

Abstract
We previously described a series of 314-helical β-peptides that bind the hDM2 protein and inhibit its interaction with a p53-derived peptide in vitro. Here we present a detailed characterization of the interaction of these peptides with hDM2 and report two new β-peptides in which non-natural side chains have been substituted into the hDM2-recognition epitope. These peptides feature both improved affinity and inhibitory potency in fluorescence polarization and ELISA assays. Additionally, one of the new β-peptides also binds the hDM2-related protein, hDMX, which has been identified as another key therapeutic target for activation of the p53 pathway in tumors.
Keywords: Protein–protein interactions, β-Peptides, Peptidomimetics, Inhibitors, Foldamer, p53, hDM2, Cancer
1. Introduction
Short β3-peptides can adopt a helical secondary structure in water when stabilized by salt-bridges and/or macrodipole effects. 1–8 This secondary structure, the 314-helix, is characterized by a periodicity of 3.1-residues/turn that arranges the β3-peptide side chains at 120° intervals when the structure is viewed down the helical axis. The periodicity of the 314-helix results in a reasonable, but imperfect, superposition of side chains along one face of the β-peptide helix (at positions i, i + 3, and i + 6) with those at positions i, i + 4, and i + 7 of an α-helix; this superposition can be improved by design.9–11 Indeed, over the past decade, we and others have reported a number of β-peptide 314-helices that bind diverse protein targets including somatostatin receptors, hDM2, and viral fusion proteins.11–14 Longer molecules containing both β- and α-amino acids have been shown to bind Bcl-xL.15 In addition, oligomers of β3-amino acids convey the advantage of near complete resistance to proteolysis.1,16
One target for β3-peptides that has received significant attention is hDM2, largely because of its recognized potential for novel oncology treatments.17,18 hDM2 is overexpressed in about 7% of human tumors, most commonly osteosarcomas and soft tissue sarcomas, and impairs the p53 stress response to allow unchecked cell growth.17–20 The structure of hDM2 bound to a short α-helical fragment of p53 has provided a virtual blueprint for inhibitor design, highlighting the importance of three p53 side chains (F19, W23, and L26) that insert into a hydrophobic cleft of hDM2.21 A number of peptidic, peptidomimetic, and small molecule p53-mimetics have been described that inhibit this interaction in vitro.17,22–33 and, in some cases, in vivo.24,27,34 In 2005, we reported a series of macrodipole- and salt bridge-stabilized 314-helical β3-peptides that each display the three critical p53 side chains along one face of the 314-helix; one of these molecules binds hDM2 with nanomolar affinity and inhibits its interaction with a p53-derived peptide.11,13 Importantly, high-resolution NMR analysis of these β-peptides indicates that their 314-helix is distorted—slightly unwound at the C-terminus—and that this distortion results in a significantly improved structural mimic of the p53 α-helix.9,11 These observations emphasize the versatility of β-peptides dominated by β3-amino acids when compared with more conformationally constrained analogs such as ACHC.35
This success encouraged us to evaluate the affinity of these β3-peptides for the hDM2-related protein, hDMX.36–38 hDMX is similar to hDM2 in that its N-terminal region binds to p53 and blocks transactivation;39 however, the expression of hDMX is not regulated by p53, nor has it been shown to target p53 for degradation, so the overall mechanism of regulation is likely to differ from that of hDM2. β3-peptides capable of inhibiting both hDMX and hDM2 might offer a unique advantage with respect to p53 activation, whereas those that selectively target one of these two proteins might have use as novel research tools. Here we build upon our initial results with β3-peptide ligands for hDM2 to characterize further the binding interaction of these peptides with hDM2 and report two β3-peptides with improved affinity and inhibitory potency. Additional binding data indicates that one of these peptides also possesses sub-micromolar affinity for hDMX.
2. Results
2.1. Design of third-generation β3-peptide ligands for hDM2: Non-natural side chains
One of the most notable features of the previously reported X-ray structure of mDM2 (the murine homolog of hDM2) bound to a p53-derived peptide (p53AD) is the inability of the p53AD Trp23 side chain to fill its binding pocket on the hDM2 surface.21 Indeed, replacement of Trp23 with 6-chlorotryptophan in the context of an α-peptide ligand reported more than a decade ago resulted in a 60-fold increase in affinity for hDM2.23 More modest (2–4-fold) improvements were observed when 6-chlorotryptophan was introduced into the equivalent position of either peptoid or β-hairpin ligands. 29,30 Similarly, a trifluoromethylphenyl substituent is found in several high affinity small molecule ligands for hDM2 (IC50 values ranging from 0.98 to >125 μM for a series of 1,4-benzodiazepine-2,5-diones).40–42 Based upon these results, we synthesized two new variants of β53-8: one containing a 3-trifluoromethylphenyl side chain in place of Trp23, (β53-12) and one containing 6-chlorotryptophan (β53-13) (Fig. 1).
Figure 1.
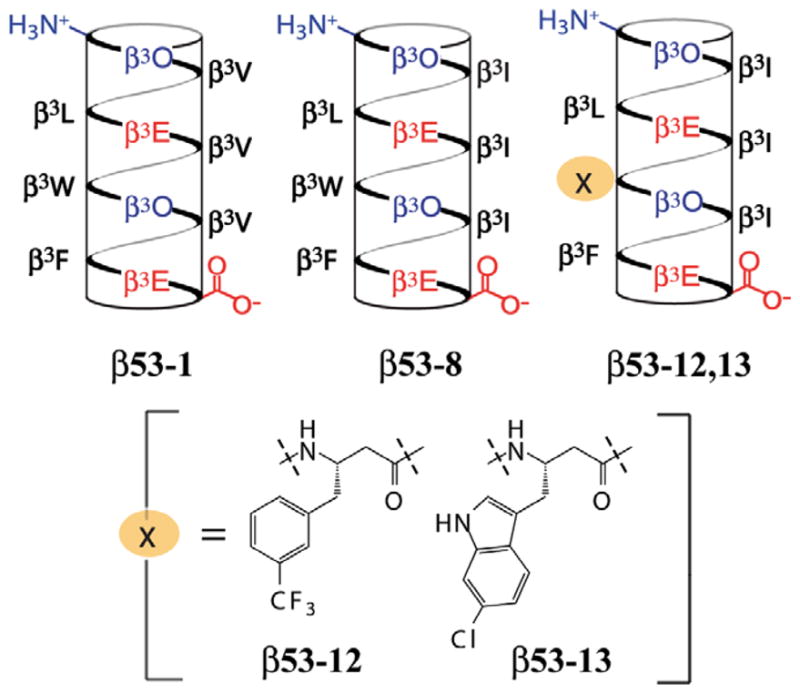
Helical net representations of β53-1, β53-8, β53-12, and β53-13. β3-Homoamino acids are identified by the one-letter code corresponding to the analogous α-amino acid.
2.2. Direct and competitive fluorescence polarization analysis of hDM2·β3-peptide interactions
We first used a direct fluorescence polarization assay to compare the hDM2 affinities of β53-12flu and β53-13flu to that of β53-8flu and assess whether the non-natural side chains improved the affinity of the β-peptide for hDM2. For this assay, β3-peptides labeled on the N terminus with fluorescein-5-EX, succinimidyl ester (Invitrogen) were equilibrated with hDM21-188 at concentrations between 1 nM and 15 μM. A fluorescently labeled α-peptide corresponding to residues 15-31 of p53 ( ) was used as a positive control.13 The Kd of the complex determined using this direct binding assay was 72.5 ± 9.12 nM, in agreement with previous work (Fig. 2A).13,17,21,43–46 The Kd values of the hDM2 complexes with β53-12flu and β53-13flu were 28.2 ± 4.79 and 30.1 ± 4.93 nM, respectively, representing a 2-fold improvement in affinity over . Moreover, the Kd values measured for β53-12flu and β53-13flu represent a 10-fold improvement over previously reported β3-peptide ligands for hDM2 (Kd = 292 ± 22.8 nM for β53-1flu and 204 ± 17.7 nM for β53-8flu).11,13
Figure 2.
