

Abstract
Neural tube defects (NTDs) are some of the most common birth defects observed in humans. The incidence of NTDs can be reduced by peri-conceptional folic acid supplementation alone and reduced even further by supplementation with folic acid plus a multivitamin. Here, we present evidence that iron maybe an important nutrient necessary for normal development of the neural tube. Following implantation of the mouse embryo, ferroportin 1 (Fpn1) is essential for the transport of iron from the mother to the fetus and is expressed in the visceral endoderm, yolk sac and placenta. The flatiron (ffe) mutant mouse line harbors a hypomorphic mutation in Fpn1 and we have created an allelic series of Fpn1 mutations that result in graded developmental defects. A null mutation in the Fpn1 gene is embryonic lethal before gastrulation, hypomorphic Fpn1ffe/ffe mutants exhibit NTDs consisting of exencephaly, spina bifida and forebrain truncations, while Fpn1ffe/KI mutants exhibit even more severe NTDs. We show that Fpn1 is not required in the embryo proper but rather in the extra-embryonic visceral endoderm. Our data indicate that loss of Fpn1 results in abnormal morphogenesis of the anterior visceral endoderm (AVE). Defects in the development of the forebrain in Fpn1 mutants are compounded by defects in multiple signaling centers required for maintenance of the forebrain, including the anterior definitive endoderm (ADE), anterior mesendoderm (AME) and anterior neural ridge (ANR). Finally, we demonstrate that this loss of forebrain maintenance is due in part to the iron deficiency that results from the absence of fully functional Fpn1.
Keywords: Neural tube defect, Iron deficiency, Forebrain patterning, Mouse, Sl40a1
INTRODUCTION
Neural tube defects (NTDs) affect ~1 out of 1000 live births (for a review, see Detrait et al., 2005). The causes of NTDs are complex, with both genetic and environmental factors influencing its penetrance (for a review, see Zohn and Sarkar, 2008). Epidemiological studies have implicated peri-conception maternal nutrition as an important factor influencing the occurrence of NTDs (for a review, see Blom et al., 2006). From these studies, folic acid has emerged as an important nutrient for normal development, and experimental data in both human and animal models demonstrate that folic acid fortification can reduce the incidence of NTDs. As a result of these findings, many countries now fortify the food supply with folic acid and observe significant reductions in the incidence of NTDs in their populations (Eichholzer et al., 2006). Other nutrients, such as iron, have also emerged from epidemiological studies as nutritional factors that are likely to influence the penetrance of NTDs (Felkner et al., 2005; Groenen et al., 2004) (for a review, see Czeizel, 2009); however, experimental evidence demonstrating a clear role for iron in the prevention of NTDs is lacking.
Iron plays essential roles in development and growth by participating in electron transport, DNA synthesis, oxygen transport and reduction-oxidation reactions. It also acts as a co-factor for many enzymes and regulates translation by binding to iron-responsive elements. Iron levels are tightly regulated, as either iron deficiency or iron overload results in diverse pathological conditions (for a review, see Andrews, 1999). Unlike with folic acid, controlled studies in humans to investigate the role of iron in suppressing NTDs have not been carried out. Additionally, mouse models with disruption of iron homeostasis either suffer from early embryonic lethality or exhibit no observable phenotype, presumably owing to functional redundancy, thus precluding the investigation of this nutrient in the etiology of NTDs (for a review, see De Domenico et al., 2008).
