopolymer/Keck Foundation Biotechnology Resource Laboratory at the Yale University School of Medicine, New Haven, CT.
Footnotes
Supporting Information Available: Tables of binding and washing conditions for selections 1 – 4, CD spectra of PPKID4P and PPKID6U, and competition analysis of PPKID4P•HisKIX complexation. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).McCormick F. Curr. Opin. Biotechnol. 2000;11:593–597. doi: 10.1016/s0958-1669(00)00149-x. [DOI] [PubMed] [Google Scholar]
- (2).Huang ZW. Pharmacol. Ther. 2000;86:201–215. doi: 10.1016/s0163-7258(00)00052-8. [DOI] [PubMed] [Google Scholar]
- (3).Ockey DA, Gadek TR. Expert Opin. Ther. Patents. 2002;12:393–400. [Google Scholar]
- (4).Specht KM, Shokat KM. Curr Opin Cell Biol. 2002;14:155–159. doi: 10.1016/s0955-0674(02)00317-4. [DOI] [PubMed] [Google Scholar]
- (5).Rutledge SE, Chin JW, Schepartz A. Curr Opin Chem Biol. 2002;6:479–485. doi: 10.1016/s1367-5931(02)00352-6. [DOI] [PubMed] [Google Scholar]
- (6).Cochran AG. Chem Biol. 2000;7:R85–94. doi: 10.1016/s1074-5521(00)00106-x. [DOI] [PubMed] [Google Scholar]
- (7).Cochran AG. Curr Opin Chem Biol. 2001;5:654–659. doi: 10.1016/s1367-5931(01)00262-9. [DOI] [PubMed] [Google Scholar]
- (8).Schreiber SL. Bioorg Med Chem. 1998;6:1127–1152. doi: 10.1016/s0968-0896(98)00126-6. [DOI] [PubMed] [Google Scholar]
- (9).Schepartz A, Kim PS. Curr Opin Chem Biol. 1998;2:9–10. doi: 10.1016/s1367-5931(98)80029-x. [DOI] [PubMed] [Google Scholar]
- (10).Lo Conte L, Chothia C, Janin J. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- (11).Jones S, Thornton JM. Proc Natl Acad Sci U S A. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zondlo NJ, Schepartz A. J. Am. Chem. Soc. 1999;121:6938–6939. [Google Scholar]
- (13).Chin JW, Grotzfeld RM, Fabian MA, Schepartz A. Bioorg Med Chem Lett. 2001;11:1501–1505. doi: 10.1016/s0960-894x(01)00139-1. [DOI] [PubMed] [Google Scholar]
- (14).Chin JW, Schepartz A. J Am Chem Soc. 2001;123:2929–2930. doi: 10.1021/ja0056668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chin JW, Schepartz A. Angew Chem Int Ed Engl. 2001;40:3806–3809. [PubMed] [Google Scholar]
- (16).Montclare JK, Schepartz A. J. Am. Chem. Soc. 2003;125:3416–3417. doi: 10.1021/ja028628s. [DOI] [PubMed] [Google Scholar]
- (17).Glover I, Haneef I, Pitts J, Wood S, Moss D, Tickle I, Blundell T. Biopolymers. 1983;22:293–304. doi: 10.1002/bip.360220138. [DOI] [PubMed] [Google Scholar]
- (18).Adams JM, Cory S. Trends Biochem.Sci. 2001;26:61–66. doi: 10.1016/s0968-0004(00)01740-0. [DOI] [PubMed] [Google Scholar]
- (19).Antonsson B, Martinou JC. Exp. Cell Res. 2000;256:50–57. doi: 10.1006/excr.2000.4839. [DOI] [PubMed] [Google Scholar]
- (20).Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SC, Fesik SW. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- (21).Petros AM, Nettesheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang HC, Thompson CB, Fesik SW. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- (23).Wang JL, Liu DX, Zhang ZJ, Shan SM, Han XB, Srinivasula SM, Croce CM, Alnemri ES, Huang ZW. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J, Zhang KYJ, Hockenbery DM. Nat. Cell Biol. 2001;3:183–191. doi: 10.1038/35055095. [DOI] [PubMed] [Google Scholar]
- (25).Kutzki O, Park HS, Ernst JT, Orner BP, Yin H, Hamilton AD. J. Am. Chem. Soc. 2002;124:11838–11839. doi: 10.1021/ja026861k. [DOI] [PubMed] [Google Scholar]
- (26).Kaneko M, Nakashima T, Uosaki Y, Hara M, Ikeda S, Kanda Y. Bioorg. Med. Chem. Lett. 2001;11:887–890. doi: 10.1016/s0960-894x(01)00094-4. [DOI] [PubMed] [Google Scholar]