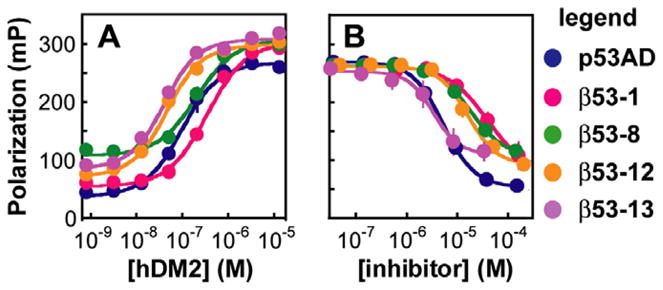
Fluorescence polarization analysis of β3-peptide·hDM2 equilibria. (A) Direct fluorescence polarization assay: plot of the polarization of the indicated fluorescently labeled β3-peptides and as a function of [hDM21-188]. (B) Competition fluorescence polarization assay: plot of the polarization of the complex with hDM21-188 as a function of the concentration of unlabeled peptide shown. Each value shown represents the average of at least four independent determinations, each performed in triplicate and averaged; error bars represent the standard error.
The relative affinities of β53-12 and β53-13 for hDM2 were also measured using a competitive fluorescence polarization assay in which unlabeled p53AD15-31 or β3-peptide competed with for binding to hDM2. In this assay, unlabeled p53AD15-31 inhibited complexation with an IC50 of 5.22 ± 0.196 μM, in line with previous reports.11,13,43 Under identical conditions β53-12 and β53-13 inhibited complexation with IC50 values of 15.9 ± 2.72 μM and 3.30 ± 0.681 μM, respectively (Fig. 2B). The inhibitory activity of β53-13 is several-fold greater than that of previously reported β-peptides β53-1 and β53-8 (IC50 = 39.2 ± 8.52 μM and 17.8 ± 0.530 μM, respectively).11 By contrast, the IC50 value for β53-12 is somewhat higher than expected, likely due to its poor solubility in aqueous buffer at concentrations greater than 100 μM. While the data from both direct and competition fluorescence polarization experiments indicate that β53-12 and β53-13 possess high affinity for hDM2, the competition data is particularly valuable as it directly measures the affinity of a ligand for the p53 binding pocket on hDM2.
2.3. Exploring the binding mode of β3-peptide ligands for hDM2
Previous work in our lab has indicated that versions of β53-1 labeled with fluorescein at the N- or C-termini possess similar affinities for hDM2 as determined by direct FP analysis.13 Nevertheless, to rule out the possibility that the appended fluorescein moiety contributes substantially to binding affinity, we prepared a second set of β3-peptides labeled with Alexa Fluor® 594 carboxylic acid, succinimidyl ester (Invitrogen). Both the structure and charge of Alexa Fluor® 594 differ from that of fluorescein (Fig. 3)—Alexa Fluor® is larger than fluorescein and exists as a zwitterion under the conditions of the binding assays (pH ~ 7.4) while fluorescein exists as a dianion. In addition, the fluorescein label contains a hydrophilic linker whereas the Alexa Fluor® label does not. With these differences in mind, we synthesized two additional β3-peptides labeled with Alexa Fluor® 594 on their N-termini. One (β53-8Al) is extended at the N-terminus by two additional β3-homogly-cine residues, whereas the other (β53-12Al) is not. The affinities of these two β3-peptides for hDM2 were evaluated by direct fluorescence polarization analysis as described above. The Kd values measured for β53-8Al and β53-12Al differed by only about 2-fold from the values measured for the respective fluorescein-labeled molecule despite the differences in linker and label (Fig. 3B and C). These results suggest that the fluorophore does not contribute more than minimally to the stability of β3-peptide·hDM2 complexes and that the labeled β3-peptides provide an accurate report of hDM2 affinity.
Figure 3.

(A) Structures of β53-8flu and β53-8Al (n = 2). β53-12flu and β53-12Al (n = 0) contain a β3-m-trifluoromethylphenylalanine residue in place of β3W. (B) Plot illustrating the polarization of the indicated β3-peptide as a function of [hDM21-188].
2.4. ELISA competition
Next we employed a validated competitive ELISA to further support a functional interaction between hDM2 and β53-1, 8, 12, and 13.28,47 Unlabeled α- and β3-peptides at concentrations between 0.012 and 200 μM were first equilibrated with 1 μM hDM21-188, and then added to streptavidin-coated microtiter plates coated with p53AD15-31 carrying a biotin label at the N-terminus ( ). The ability of unlabeled α- and β3-peptides to inhibit the interaction between hDM21-188 and immobilized was measured by detecting bound hDM2 with an hDM2-specific primary antibody (N-20, Santa Cruz Biotechnology), a horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit IgG-HRP, Santa Cruz Biotechnology) and QuantaBlu™ Fluorogenic Peroxidase Substrate (Pierce) (Fig. 4A). As expected, p53AD15-31 itself as well as β53-1, 8, 12, and 13 reduced the amount of hDM2 captured by immobilized in a dose-dependent manner, indicating effective inhibition of p53·hDM2 complexation. A related β3-peptide containing β3-homoalanine residues in place of the β3-homophenylalanine, β3-homotryptophan, and β3-homoleucine residues in β53-8 (βNEG, Fig. 4B) did not inhibit the fraction of hDM2 captured at concentrations as high as 200 μM.11 The relative IC50 values determined for β53-1, 8, 12, and 13 and for p53AD15-31 in the ELISA were similar to those determined using the fluorescence polarization competition assay, but the absolute values were generally 2–3-fold lower in the ELISA (Table 1). Despite these small systematic differences, the ELISA results provide further evidence for the inhibitory potency of the β3-peptides studied herein and supports the conclusion that the fluorophore used in the fluorescence polarization assays does not play an appreciable role in the observed affinity.
Figure 4.
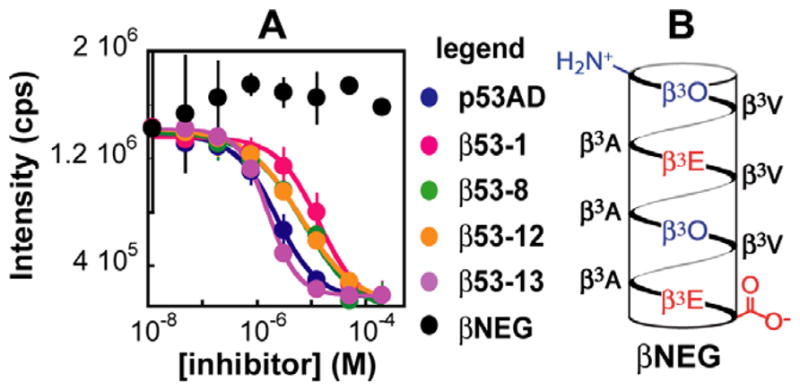
ELISA analysis of β3-peptide·hDM2 affinity. (A) Plot of the fluorescence intensity of QuantaBlu™ Fluorogenic Peroxidase Substrate (Pierce) as a function of the concentration of the indicated inhibitor. Each value represents the average of at least two independent experiments of three separate trials; error bars represent the standard error. (B) Helical net representation of βNEG. β3-Homoamino acids are identified by the one-letter code corresponding to the analogous α-amino acid.
Table 1.