In the mouse embryo, neural tube closure begins around embryonic day 8.5 (E8.5) and is complete by E10.5 when the posterior neuropore finally closes. During this time and until the establishment of the placenta between E9.5 and E10.5, the visceral endoderm (VE) and yolk sack (YS) provide for the nutritional support (including the uptake of iron) and exchange of waste products between the maternal circulation and the developing embryo (for a review, see Bielinska et al., 1999). Following implantation, the conceptus or egg cylinder is shaped like an elongated cup (see Fig. 10). The distal half (from the implantation site) of the egg cylinder is the epiblast, which will develop into the embryo proper; and the proximal half (the extra-embryonic region) will develop into the placenta. The entire egg cylinder is encased within the VE, a polarized epithelial layer of cells morphologically and functionally similar to gut endoderm. The apical surface of the VE has microvilli to allow absorption of nutrients, including iron, from the maternal environment. These nutrients are then transferred to the epiblast on the basal side of the VE. As gastrulation proceeds, the VE is displaced from the distal half of the epiblast as the definitive endoderm (DE) emerges from the primitive streak. At this time the VE forms one of the two cell layers that make up the YS, which forms as the extra-embryonic mesoderm migrates beneath the VE. At the end of gastrulation, the YS assumes the role of nutrient and waste exchange for the embryo until the placenta begins to take over starting around E9.5.
Fig. 10.

Model for the role of Fpn1 in anterior neural development. In wild-type embryos (top), multiple transporters (pink) are involved in the uptake of iron (yellow) from the maternal environment into the VE (blue). In the VE, ferritin (green) binds to free iron to buffer its oxidative potential. Fpn1 (red) is the only exporter capable of transporting iron out of the VE and to the developing epiblast. Mutation of Fpn1 (bottom) would result in accumulation of iron in the VE and iron deficiency in the epiblast owing to reduced transport of iron to the developing embryo. In wild-type embryos (top), by E5.5 the DVE (pink) is induced in the distal region of the egg cylinder. By E6.5, the AVE migrates to the anterior region of the embryo where it participates in induction of the anterior neuroectoderm (green). By E7.5, the VE has been replaced by definitive endoderm (white) and the prospective forebrain (green) begins to express markers of anterior neural identity such as Six3. By E8.5 the neural domain is further stabilized and refined. In Fpn1 mutants (bottom), the AVE is diffusely localized to the anterior region of the embryo resulting in the diffuse and ectopic induction of tissue of anterior neural character at E7.5. However, this anterior neural tissue is not maintained, because by E8.5 the expression of markers of the forebrain are disproportionately reduced. Between E7.5 and E8.5, iron that is transported from the VE to the embryo is required to maintain forebrain development.
In addition to its role in supporting the nutritional requirements of the embryo, the VE provides patterning information to establish the anterior-posterior axis of the fetus. The VE in the posterior region of the embryo is required for induction of the hematopoietic lineage (Belaoussoff et al., 1998). The amnionless gene is required in the VE for development of the non-midline trunk derivatives of the primitive streak, including the lateral plate, intermediate and paraxial mesoderm (Tomihara-Newberger et al., 1998). Perhaps the most well-characterized signaling center in the VE is the anterior visceral endoderm (AVE), which is required for induction of the head and positioning of the primitive streak at the posterior end of the embryo (for a review, see Martinez-Barbera and Beddington, 2001; Srinivas, 2006). By E5.5, reciprocal interactions between the epiblast and the VE induce the formation of a specialized group of VE cells at the distal tip of the egg cylinder called the distal visceral endoderm (DVE). Following its induction, the DVE migrates to one side of the egg cylinder, forming the AVE and defining the anterior pole of the embryo. The DVE and AVE express a panel of transcription and secreted factors, including Hex1, Lim1 and Cerl, which are required for the proper specification, migration and signaling activities of this organizing center.
Following gastrulation, several signaling centers in the embryo proper maintain and refine the anterior-posterior pattern of the neuroectoderm (for a review, see Martinez-Barbera and Beddington, 2001). Signals from the AVE induce an unstable pre-forebrain, which is stabilized by interaction with the anterior definitive endoderm (ADE) and anterior mesendoderm (AME). Both the ADE and AME promote neuralization and protect the anterior neural tissue from caudalizing signals secreted from the node. For example, Hex1 is expressed in the ADE and deletion of Hex1 in the epiblast results in forebrain truncations (Martinez Barbera et al., 2000). Similarly, Foxa2 is expressed in the AME underlying the prospective forebrain and, in its absence, the forebrain is reduced (Hallonet et al., 2002). Another important signaling center for stabilizing and refining the anterior forebrain is the anterior neural ridge (ANR) at the rostral tip of the neural plate (for a review, see Martinez-Barbera and Beddington, 2001; Rubenstein et al., 1998). Fgf8 is expressed in the ANR where it is essential for the induction of Foxg1, a telencephalic marker expressed beginning around E8.5 (Shimamura and Rubenstein, 1997).