- (27).Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu XH, Cao YY, Guo RB, Li BH, Zhu XF, Huang Y, Long YQ, Roller PP, Yang DJ, Wang SM. J. Med. Chem. 2001;44:4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- (28).Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan JY. Nat. Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- (29).Huang ZW. Chemistry & Biology. 2002;9:1059–1072. doi: 10.1016/s1074-5521(02)00247-8. [DOI] [PubMed] [Google Scholar]
- (30).Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- (31).Mestas SP, Lumb KJ. Nat Struct Biol. 1999;6:613–614. doi: 10.1038/10655. [DOI] [PubMed] [Google Scholar]
- (32).Zor T, Mayr BM, Dyson HJ, Montminy MR, Wright PE. J Biol Chem. 2002;277:42241–42248. doi: 10.1074/jbc.M207361200. [DOI] [PubMed] [Google Scholar]
- (33).Parker D, Jhala US, Radhakrishnan I, Yaffe MB, Reyes C, Shulman AI, Cantley LC, Wright PE, Montminy M. Mol Cell. 1998;2:353–359. doi: 10.1016/s1097-2765(00)80279-8. [DOI] [PubMed] [Google Scholar]
- (34).Du K, Asahara H, Jhala US, Wagner BL, Montminy M. Mol Cell Biol. 2000;20:4320–4327. doi: 10.1128/mcb.20.12.4320-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).See Supporting Information.
- (36).PPKID2 and PPKID3 each contain a spurious mutation not encoded in the original library pool, but the mutation is different in each peptide (Tyr to Asp at position 21 for PPKID2, Leu to Arg at position 24 for PPKID3). Further experimentation will be required to understand the functional consequences of these mutations.
- (37).Although the selected peptides contain at most one proline residue in this region, previous work has shown that the polyproline II (PPII) conformation can form in regions that lack proline residues.48-50
- (38).Parker D, Ferreri K, Nakajima T, LaMorte VJ, Evans R, Koerber SC, Hoeger C, Montminy MR. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Shaywitz AJ, Dove SL, Kornhauser JM, Hochschild A, Greenberg ME. Mol Cell Biol. 2000;20:9409–9422. doi: 10.1128/mcb.20.24.9409-9422.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Munson PJ, Rodbard D. J Recept Res. 1988;8:533–546. doi: 10.3109/10799898809049010. [DOI] [PubMed] [Google Scholar]
- (41).Parker D, Rivera M, Zor T, Henrion-Caude A, Radhakrishnan I, Kumar A, Shapiro LH, Wright PE, Montminy M, Brindle PK. Mol Cell Biol. 1999;19:5601–5607. doi: 10.1128/mcb.19.8.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Radhakrishnan I, Perez-Alvarado GC, Dyson HJ, Wright PE. FEBS Lett. 1998;430:317–322. doi: 10.1016/s0014-5793(98)00680-2. [DOI] [PubMed] [Google Scholar]
- (43).Hua QX, Jia WH, Bullock BP, Habener JF, Weiss MA. Biochemistry. 1998;37:5858–5866. doi: 10.1021/bi9800808. [DOI] [PubMed] [Google Scholar]
- (44).Campbell KM, Lumb KJ. Biochemistry. 2002;41:13956–13964. doi: 10.1021/bi026222m. [DOI] [PubMed] [Google Scholar]
- (45).Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. J Biol Chem. 2002;277:43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- (46).Frangioni JV, LaRiccia LM, Cantley LC, Montminy MR. Nat Biotechnol. 2000;18:1080–1085. doi: 10.1038/80280. [DOI] [PubMed] [Google Scholar]
- (47).Yan JP, Garrus JE, Giebler HA, Stargell LA, Nyborg JK. J Mol Biol. 1998;281:395–400. doi: 10.1006/jmbi.1998.1951. [DOI] [PubMed] [Google Scholar]
- (48).Rucker AL, Creamer TP. Protein Sci. 2002;11:980–985. doi: 10.1110/ps.4550102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Shi Z, Olson CA, Rose GD, Baldwin RL, Kallenbach NR. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9190–9195. doi: 10.1073/pnas.112193999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Stapley BJ, Creamer TP. Protein Sci. 1999;8:587–595. doi: 10.1110/ps.8.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.