Comparison of IC50 values for β3-peptides and p53AD as determined by competitive fluorescence polarization and ELISA.
| Ligand | IC50 (FP) (μM) | IC50 (ELISA) (μM) |
|---|---|---|
| p53AD | 5.22 ± 0.196 | 2.29 ± 0.265 |
| β53-1 | 39.2 ± 8.52 | 13.5 ± 3.20 |
| β53-8 | 17.8 ± 0.530 | 7.17 ± 2.52 |
| β53-12 | 15.9 ± 2.72 | 6.32 ± 0.316 |
| β53-13 | 3.30 ± 0.681 | 1.60 ± 0.0360 |
2.5. Cellular entry of β3-peptides
Recent work has suggested that certain peptides, even those lacking appreciable positive charge, can enter the cytosol of living mammalian cells.48 With this in mind, we investigated whether β53-12 might show even modest uptake by HCT116 cells. β53-12 was chosen because of its relative ease of synthesis compared to β53-13, which is complicated by optical resolution of 6-chlorotrytophan. β53-12flu and fluorescein-labeled control peptides (at concentrations ranging from 5 to 20 μM) were incubated with HCT116 cells for either 1 h or 4 h, and the resulting mean cellular fluorescence was quantified by flow cytometry (Fig. 5A and B). The p53ADflu peptide served as a negative control and showed only slight uptake at both time points studied. The positive controls, (PRR)3flu49 and Tatflu50 were taken up by the cells at both time points, but the signal for both decreased over time.49,50 Though the signal for β53-12flu was lower than that observed for arginine-rich (PRR)3flu49 and Tatflu50, β53-12flu showed clearly increased uptake relative to p53ADflu. Additionally, the fluorescence signal for β53-12flu increased over time, highlighting its resistance to cellular degradation. Although a trypsin wash was included in the flow cytometry protocol in an effort to digest membrane proteins that could bind the arginine-rich peptides,51 live-cell confocal microscopy was used to confirm the internalization and characterize the intracellular distribution of β53-12flu. As shown in Figure 5C, addition of β53-12flu to HCT116 cells resulted in a punctate staining pattern that colocalized with the endocytotic marker dextran, indicating internalization via endosomes. Western blot experiments reveal that β53-12flu modestly upregulates p53 activity in HCT116 and JAR cells but not HCT116 p53-null cells (E. Harker, unpublished results), suggesting that endosomal leakage occurs. Current work is focused on the application of Kodadek’s cell permeability screen48,52 to identify variants of β53-12 that achieve higher levels of cytosolic localization.
Figure 5.

Entry of β53-12flu and controls quantified by flow cytometry (A and B) and observed by confocal microscopy (C). Flow cytometry: mean cellular fluorescence was calculated from the histogram of fluorescence intensity and was corrected for background cellular fluorescence by subtracting the geometric mean of cells treated with only PBS. Each value represents the average of three independent trials. Error bars represent the standard error. (A) HCT116 cell uptake of fluorescein-labeled peptides after 1 h. (B) Uptake after 4 h. Confocal microscopy: HCT116 cells were incubated with 20 μM fluorescein-labeled peptide (green) for 2 h in McCoy’s 5A medium supplemented with 10% FBS. Endosomes were visualized using 10 μM 10 kDa dextran labeled with AlexaFluor 647 (red). Panel 1 shows β53-12flu only, panel 2 dextran only, panel 3 bright-field cell image only, panel 4 superposition of previous 3 panels.
2.6. Affinity of β3-peptides for hDMX
As described earlier, the p53-binding surfaces on hDM2 and the related protein hDMX are quite similar, with about 80% identity between the two.53 Despite this close relationship, and the fact that α-peptides that compete with p53 to bind hDM2 also bind hDMX,54–56 a number of small molecule ligands for hDM2, including Nutlin-3, do not bind hDMX.53,57 Structural work has indeed shown that the hDMX binding pocket for p53 is somewhat obstructed compared to the hDM2 binding pocket.58,59 We therefore tested our existing β3-peptide ligands to see if their extended binding surface, similar to that of an α-helix, would render them suitable for inhibition of the p53·hDMX interaction.
We used a direct fluorescence polarization assay to measure the affinities of β53-1flu, β53-8flu, β53-12flu, and β53-13flu for hDMX1-200. As a positive control, we used the α-peptide p53 12/1flu54 which possesses higher affinity for hDMX (Kd = 237 ± 21.5 nM) than (Kd = 841 ± 28.8 nM) (Fig. 6A). Interestingly, of all β3-peptides tested, only β53-12flu bound hDMX with submicromolar affinity (Kd = 518 ± 41.3 nM). β53-13flu, which was comparable to β53-12flu in affinity for hDM2, bound more weakly to hDMX (Kd = 1600 ± 79.9 nM). β53-8flu and β53-1flu also displayed low affinity for hDMX (Kd = 2130 ± 139 and 9090 ± 9937 nM, respectively).
Figure 6.
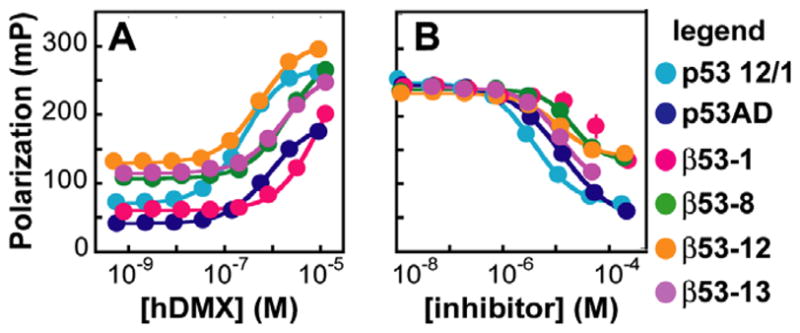
Fluorescence polarization analysis of β3-peptide·hDMX equilibria. (A) Direct fluorescence polarization assay: plot of the polarization of the indicated fluorescently labeled β3-peptides, p53 12/1flu, and as a function of [hDMX1-200]. (B) Competition fluorescence polarization assay: plot of the polarization of the p53 12/1flu complex with hDMX1-200 as a function of the concentration of unlabeled peptide shown. Each value shown represents the average of at least four independent determinations, each performed in triplicate and averaged; error bars represent the standard error.
Next we used a competitive fluorescence polarization assay to determine whether the β3-peptides competed with p53 12/1flu for binding to hDMX. Unlabeled p53 12/1 inhibited the binding of p53 12/1flu and hDMX with an IC50 of 3.87 ± 0.389 μM (Fig. 6B). Similarly, β53-12 inhibited p53 12/1flu·hDMX complexation with an IC50 of 10.9 ± 1.13 μM; the IC50 values for β53-13 and β53-8 were 12.0 ± 0.748 and 20.1 ± 2.51 μM, respectively, whereas β53-1 was too weak as an inhibitor to be quantified. Overall, the data indicates that β3-peptides designed to bind hDM2 also efficiently bind the hDMX protein, albeit with some variations in affinity.
3. Discussion
The development of general chemical strategies to inhibit transient interactions between proteins—both inside and outside the cell—represents one of the paramount goals of modern chemistry. Historically, it has been challenging to develop drug-like inhibitors of proteins whose natural ligands are other proteins, highlighting the need for novel approaches.60,61 In this work we describe β3-peptides containing non-natural side chains that possess significantly improved affinity for hDM2, a protein widely recognized for its interaction with the tumor suppressor p53.17,18 Introduction of either a 3-trifluoromethylphenyl or a 6-chlorotryptophan side chain in place of the central tryptophan of the hDM2-recognition epitope resulted in molecules that bind hDM2 with Kd values of approximately 30 nM, an improvement of nearly 10-fold over previously reported β3-peptide ligands. Furthermore, we completed a detailed study of the binding mode of these peptides using a series of fluorescence and ELISA assays to confirm their inhibitory activity towards the p53·hDM2 interaction. The results provide added evidence that these β3-peptides interact with hDM2 in much the same manner as p53AD. Based on previous reports, it is likely that the observed gain in affinity with the 3-trifluoromethylphenyl and 6-chlorotryptophan side chains is due to the ability of these groups to better fill the hydrophobic pocket on the hDM2 surface compared to the tryptophan side chain of the parent peptide.21,23,29,30 It is also interesting to note that both the trifluoromethyl and chloro groups are electron withdrawing; therefore it is possible that this electrostatic effect leads to a favorable aromatic donor–acceptor interaction with a side chain on the hDM2 surface. Structural information on the β3-peptide·hDM2 interaction is still needed to further explore these possibilities and would provide useful information for future designs.