In an ENU mutagenesis screen to identify genes required for neural tube closure, we isolated the flatiron (ffe) mouse mutant line that carries a point mutation in the Scl40a1 gene encoding the iron transporter ferroportin 1 (Fpn1) (Zohn et al., 2007). Fpn1 is the only iron exporter in eukaryotes and is expressed in the VE, placenta and intestine, where it plays an essential role in the absorption of iron (Donovan et al., 2000; Donovan et al., 2005). The ffe mutation results in altered cell surface localization of the transporter and reduces its ability to transport iron out of the cell (Zohn et al., 2007). Our previous study demonstrated that the ffe mouse provides a model for the iron overload disorder Hemochromatosis Type IV in heterozygous animals (Zohn et al., 2007). Here, we characterize the neural tube closure and forebrain patterning defects observed in homozygous mutant embryos. We demonstrate that Fpn1 is required in the extra-embryonic lineage for normal development of the neural tube. NTDs in Fpn1 mutants are due to both a cell-autonomous requirement for Fpn1 in the VE, presumably to prevent the accumulation of iron in these cells, and a non-autonomous requirement for Fpn1 in the establishment of signaling centers in the epiblast that refine and maintain the initial anterior-posterior pattern of the neural tube. Significantly, our data indicate that this cell non-autonomous role for Fpn1 is probably related to its iron transport activity and to the severe iron deficiency that is predicted to result in the embryo in its absence.
MATERIALS AND METHODS
Mouse strains and genotyping
The Fpn1ffe mouse line was generated by ENU mutagenesis and genotyped as described (Zohn et al., 2007). MORE- (Tallquist and Soriano, 2000), Sox2-Cre (Hayashi et al., 2002) and BAT-Gal (Maretto et al., 2003) transgenic lines were purchased from Jackson Laboratories and genotyped as indicated. The Ttr-Cre transgenic line was generously provided by Dr Anna-Katerina Hadjantonakis and genotyped as described (Kwon and Hadjantonakis, 2009; Kwon et al., 2008). The Fpn1KI mouse line was generated by Deltagen, purchased from Jackson Laboratories and genotyped as indicated (http://jaxmice.jax.org/strain/005840.html). For conditional deletion experiments, the Fpn1flox allele was generously provided by Dr Nancy Andrews and genotyped as described (Donovan et al., 2005). All mouse lines were crossed onto the C3H/HeJ genetic background for 2-5 generations before crossing with the Fpn1ffe allele.
Embryo culture with iron chelators
Embryos were dissected from deciduas at E7.5 and cultured by standard techniques (Nagy et al., 2003). Briefly, embryo dissections were carried out in culture media (DMEM (#11054-020, GIBCO) supplemented with 1× Pen/strep (#15070-063, GIBCO), Glutamax (#350505-061, GIBCO) and HEPES (#15620-080, GIBCO). Embryos were dissected with the YS intact and the ectoplacental cone still attached to uterine tissue. Following dissection, embryos were transferred to roller bottles containing complete culture media [culture media supplemented with 50% whole embryo culture rat serum (BT-4520, Harlan)] that was spiked with either water (10 μl, vehicle control) or 1,2 Dimethyl-3-hydroxy-4-pyridone (200 μM; ACROS #30652-11-0). Prior to the addition of embryos, the complete culture media was equilibrated in a roller culture apparatus (BTC engineering) at 37°C with a constant flow of gas (90% N2, 5% O2 and 5% CO2) for at least 2 hours. Embryos were cultured in the roller culture apparatus at 37°C with a constant flow of gas for ~36 hours until they developed to E8.5.
Analysis of mutant phenotypes
Whole-mount RNA in situs were performed as described (Zohn et al., 2006) using the following probes: Foxg1 (Tao and Lai, 1992), Pax6 (Grindley et al., 1995), Six3 (Oliver et al., 1995), Wnt1 (Parr et al., 1993), Otx2 (Ang et al., 1994), Gbx2 (Bouillet et al., 1995), Cerl (Belo et al., 1997), Lim1 (Barnes et al., 1994), Fgf8 (Crossley and Martin, 1995), Shh (Echelard et al., 1993) and Krox20 (Wilkinson et al., 1989). Fpn1 transcript expression was determined using an antisense RNA probe synthesized from IMAGE clone: 2647365 or by expression of the lacZ transcript in Fpn1KI/+ embryos. All in situs were performed on at least two mutant and two control embryos, with similar results unless otherwise indicated in the figure legend.
RESULTS
Hypomorphic Fpnffe/ffe mutant embryos exhibit NTDs
We have previously isolated the flatiron mutation in ferroportin 1 (Fpnffe) from an ENU mutagenesis screen to identify genes required for neural tube closure (Zohn et al., 2005; Zohn et al., 2007). Fpnffe/ffe mutants exhibit NTDs on a C3H/HeJ genetic background, including exencephaly and spina bifida, as well as developmental delays, microphthalmia and severe iron deficiency (Fig. 1; data not shown). In addition, the size of the forebrain in Fpnffe/ffe mutants appeared smaller than that of wild-type controls, an observation confirmed by examination of the expression of molecular markers of the forebrain (Fig. 2). At E9.5, Foxg1 is expressed in the telencephalon of wild-type embryos and its expression is reduced in Fpnffe/ffe mutants. Pax6 is also reduced in the telencephalon and less so in the diencephalon of Fpnffe/ffe mutants. Reductions in telencephalic marker expression are evident by E8.5, as shown by decreased Six3 expression in Fpnffe/ffe mutants when compared with sibling controls. Fpnffe/ffe mutants also exhibit a mild holoprosencephaly evident at E9.5 by the BAT-Gal reporter that marks a narrower Wnt-activation domain in the dorsal midline of a narrower telencephalon in Fpnffe/ffe mutants. In contrast to the anterior forebrain defects, the expression domains of Wnt1, Otx2 and Gbx2 indicate that the midbrain/hindbrain boundary is properly positioned along the anterior-posterior axis of the embryo (Fig. 2).
Fig. 1.
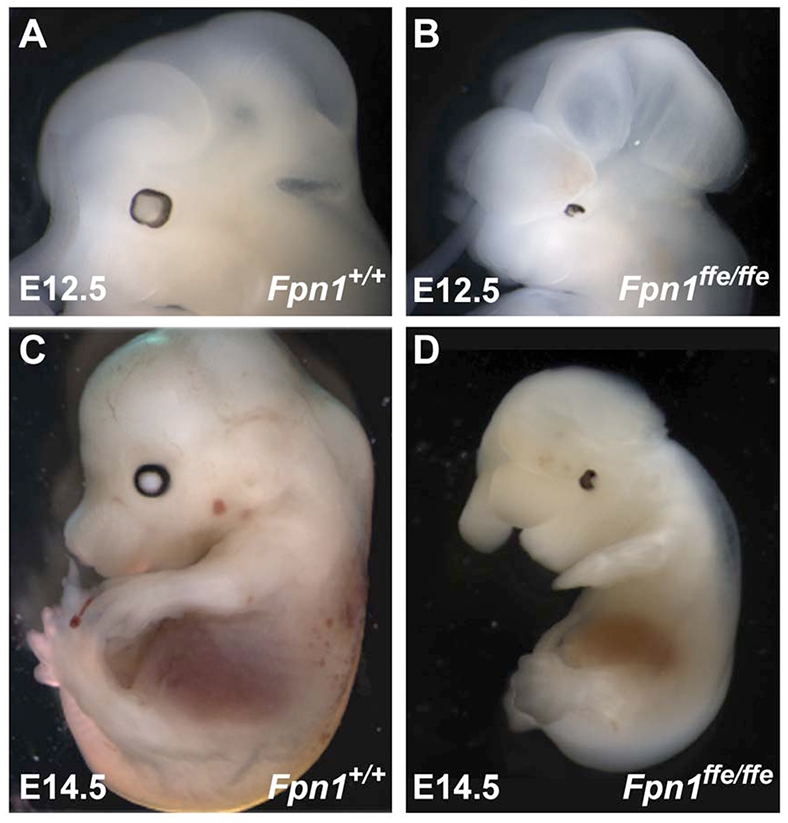