Recent studies of p53 activation as a therapeutic strategy for cancer have pointed to the importance of inhibiting the interaction between p53 and the hDM2-related protein hDMX. As mentioned above, these two proteins have both been implicated in the regulation of p53, but likely function through different mechanisms. 36,37,62 Both proteins bind to the p53 activation domain through their N-terminal regions; in fact, 10 of the 13 residues of hDM2 determined to contribute to its interaction with p53 are conserved in the hDMX protein.39 Previous research has shown that optimized α-peptides designed to bind hDM2 also inhibit the p53·hDMX interaction with IC50 values of 1.65 μM (12/1: MRPFMDYWEGLN) and 0.10 μM (pDI: LTFEHYWAQLTS) reported for the most potent.54,55 By contrast, similar experiments have shown that potent small molecule inhibitors of the p53·hDM2 interaction (e.g., Nutlin-3, IC50 of 0.09 μM) do not inhibit p53·hDMX at concentrations as high as 30 μM. These results indicate that although the p53-binding surfaces on hDM2 and hDMX are similar enough to allow for targeting by longer peptides, they are not readily bound by the same small molecule ligand, findings that have been recently substantiated by structural data reported for the p53·hDMX interaction.58,59 Interestingly, it has been demonstrated that high hDMX levels or an inability to degrade hDMX leads to Nutlin-resistance in certain cell lines.53,57 As a result, there exists a subset of tumors that is not responsive to treatment with Nutlin-3. We hoped that our β3-peptides designed to bind hDM2 would also bind hDMX based upon their structural similarity to the α-helical structure already shown to target hDMX efficiently. Excitingly, one of these β3-peptides (β53-12) does in fact bind hDMX with submicromolar affinity and effectively inhibits its interaction with a p53-derived peptide. Interestingly, we saw some variation in affinity between hDM2 and hDMX for the series of β3-peptides studied, which can likely be traced to the observed differences in the p53-binding pockets of each protein. Further optimization of β3-peptide affinity for hDMX is a clear progression from this promising starting point given the limited number of molecules available to bind this new oncology target and could provide an advantage for the application of the β3-peptide scaffold compared to a small molecule.
Ultimately, we are interested in determining if these peptides are able to disrupt the p53·hDM2 and/or the p53·hDMX interaction in cells. β3-peptides would provide a useful complement to existing molecules aimed at p53 activation since they are more proteolytically stable than α-peptides and cover more surface area than small molecules (this latter feature may be particularly advantageous in the case of hDMX). The flow cytometry and confocal microscopy data presented here clearly demonstrate a modest degree of uptake for β53-12flu; however, compared to the positive control peptides (PRRflu)3 and Tatflu the cellular entry of β53-12flu stands to be significantly improved upon. At this point, we are pursuing strategies of arginine incorporation into the β3-peptide 314-helix based upon observations that the positively charged guanidinium group of arginine can promote cellular entry.63 This builds upon earlier work in our lab in which we incorporated arginine residues into a well-folded miniature protein to improve its cellular permeability. 49,64 Additionally, this strategy has been implemented previously in the Gellman and Seebach labs;65,66 however, there has not yet been a comprehensive analysis of the effects of arginine incorporation on a functional 314-helical β3-peptide.
4. Conclusion
We have significantly improved upon the affinity of our first-and second-generation β3-peptide ligands for hDM2 through the incorporation of non-natural side chains into the 314-helical scaffold. The use of a 3-trifluoromethylphenyl or a 6-chlorotryptophan side chain in place of the central tryptophan residue of the hDM2-recognition epitope led to molecules with Kd values of 30 nM for binding hDM2, a 10-fold improvement over our previously reported β3-peptide ligands. A detailed analysis of the binding mode of these peptides indicates that they effectively compete with a p53-derived peptide for its binding site on the hDM2 protein. Additionally, we have demonstrated that β3-peptides effectively target the hDM2-related protein, hDMX, a target of growing importance in oncology. Current work is focused on improving the cellular permeability of these β3-peptides and on further optimization of their binding affinity, particularly in the case of hDMX for which only a limited number of potent ligands have been reported.
5. Experimental
5.1. General
Fmoc-protected α-amino acids, PYBOP®, HOBt, and Wang resin were purchased from Novabiochem (San Diego, CA). Dimethylformamide (DMF), trifluoroacetic acid (TFA), and piperidine were purchased from American Bioanalytical (Natick, MA). All other reagents were purchased from Sigma–Aldrich. Fmoc-(S)-3-amino-4-(3-trifluoromethylphenyl)butyric acid was purchased from Ana-Spec, Inc. (San Jose, CA). The remaining Fmoc-β3-(L)-amino acids were synthesized from enantiomerically pure α-amino acids via the Arndt-Eistert procedure1 with the exception of Fmoc-(S)-3-amino-4-(6-chloroindole)butyric acid, the enantiomeric resolution and homologation of which is reported here.
5.2. Enantiomeric resolution of 6-chlorotryptophan
D,L-6-Chlorotryptophan was resolved by esterification and selective hydrolysis in the presence of α-chymotrypsin, a procedure adapted from that for tryptophan and α-methyltryptophan.67,68 Thionyl chloride (1.6 mL, 22.0 mmol) was added dropwise to D,L-6-chlorotryptophan (Biosynth AG, Switzerland, 1.048 g, 4.39 mmol) in 100 mL of chilled, anhydrous methanol with stirring. The mixture was warmed to room temperature over 1 h and then brought to reflux at 65 °C. The reaction was monitored by thin layer chromatography (85:15 acetonitrile/water), and, after 19 h, cooled and solvent removed under reduced pressure. D,L-6-chlorotryptophan methyl ester was recrystallized in four crops from methanol with diethyl ether to yield, after lyophilization, 1.22 g (96%) of the hydrochloride salt. 1H NMR (400 MHz, CD3OD): δ 7.49 (d, J = 8.5 Hz, 1H), 7.40 (s, 1H), 7.21 (s, 1H), 7.06 (d, J = 8.5 Hz, 1H), 4.32 (t, J = 6.5 Hz, 1H), 3.79 (s, 3H), 3.40 (d, J = 5.5 Hz, 1H). Calcd Mass 252.697, obsd 253.09.
D,L-6-Chlorotryptophan methyl ester hydrochloride (1.0691 g, 3.7 mmol) was dissolved in 11 mL DMSO and diluted with 320 mL water. Sodium citrate, pH 5.5 (22.2 mL, 500 mM) was added and hydrolysis initiated by the addition of α-chymotrypsin (97.9 mg). The reaction was monitored by HPLC, and after 22 h, stopped by the addition of 150 mL of ethanol. The mixture was concentrated under reduced pressure, extracted three times with ethyl acetate and back-extracted with water. The combined aqueous layers were concentrated under reduced pressure, diluted 2-fold with acetonitrile and purified by flash chromatography (85:15 acetonitrile/water). The recovered solid was washed sparingly with cold water to yield 213 mg of L-6-chlorotryptophan, and optical purity assessed by N-α-(2,4-dinitro-5-fluorophenyl)-L-valine amide derivatization69 (63% yield, 94% ee). 1H NMR (400 MHz, CD3OD): δ 7.66 (d, J = 8.5 Hz, 1H), 7.36 (s, 1H), 7.21 (s, 1H), 7.02 (d, J = 8.5 Hz, 1H), 3.83 (t, J = 4.5 Hz, 1H), 3.46 (d, J = 15.3 Hz, 1H), 3.17 (d, J = 9.2 Hz, 1H). Calcd Mass 238.670, obsd 239.00.