Fpn1ffe/ffe mutant embryos exhibit NTDs. (A-D) Lateral view of wild-type (A,C) and Fpn1ffe/ffe mutant (B,D) embryos at E12.5 (A,B) and E14.5 (C,D). Mutant embryos are developmentally delayed, and exhibit exencephaly, microphthalmia and generalized edema. Lateral views of embryos with anterior towards the left.
Fig. 2.

Reduction in the expression of molecular markers of the forebrain in Fpn1ffe/ffe mutants. (A-F) Expression of molecular markers of the forebrain Foxg1 (A,B), Pax6 (C,D) and Six3 (E,F) at E9.5 (A-D) and E8.5 (E,F) in wild-type (A,C,E) and Fpn1ffe/ffe mutant (B,D,F) embryos. At E9.5, expression of Foxg1 and Pax6 in the telencephalon (T) is reduced and Pax6 expression in the diencephalon (D) is reduced to a lesser extent in Fpn1 mutants compared with sibling controls. (E,F) The reduction in Six3 expression in the telencephalon is evident by E8.5 (n=4). (G,H) Fpn1 mutants exhibit a mild holoprosencephaly evident by a narrower domain of BAT-Gal reporter activation in the dorsal telencephalon of a mutant (H) when compared with wild-type embryo (G) at E9.5. (I-N) Expression of molecular markers of the midbrain-hindbrain boundary (arrows) Wnt1 (I,J), Otx2 (K,L) and Gbx2 (M,N) at E9.5 in wild-type (I,K,M) and Fpn1ffe/ffe mutant (J,L,N) embryos. (A-D,I-N) Lateral views with anterior towards the left. (E-H) Frontal views.
An Fpn1 null allele fails to complement the Fpn1ffe mutation
Using a positional cloning strategy, the ffe phenotype was mapped to a 2.6 Mb interval on mouse chromosome 1 that contains 16 transcripts, including the Slc40a1 gene encoding the Fpn1 protein (Zohn et al., 2007). Our previous studies identified a point mutation in the Fpn1 gene in ffe mutants that changes a conserved histidine to an arginine (H32R), and demonstrated that this amino acid substitution results in a reduction in the iron export activity of Fpn1 (Zohn et al., 2007). However, the involvement of the Fpn1 gene in neural tube closure and forebrain patterning was unexpected. As the possibility remained that NTD phenotypes were due to mutation of another gene in this 2.6 Mb interval, we obtained a targeted β-galactosidase knock-in allele of Fpn1 that expresses lacZ from the Fpn1 locus (Fpn1KI) and used it in a complementation test cross. Although a null allele of Fpn1 is embryonic lethal before neural tube closure (Donovan et al., 2005), transheterozygous Fpn1ffe/KI mutant embryos exhibit NTDs, which were more severe than in Fpn1ffe/ffe mutants (Fig. 3). Whereas Fpn1ffe/ffe mutants primarily exhibit exencephaly and, rarely, spina bifida, Fpn1ffe/KI mutant embryos consistently exhibit both. In addition, in many embryos the neural tube failed to close along the entire anterior/posterior neural axis, resulting in a condition known as craniorachischisis (Fig. 3G). Furthermore, Fpn1ffe/KI mutants are more significantly developmentally delayed when compared with wild-type or Fpn1ffe/ffe mutants. For example, by E9.5, Fpn1ffe/KI mutants fail to turn and are markedly smaller than littermate controls (Fig. 3E,F). Forebrain truncations in Fpn1ffe/KI mutants are also more pronounced than in wild-type or Fpn1ffe/ffe mutants (Figs 3 and 4). Expression of telencephalic specific markers Foxg1 and Six3 at E9.5 and E8.5, respectively, are significantly reduced when compared with wild type or even Fpn1ffe/ffe mutants (compare Fig. 2 with Fig. 4). Similar to Fpn1ffe/ffe mutants, positioning of the midbrain-hindbrain boundary, which is marked by Gbx2 expression, is relatively normal in Fpn1ffe/KI mutants. These results indicate that Fpn1 is required for neural tube closure along the entire anterior-posterior axis and that development of the forebrain is most sensitive to Fpn1 loss.
Fig. 3.

An FpnKI allele fails to complement the Fpn1ffe mutation. (A-H) Lateral view of wild-type (A,C,E), Fpn1ffe/ffe (B) and Fpn1ffe/KI mutant (D,F-H) embryos at E10.5 (A-D,G,H) and E9.5 (E,F). Fpn1ffe/KI mutants are developmentally delayed and exhibit severe NTDs that consists of exencephaly (between arrowheads in B), spina bifida and craniorachischisis. (A-F) Lateral views with anterior towards the left. (G) Dorsal view. (H) Frontal view.
Fig. 4.
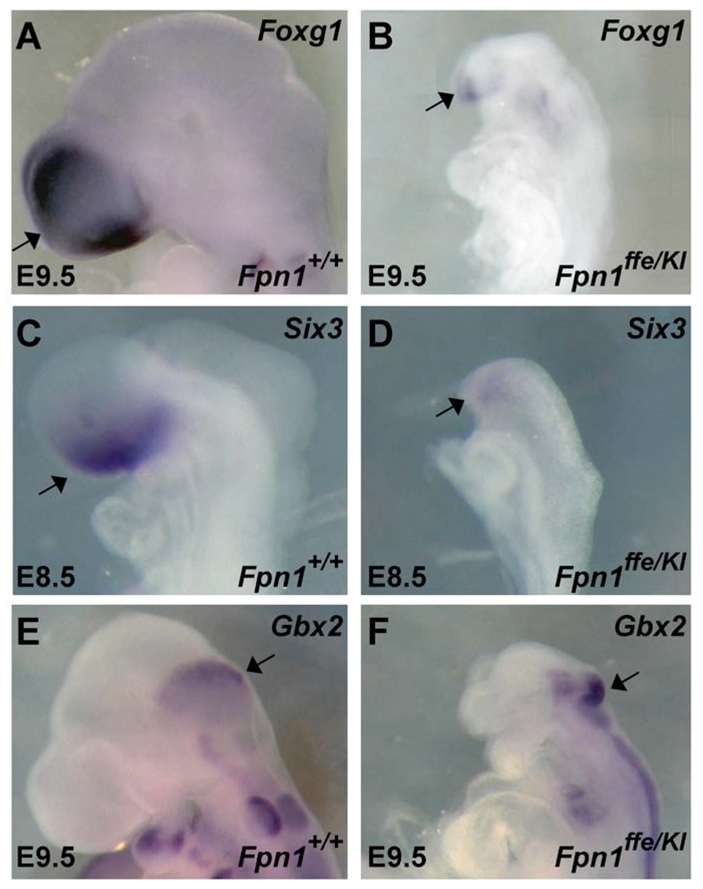
Severe reduction in the expression of molecular markers of the telencephalon in Fpn1ffe/KI mutants. (A-D) Expression of the telencephalic markers (arrows) Foxg1 at E9.5 (A,B) and Six3 at E8.5 (C,D) are reduced in Fpn1ffe/KI mutants (B,D) when compared with wild-type controls (A,C). (E,F) Expression of the midbrain-hindbrain boundary marker Gbx2 (arrows) is relatively normal in wild-type (E) versus mutant (F) embryos at E9.5. Lateral views of embryos with anterior towards the left.