5.3. Synthesis of Fmoc-(S)-3-amino-4-(6-chloroindole)butyric acid
The base labile 9-fluorenylmethyloxycarbonyl (Fmoc-) amino protective group was coupled to the free N terminus of L-6-chlorotryptophan according to previously published methods.70,71 The amino acid (25 mmol) was dissolved in water (25 mL) in the presence of 1 equiv of triethylamine. Simultaneously, the Fmoc succinimidyl ester (24 mmol) was dissolved in acetonitrile (25 mL) and gently heated to promote dissolution. The ester solution was added to the amino acid solution in one portion and allowed to stir at room temperature for 30 min. The pH of the solution was maintained in the 8.5–9.0 range by dropwise addition of triethylamine as the pH of the reaction mixture drops sharply initially due to liberation of free N-hydroxysuccinimate. When complete, the solution was concentrated via rotary evaporation and the resulting residue was poured over a solution of hydrochloric acid (1.5 N). A spontaneously crystallized product was separated, washed and set aside. The oil precipitate was isolated by two ethyl acetate extractions followed by a wash with brine. The resulting solution was dried over sodium sulfate and rotary evaporated to dryness. The product was then brought up in acetonitrile and water and dried over vacuum. The yield obtained was 87%. 1H NMR (400 MHz, CDCl3): δ 7.78 (d, 2H), 7.30–7.52 (8H), 7.06 (d, 1H), 6.93 (s, 1H), 5.26 (d,1H), 4.73 (s, 1H), 4.43 (t, 2H), 4.19 (d, 1H), 3.32 (s, 2H), 2.83 (s, 1H). Calcd Mass 461.00, obsd 461.30 and 483.30 (+Na).
The corresponding Fmoc-protected β-amino acid was synthesized using the Arndt-Eistert procedure to yield 45 mg (22%) of Fmoc-(S)-3-amino-4-(6-chloroindole)butyric acid.1 1H NMR (400 MHz, CD3OD): δ 9.29 (d, J = 7.5 Hz, 2H), 9.09 (t, J = 6.6 Hz, 3H), 8.89 (t, J = 7.3 Hz, 3H), 8.79 (dd, J = 8.6 Hz, 2H), 8.58 (s, 1H), 8.47 (d, J = 8.4 Hz, 1H), 5.77 (m, J = 7.2 Hz, 3H), 5.65 (t, J = 6.8 Hz, 1H), 4.46 (dd, J = 3.1 Hz, 2H), 4.02 (t, J = 6.6 Hz, 2H), 2.79 (s, 1H), 2.41 (s, 1H). Calcd Mass 474.7, obsd 497.4 (product + Na+).
5.4. β3-Peptide synthesis, general procedure
β3-Peptides were synthesized manually in a glass peptide-synthesis vessel with fritted glass at the top and bottom and a sidearm for addition of reagents. Peptides were synthesized on a 25 μmol scale using standard Fmoc chemistry with Wang resin and Fmocprotected β3-amino acid monomers.
5.4.1. β3-Peptide synthesis, Wang resin loading
Wang resin (1.0 mmol-OH/g, 380 mg, 0.38 mmol) was swelled in DMF for 30 min in a glass peptide-synthesis vessel. Excess DMF was removed by filtration. Fmoc-β3-hGlu(tBu) (500 mg, 1.14 mmol) was added to 10 mL dry CH2Cl2 at 0 °C and stirred under argon. 1,3-Dicyclohexylcarbodiimide (78.4 mg, 0.38 mmol) was dissolved in 3 mL DMF and added to the solution. After 30 min, the solution was warmed to room temperature and concentrated to a white slurry by rotary evaporation. The slurry was dissolved in 5 mL DMF and added directly to the resin. Dimethyl-aminopyridine (46.5 mg, 0.38 mmol) was added and the mixture was shaken vigorously on a moving tray for 3 h. The reaction mixture was drained and washed with DMF (2 × 30 s). Unreacted amino groups were acetylated by treatment with 6% v/v acetic anhydride and 6% v/v NMM in DMF (30 min). The capped resin was washed with DMF (8 × 30 s), CH2Cl2 (8 × 30 s), and MeOH (4 × 30 s) and dried for 30 min under N2. The extent of resin loading was determined as described.8
5.4.2. β3-Peptide synthesis, microwave procedure
The protocol for the synthesis of β3-peptides was adapted from reports by Murray and Gellman.72 Fmoc-β3-hGlu(tBu)-loaded Wang resin was placed in a glass peptide-synthesis vessel and swelled with DMF for 20 min. A solution of 20% piperidine/DMF was added to the vessel along with a stir bar for agitation, and the vessel was placed in the microwave reactor (CEM MARS) and irradiated (200W maximum power, 70 °C, ramp 2 min, hold 4 min, cool 5 min). The vessel was removed from the microwave reactor, and the deprotected resin washed with DMF (6 × 30 s). The appropriate β3-homoamino acid (3 equiv), PyBOP® (3 equiv), HOBt (3 equiv), and diisopropylethylamine (DIEA, 8 equiv) were dissolved in 1.5 mL DMF and added to the resin. The vessel was again placed in the microwave reactor and irradiated (200W maximum power, 60 °C, ramp 2 min, hold 6 min, cool 5 min). Upon removal from the microwave reactor, the resin was washed with DMF (6 × 30 s), and the above steps were repeated until the β3-peptide sequence was complete. Following removal of the final Fmoc protecting group, the resin was washed with DMF (8 × 30 s) and CH2Cl2 (8 × 30 s), dried for 20 min under N2, and treated for 90 min with a cleavage cocktail of 3% v/v water and 3% v/v triisopropylsilane in TFA. The cleaved peptide was collected and the resin washed with the cleavage cocktail (1 × 30 s). The resulting solution was concentrated by rotary evaporation and reconstituted in CH3CN/H2O (1:1).
5.4.3. Preparation of labeled variants
Peptides were prepared with an N-terminal label by synthesizing the peptide on-resin as described above. For the addition of a β3-hGly linker as in β53-8Al594, the deprotection and coupling steps were repeated as above to add two Fmoc-β3-hGly residues. After removal of the final Fmoc protecting group, the resin was washed with DMF (6 × 30 s), and fluorescein-5-EX succinimidyl ester (5 mg, Invitrogen) was added to the resin in 1.5 mL DMF with 15 μL DIEA. The vessel was placed in the microwave reactor and irradiated (200W maximum power, 60 °C, ramp 2 min, hold 6 min, cool 5 min). The resin was washed with DMF (8 × 30 s) and CH2Cl2 (8 × 30 s), dried for 20 min under N2, and cleaved as described above. Alternatively, Alexa Fluor® 594 carboxylic acid, succinimidyl ester (2 mg, Invitrogen) was used in 1 mL DMF with 10 μL DIEA following the same protocol.
5.4.4. β3-Peptide purification and characterization
Peptides were purified by reverse-phase HPLC. Identity and purity were assessed by analytical HPLC and MALDI-TOF (matrix-assisted laser desorption-ionization time-of-flight) mass spectrometry on a Voyager (Applied Biosystems) MALDI-TOF spectrometer with a 337 nm laser using the α-cyano-4-hydroxycinnaminic acid matrix. Following purification, peptides were lyophilized, stored at −20 °C, and reconstituted just prior to use. Theoretical and observed molecular weights are listed in Table 2.
Table 2.
Theoretical and MALDI-TOF MS-observed molecular weights for β53-12 and β53-13 and labeled variants
| β-Peptide | Formula | Calcd mass |
|
|---|---|---|---|
| (M+H+) | Obsd mass | ||
| β53-12 | C73H117F3N12O15 | 1459.8 | 1458.7 (M+H+), 1480.8 (M+Na+) |
| β53-13 | C74H118ClN13O15 | 1465.3 | 1463.6 (M+H+), 1485.3 (M+Na+) |
| β53-12flu | C98H134F3N13O22S | 1935.3 | 1937.9 (M+H+), 1959.8 (M+Na+) |
| β53-13flu | C99H135ClN14O22S | 1940.7 | 1939.9 (M+H+), 1960.8 (M+Na+) |
| β53-8A594Al | C116H163N17O27S2 | 2277.7 | 2279.9 (M+H+), 2292.6 (M+Na+) |
| β53-12A594Al | C109H151F3N14O25S2 | 2164.5 | 2164.2 (M+H+), 2186.5 (M+Na+) |
5.5. Overexpression of hDMX1-200
BC21 cells containing the pEGX-MDMX(1-200) plasmid were obtained from the lab of Professor Jiandong Chen. The sequence of the human MDMX protein N-terminal domain (residues 1-200) was cloned into the glutathione S-transferase fusion vector to obtain this plasmid. A single colony was used to inoculate a 1 L culture of 2XYT media containing 0.1 mg/mL carbenicillin. The culture was incubated at 37 °C with shaking at 220 rpm until the optical density at 600 nm reached 0.9 absorbance units. After cooling the culture at 15 °C for 30 min, protein expression was induced by addition of isopropyl β-D-thiogalactoside (IPTG) to a final concentration of 0.1 mM. Cells were harvested after 15 h by centrifugation at 5000g for 20 min, resuspended in PBS buffer (0.5 M NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 (pH 7.4), 1 mM EDTA, and 2 mM DTT) and lysed using a French press. Cell debris was pelleted by centrifugation for 20 min at 18,000g and the supernatant was incubated with glutathione sepharose at 4 °C for 8 h to immobilize the fusion protein. The resin was washed with three bed volumes of chilled PBS buffer and two bed volumes of room temperature thrombin cleavage buffer (Novagen, 200 mM Tris–HCl, pH 8.4, 1.5 M NaCl, 25 mM CaCl2 with 2 mM DTT added) before treatment with 16 units biotinylated thrombin for 12–15 h at room temperature to release hDMX1-200 while leaving GST bound to the solid phase. The biotinylated thrombin was removed from hDMX1-200 upon incubation with streptavidin-agarose and subsequent filtration. The resulting solution was characterized by SDS–PAGE and amino acid analysis to verify identity, purity, and protein concentration. Pure protein was distributed into aliquots and stored at −80 °C until use.
5.5.1. Fluorescence polarization assays, direct binding
The synthesis and purification of p53AD, p53ADflu, β53-1, β53-1flu, β53-8, β53-8flu, and βNEG as well as the overexpression and purification of hDM21-188 have been described previously.11,13 All binding experiments were performed in 384-well plates (Corning Life Sciences Plastic non-binding 384-well microplates, Fisher Scientific) with an Analyst™ AD automated fluorescence-plate reader (LJL Bio-Systems, Inc.). Polarization was measured by excitation at 485 nm and subsequent measurement of the fluorescence emission at 530 nm using a 505 dichroic mirror.
Serial dilutions of hDM21-188 were prepared in Tris-buffered saline with detergent (10 mM Tris, 100 mM NaCl, 0.01% Tween-20, pH 7.4) or phosphate-buffered saline (138 mM NaCl, 2 mM KCl, 13 mM Na2HPO4, 1.9 mM KH2PO4) with final concentrations ranging from 0.001 to 15 μM. An aliquot of fluorescently labeled peptide was added to a final concentration of 25 nM (20 μL total volume per well). The binding reaction was incubated for 30 min at room temperature. This was determined to be a sufficient length of time for the binding reaction to reach equilibrium, as there was no change in observed polarization values after 24 h. Measurements from three or more independent sets of samples were averaged for each determination. The average fluorescence polarization values were plotted as a function of hDM2 concentration. The equation used to determine the Kd for each labeled peptide is73
where
F measured average fluorescence polarization
FL fluorescence polarization of free labeled peptide
FLP maximum fluorescence polarization of the peptide–protein complex
Kd equilibrium dissociation constant of the peptide–protein complex
[L]T total concentration of labeled peptide
[P]T total concentration of protein
This assay was performed for p53ADflu, β53-1flu, β53-8flu, β53-12flu, β53-13flu, β53-8A594Al, and β53-12Al.
5.5.2. Fluorescence polarization assays, competition
A solution of 250 nM p53ADflu and 2.5 μM hDM21-188 (15 μL total volume per well) was prepared in Tris-buffered saline or phosphate-buffered saline and incubated for 30 min at room temperature. Serial dilutions of unlabeled peptide were prepared with final concentrations ranging from 12 nM to 200 μM and added to the initial solution (30 μL total volume per well with final concentrations of p53ADflu and hDM21-188 of 25 nM and 1 μM, respectively). After 30 min, the fluorescence polarization of each well was measured as above. Measurements from three or more independent sets of samples were averaged for each determination. The average fluorescence polarization values were plotted as a function of inhibitor concentration. The equation used to determine the IC50 for each labeled peptide is:74
where
F measured average fluorescence polarization
FL fluorescence polarization of free labeled peptide
FLP maximum fluorescence polarization of the peptide–protein complex
[I] concentration of unlabeled peptide
n Hill coefficient
The apparent affinity of the unlabeled peptide for hDM2 (Ki) can be calculated using the equation11
where
[L]T total concentration of labeled peptide
Kd,L measured Kd of L for hDM2
5.6. ELISA competition assay
The protocol for this assay was adapted from methods described by Stoll et al. and Sakurai et al. with all incubations at room temperature.28,47 The p53AD control peptide was synthesized as described previously13 and biotinylated using Biotin Polyethyleneoxide Iodoacetamide (Sigma). A black, Reacti-Bind™ Streptavidin High Binding Capacity Coated 96-well plate pre-blocked with SuperBlock® Blocking Buffer (Pierce) was washed three times with TBS-T (Tris-buffered saline/0.05% Tween 20). To each well was added 50 μL of a 125 nM solution of biotinylated p53AD. The plate was incubated for 1 h and the wells were washed with TBS-T five times. During this time, a solution of 1 μM hDM2 in TBS-T with 2% BSA and 10 mM DTT (15 μL per well) was incubated with varying concentrations of unlabeled peptide inhibitor in TBS-T with final concentrations ranging from 12 nM to 200 μM for 30 min. The solutions of peptide and protein were then distributed into the wells of the plate (30 μL per well) and incubated for 1 h. Each well was washed five times with TBS-T. The anti-MDM2 N20 antibody (Santa Cruz Biotechnology) was added to each well (100 μL per well) at a dilution of 1:250 in TBS-T with 1% BSA. After 1 h incubation on amicrotiter shaker, the wells were washed five times with TBS-T, and 100 μL of anti-rabbit-IgG-HRP (Santa Cruz Biotechnology) diluted 1:1000 in TBS-T with 1% BSA was added to each well. While the plate was incubated for 1 h on a microtiter shaker, the QuantaBlu™ working solution was prepared by mixing 9 parts QuantaBlu™ Substrate Solution to 1 part QuantaBlu™ Stable Peroxide Solution (Pierce) and allowed to equilibrate to room temperature. At the end of the incubation, the wells were washed five times with TBS-T. The QuantaBlu™ working solution (100 μL per well) was added, and the plate was incubated for 1 h. The fluorescence intensity (counts per second, CPS) for each well was measured with an Analyst™ AD automated fluorescence-plate reader (LJL BioSystems, Inc.) by excitation at 325 nm and subsequent measurement of the fluorescence emission at 420 nm using a 50/50 dichroic mirror.