Fpn1 is required in the visceral endoderm for development of the neural tube
Previous studies indicated that Fpn1 is expressed in the syncytiotrophoblast cells of the placenta and visceral endoderm at E7.5 (Donovan et al., 2000; Donovan et al., 2005), but a comprehensive analysis of Fpn1 expression has not been carried out during neurulation. Therefore, we analyzed Fpn1 expression at E6.5, 7.5, 8.5 and 9.5 of mouse development by detecting expression of lacZ in the FpnKI/+ embryos (Fig. 5). Expression of Fpn1 at E6.5 is restricted to the VE surrounding the developing epiblast. At E7.5, as the VE is displaced by the DE, Fpn1-driven lacZ expression remains exclusively in the VE-derived cell layer of the YS surrounding the middle region of the egg cylinder. By E8.5 and E9.5, strong staining is detected in the YS and weak staining in ventral regions of the embryo proper (Fig. 5C,D). Similar results were obtained when an in situ probe against Fpn1 was used in wild-type embryos (Fig. 5E,F). Sectioning of YS stained for Fpn1-lacZ expression reveals expression exclusively in the cell layer derived from the VE not the extra-embryonic mesoderm. As the major sites of Fpn1 expression were not detected in the embryo proper, these results suggest that NTDs in Fpn1 mutants are probably due to a cell non-autonomous role for Fpn1 in the VE.
Fig. 5.

Fpn1 is expressed in the visceral endoderm during gastrulation and neurulation. (A-F) Expression of lacZ-Fpn1 in Fpn1KI/+ embryos (A-D) or Fpn1 in wild-type embryos (E,F) at E6.5 (A), E7.5 (B,E), E8.5 (C,F) and E9.5 (D) determined by in situ hybridization analysis. Inset shows sectioned embryos, demonstrating expression in the VE lineages. VE, visceral endoderm; M, mesoderm; E, ectoderm. Lateral views of embryos with anterior towards the left.
To investigate the hypothesis that Fpn1 is required in the VE lineage for neural tube closure, we performed conditional gene ablation experiments using Cre-deleter lines to remove Fpn1 in either the epiblast (MORE- and Sox2-Cre) or visceral endoderm (Ttr-Cre) lineages. Previous studies have demonstrated that deletion of Fpn1 using the MORE-Cre transgenic deleter strain on a mixed B6;129 genetic background resulted in the birth of morphologically normal embryos (Donovan et al., 2005). These Fpn1flox/flox;MORE-Cre animals die within a couple of weeks of birth owing to severe iron deficiency that results from an inability to absorb iron through intestinal enterocytes. However, our previous studies suggest that the penetrance of NTDs in Fpn1ffe mutants depends on the genetic background (Zohn et al., 2007), so the possibility remained that the lack of a NTDs is due to genetic background effects. In addition, as Cre-mediated recombination by the MORE-Cre driver occurs in a mosaic pattern (Hayashi et al., 2002; Tallquist and Soriano, 2000), NTDs were potentially not observed because of incomplete Cre-mediated recombination of the Fpn1flox allele. To investigate these possibilities, we performed the conditional deletion experiments on the C3H background using the Sox2-Cre transgenic deleter strain that allows complete recombination in the same cell types as MORE-Cre (Hayashi et al., 2002; Tallquist and Soriano, 2000). Deletion of Fpn1 with either the MORE- or Sox2-Cre drivers resulted in embryos without NTDs (Fig. 6). Furthermore, identical results were obtained when Fpn1ffe/flox or Fpn1flox/KI alleles were used in this experiment (data not shown). Fpn1ffe/flox;Sox2-Cre and Fpn1flox/KI;Sox2-Cre embryos were smaller than littermate controls (Fig. 6D,E) probably owing to a role for Fpn1 in the epiblast lineages. Nevertheless, as these smaller embryos did not exhibit NTDs, these experiments suggest that Fpn1 is required in the VE and not the epiblast during neural tube closure. To further test the idea that Fpn1 is required in the VE for neural tube closure, the Ttr-Cre transgenic deleter strain was used to inactivate Fpn1 in the VE lineage. Ttr-Cre is expressed in a mosaic fashion in the VE by pre-primitive streak stages (Kwon and Hadjantonakis, 2009). Significantly, Fpn1flox/KI;Ttr-Cre embryos exhibited exencephaly and forebrain truncations similar to that observed in hypomorphic Fpn1ffe/ffe mutants (Fig. 6G-J). These results confirm that Fpn1 is required cell non-autonomously in the VE lineage for neural tube closure and forebrain patterning.