5.7. Flow cytometry
HCT116 cells (American Type Culture Collection, Manassas, VA) were grown in T-75 culture flasks containing McCoy’s 5A media supplemented with 10% fetal bovine serum (Invitrogen) in a humidified environment with 5% CO2 to ~80% confluency. The cells were then washed twice with 37 °C PBS and incubated with 10 mL of 37 °C PBS-based non-enzymatic cell dissociation solution (Chemicon International, Temecula, CA) for 15 min. Cells were centrifuged at 500g, resuspended in media, counted by hemocytometer, and diluted to 2200 cells/μL with media. Aliquots of cells (230 μL) were added to fluorescein-labeled peptides (20 μL, 125 μM in PBS). Cells were incubated with the peptides for 1–4 h at 37 °C and then washed twice with 750 μL 37 °C PBS to remove extracellular peptide. To ensure removal of any surface-bound peptide,51 cells were then incubated with 500 μL 0.25% trypsin at 37 °C for 10 min, washed once with 750 μL 4°C media and once with 750 μL 4 °C PBS. Cells were suspended in 500 μL PBS with 1 μg/mL propidium iodide and analyzed on a BD FACScan (BD Biosciences, San Jose, CA) equipped with a 488 nm Argon laser. A total of 10,000 events were collected monitoring fluorescein and propidium iodide with 530/30 nm bandpass and 650 nm longpass filters, respectively. Events corresponding to cellular debris were removed by gating on forward and side scatter, while dead cells were removed by propidium iodide staining. Geometric means were then calculated from the histogram of fluorescence intensity and corrected for background cellular fluorescence by subtracting the geometric mean of cells treated only with PBS.
5.8. Confocal microscopy
Approximately 105 HCT116 cells were seeded in 2 mL media in 6-well plates containing cover glasses. After allowing the cells to adhere for 48 h, media was removed by aspiration and the cells were washed twice with 37 °C PBS. Inverted cover glasses were floated on 200 μL of media containing 20 μM peptide labeled with fluorescein for 2 h and/or 10 μM 10 kDa dextran labeled with Alexa Fluor® 647 (Invitrogen) for 15 min at 37 °C. Cover glasses were then washed with 37 °C media and PBS and mounted on microscope slides. Cells were imaged on an LSM 510 Meta (Carl Zeiss MicroImaging, Thornwood, NY) using a 488 nm Ar laser line with a 525/25 nm filter or 633 nm HeNe laser line with a 680/30 nm filter for visualizing fluorescein and Alexa Fluor® 647, respectively.
Acknowledgments
We thank Dr. Bert Vogelstein for providing HCT116 −/− cells and Dr. Jiandong Chen for providing the hDMX expression plasmid. This work was supported by the National Institutes of Health (GM 74756) and the National Foundation for Cancer Research.
References and notes
- 1.Seebach D, Overhand M, Kuhnle FNM, Martinoni B, Oberer L, Hommel U, Widmer H. Helv Chim Acta. 1996;79:913. [Google Scholar]
- 2.Appella DH, Christianson LA, Karle IL, Powell DR, Gellman SH. J Am Chem Soc. 1996;118:13071. [Google Scholar]
- 3.Seebach D, Ciceri PE, Overhand M, Jaun B, Rigo D, Oberer L, Hommel U, Amstutz R, Widmer H. Helv Chim Acta. 1996;79:2043. [Google Scholar]
- 4.Cheng RP, Gellman SH, DeGrado WF. Chem Rev. 2001;101:3219. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- 5.Raguse TL, Lai JR, Gellman SH. J Am Chem Soc. 2003;125:5592. doi: 10.1021/ja0341485. [DOI] [PubMed] [Google Scholar]
- 6.Arvidsson PI, Rueping M, Seebach D. Chem Commun. 2001:649. [Google Scholar]
- 7.Cheng RP, DeGrado WF. J Am Chem Soc. 2001;123:5162. doi: 10.1021/ja010438e. [DOI] [PubMed] [Google Scholar]
- 8.Hart SA, Bahadoor ABF, Matthews EE, Qiu XJ, Schepartz A. J Am Chem Soc. 2003;125:4022. doi: 10.1021/ja029868a. [DOI] [PubMed] [Google Scholar]
- 9.Kritzer JA, Hodsdon ME, Schepartz A. J Am Chem Soc. 2005;127:4118. doi: 10.1021/ja042933r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kritzer JA, Stephens OM, Guarracino DA, Reznik SK, Schepartz A. Bioorg Med Chem. 2005;13:11. doi: 10.1016/j.bmc.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kritzer JA, Luedtke NW, Harker EA, Schepartz A. J Am Chem Soc. 2005;127:14584. doi: 10.1021/ja055050o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gademann K, Kimmerlin T, Hoyer D, Seebach D. J Med Chem. 2001;44:2460. doi: 10.1021/jm010816q. [DOI] [PubMed] [Google Scholar]
- 13.Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. J Am Chem Soc. 2004;126:9468. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]
- 14.Stephens OM, Kim S, Welch BD, Hodsdon ME, Kay MS, Schepartz A. J Am Chem Soc. 2005;127:13126. doi: 10.1021/ja053444+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadowsky JD, Fairlie WD, Hadley EB, Lee H-S, Umezawa N, Nikolovska-Coleska Z, Wang S, Huang DCS, Tomita Y, Gellman SH. J Am Chem Soc. 2007;129:139. doi: 10.1021/ja0662523. [DOI] [PubMed] [Google Scholar]
- 16.Frackenpohl J, Arvidsson PI, Schreiber JV, Seebach D. ChemBioChem. 2001;2:445. doi: 10.1002/1439-7633(20010601)2:6<445::aid-cbic445>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 17.Chene P. Nat Rev Cancer. 2003;3:102. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- 18.Zheleva DI, Lane DP, Fischer PM. Mini-Rev Med Chem. 2003;3:257. doi: 10.2174/1389557033488178. [DOI] [PubMed] [Google Scholar]
- 19.Momand J, Jung D, Wilczynski S, Niland J. Nucleic Acids Res. 1998;26:3453. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freedman DA, Wu L, Levine AJ. Cell Mol Life Sci. 1999;55:96. doi: 10.1007/s000180050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 22.Duncan SJ, Cooper MA, Williams DH. Chem Commun. 2003:316. doi: 10.1039/b211889k. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Echeverria C, Chene P, Blommers MJJ, Furet P. J Med Chem. 2000;43:3205. doi: 10.1021/jm990966p. [DOI] [PubMed] [Google Scholar]
- 24.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 25.Fotouhi N, Graves B. Curr Top Med Chem. 2005;5:159. doi: 10.2174/1568026053507705. [DOI] [PubMed] [Google Scholar]
- 26.Vassilev LT. J Med Chem. 2005;48:4491. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]
- 27.Shangary S, et al. Proc Natl Acad Sci USA. 2008;105:3933. [Google Scholar]
- 28.Sakurai K, Chung HS, Kahne D. J Am Chem Soc. 2004;126:16288. doi: 10.1021/ja044883w. [DOI] [PubMed] [Google Scholar]
- 29.Hara T, Durell SR, Myers MC, Appella DH. J Am Chem Soc. 2006;128:1995. doi: 10.1021/ja056344c. [DOI] [PubMed] [Google Scholar]
- 30.Fasan R, Dias RLA, Moehle K, Zerbe O, Obrecht D, Mittl PRE, Grutter MG, Robinson JA. ChemBioChem. 2006;7:515. doi: 10.1002/cbic.200500452. [DOI] [PubMed] [Google Scholar]
- 31.Yin H, Lee G, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD. Angew Chem, Int Ed. 2005;44:2704. doi: 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]
- 32.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. J Am Chem Soc. 2007;129:2456. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C, Liu M, Monbo J, Zou G, Li C, Yuan W, Zella D, Lu W-Y, Lu W. J Am Chem Soc. 2008;130:13546. doi: 10.1021/ja8042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shangary S, Ding K, Qiu S, Nikolovska-Coleska Z, Bauer JA, Liu M, Wang G, Lu Y, McEachern D, Bernard D, Bradford CR, Carey TE, Wang S. Mol Cancer Ther. 2008;7:1533. doi: 10.1158/1535-7163.MCT-08-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Appella DH, Christianson LA, Klein DA, Powell DR, Huang X, Barchi JJJ, Gellman SH. Nature. 1997;387:381. doi: 10.1038/387381a0. [DOI] [PubMed] [Google Scholar]
- 36.Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RCA, van der Houven van Oordt W, Hateboer G, van der Eb AJ, Jochemsen AG. EMBO J. 1996;15:5349. [PMC free article] [PubMed] [Google Scholar]
- 37.Shvarts A, Bazuine M, Dekker P, Ramos YFM, Steegenga WT, Merckx G, van Ham RCA, van der Houven van Oordt W, van der Eb AJ, Jochemsen AG. Genomics. 1997;43:34. doi: 10.1006/geno.1997.4775. [DOI] [PubMed] [Google Scholar]
- 38.Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci PG, Jochemsen AG, Marine J-C. Mol Cell Biol. 2004;24:5835. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toledo F, Wahl GM. Int J Biochem Cell Biol. 2007;39:1476. doi: 10.1016/j.biocel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer PM, Lane DP. Trends Pharmacol Sci. 2004;25:343. doi: 10.1016/j.tips.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Gupta V, Lattanze J, Ramachandren K, Carver TE, Petrella EC, Cummings MD. Bioorg Med Chem Lett. 2005;15:765. doi: 10.1016/j.bmcl.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 42.Parks DJ, et al. Bioorg Med Chem Lett. 2006;16:3310. doi: 10.1016/j.bmcl.2006.03.055. [DOI] [PubMed] [Google Scholar]
- 43.Knight SMG, Umezawa N, Lee H-S, Gellman SH, Kay BK. Anal Biochem. 2002;300:230. doi: 10.1006/abio.2001.5468. [DOI] [PubMed] [Google Scholar]
- 44.Schon O, Friedler A, Bycroft M, Freund SMV, Fersht AR. J Mol Biol. 2002;323:491. doi: 10.1016/s0022-2836(02)00852-5. [DOI] [PubMed] [Google Scholar]
- 45.Lai Z, Auger KR, Manubay CM, Copeland RA. Arch Biochem Biophys. 2000;381:278. doi: 10.1006/abbi.2000.1998. [DOI] [PubMed] [Google Scholar]
- 46.Bottger A, Bottger V, Garcia-Echeverria C, Chene P, Hochkeppel H-K, Sampson W, Ang K, Howard SF, Picksley SM, Lane DP. J Mol Biol. 1997;269:744. doi: 10.1006/jmbi.1997.1078. [DOI] [PubMed] [Google Scholar]
- 47.Stoll R, et al. Biochemistry. 2001;40:336. doi: 10.1021/bi000930v. [DOI] [PubMed] [Google Scholar]
- 48.Yu P, Liu B, Kodadek T. Nat Biotechnol. 2005;23:746. doi: 10.1038/nbt1099. [DOI] [PubMed] [Google Scholar]
- 49.Daniels DS, Schepartz A. J Am Chem Soc. 2007;129:14578. doi: 10.1021/ja0772445. [DOI] [PubMed] [Google Scholar]
- 50.Vives E, Brodin P, Lebleu B. J Biol Chem. 1997;272:16010. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 51.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. J Biol Chem. 2003;278:585. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 52.Tan NC, Yu P, Kwon Y-U, Kodadek T. Bioorg Med Chem. 2008;16:5853. doi: 10.1016/j.bmc.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. J Biol Chem. 2006;281:33030. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 54.Bottger V, Bottger A, Garcia-Echeverria C, Ramos YFM, van der Eb AJ, Jochemsen AG, Lane DP. Oncogene. 1999;18:189. doi: 10.1038/sj.onc.1202281. [DOI] [PubMed] [Google Scholar]
- 55.Hu B, Gilkes DM, Chen J. Cancer Res. 2007;67:8810. doi: 10.1158/0008-5472.CAN-07-1140. [DOI] [PubMed] [Google Scholar]
- 56.Caputo GA, Litvinov RI, Li W, Bennett JS, DeGrado WF, Yin H. Biochemistry. 2008;47:8600. doi: 10.1021/bi800687h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wade M, Wong ET, Tang M, Stommel JM, Wahl GM. J Biol Chem. 2006;281:33036. doi: 10.1074/jbc.M605405200. [DOI] [PubMed] [Google Scholar]
- 58.Popowicz GM, Czarna A, Rothweiler U, Szwagierczak A, Krajewski M, Weber L, Holak TA. Cell Cycle. 2007;6:2386. doi: 10.4161/cc.6.19.4740. [DOI] [PubMed] [Google Scholar]
- 59.Popowicz GM, Czarna A, Holak TA. Cell Cycle. 2008;7:2441. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 60.Cochran AG. Chem Biol. 2000;7:R85. doi: 10.1016/s1074-5521(00)00106-x. [DOI] [PubMed] [Google Scholar]
- 61.Arkin MR, Wells JA. Nat Rev Drug Discovery. 2004;3:301. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 62.Toledo F, Krummel KA, Lee CJ, Liu C-W, Rodewald L-W, Tang M, Wahl GM. Cancer Cell. 2006;9:273. doi: 10.1016/j.ccr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 63.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci USA. 2000;97:13003. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith BA, Daniels DS, Coplin AE, Jordan GE, McGregor LM, Schepartz A. J Am Chem Soc. 2008;130:2948. doi: 10.1021/ja800074v. [DOI] [PubMed] [Google Scholar]
- 65.Rueping M, Mahajan YR, Sauer M, Seebach D. ChemBioChem. 2002;3:257. doi: 10.1002/1439-7633(20020301)3:2/3<257::AID-CBIC257>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 66.Umezawa N, Gelman MA, Haigis MC, Raines RT, Gellman SH. J Am Chem Soc. 2002;124:368. doi: 10.1021/ja017283v. [DOI] [PubMed] [Google Scholar]
- 67.Anantharamaiah GM, Roeske RW. Tetrahedron Lett. 1982;23:3335. [Google Scholar]
- 68.Mzengeza S, Venkatachalam TK, Rajagopal S, Diksic M. Appl Radiat Isot. 1993;44:1167. doi: 10.1016/0969-8043(93)90059-j. [DOI] [PubMed] [Google Scholar]
- 69.Bhushan R, Bruckner H. Amino Acids. 2004;27:231. doi: 10.1007/s00726-004-0118-0. [DOI] [PubMed] [Google Scholar]
- 70.Chang CD, Waki M, Ahmad M, Meienhofer J, Lundell EO, Haug JD. Int J Pept Protein Res. 1980;15:59. doi: 10.1111/j.1399-3011.1980.tb02550.x. [DOI] [PubMed] [Google Scholar]
- 71.Ten Kortenaar PBW, Vandijk BG, Peeters JM, Raaben BJ, Adams P, Tesser GI. Int J Pept Protein Res. 1986;27:398. [Google Scholar]
- 72.Murray JK, Gellman SH. J Comb Chem. 2005;8:58. doi: 10.1021/cc0501099. [DOI] [PubMed] [Google Scholar]
- 73.Heyduk T, Lee JC. Proc Natl Acad Sci USA. 1990;87:1744. doi: 10.1073/pnas.87.5.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wohland T, Friedrich K, Hovius R, Vogel H. Biochemistry. 1999;38:8671. doi: 10.1021/bi990366s. [DOI] [PubMed] [Google Scholar]
- 75.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]