Fig. 6.

Fpn1 is required in the extra-embryonic lineage. (A,F) Schematics illustrating the lineage-restricted expression of (A) epiblast-specific Cre-recombinase (MORE-Cre or Sox2-Cre) when compared with (F) VE-specific Cre-recombinase (Ttr-Cre). (B-E) Conditional deletion of Fpn1 using either MORE-Cre (B,C) or Sox2-Cre (D,E) drivers in the epiblast lineage results in morphologically normal embryos in Fpn1ffe/flox;MORE-Cre (not shown), Fpnflox/KI;MORE-Cre (C), Fpn1ffe/flox;Sox2-Cre (not shown) and Fpnflox/KI;Sox2-Cre (E) embryos at E10.5 compared with sibling controls (B,D). Fpn1ffe/flox;Sox2-Cre embryos are smaller than wild-type controls. (G-J) By contrast, conditional deletion of Fpn1 with the VE-specific Ttr-Cre (H,J) resulted in exencephaly (between arrowheads in H) in Fpn1flox/KI;Ttr-Cre embryo (H; n=6) at E10.5 and reduction in Six3 expression at E8.5 compared with sibling control embryo (G,I). Lateral views of embryos with anterior towards the left.
Fpn1 is required for proper morphogenesis of the AVE
Fpn1 is expressed on the basal surface of the VE, where it is required for transport of iron out of these cells to the epiblast (Donovan et al., 2005). Mutation of Fpn1 would result in two likely outcomes that could explain NTDs in Fpn1 mutants (see Fig. 10): (1) iron would accumulate in the visceral endoderm, possibly disrupting signaling centers in the VE that pattern the embryo; and/or (2) iron is not transported to the embryo resulting in severe iron deficiency. As Fpn1 mutants exhibit anterior truncations and the AVE is required for induction of this anterior tissue, we investigated whether forebrain truncations are due to alterations in AVE formation or function by examining expression of molecular markers of the AVE in Fpn1ffe/KI mutants. As shown in Fig. 7, the AVE appears to be properly specified in Fpn1 mutants, as evident by the expression of Cerl in the AVE at E6.5; however, the expression domain of Cerl appears diffusely positioned in the anterior region of mutant embryos. These results indicate that morphogenesis of the AVE in Fpn1 mutants is abnormal, possibly owing to the accumulation of iron that is predicted to occur in the VE in the absence of Fpn1 exporter activity.
Fig. 7.

Diffuse and ectopic AVE and neural induction in Fpn1ffe/KI mutants. (A-D) Expression of Cerl in E6.5 wild-type (A,C) and Fpn1ffe/KI mutant (B,D) embryos shows ectopic (asterisk) and diffuse localization of the AVE in the anterior region in Fpnflox/KI mutant embryos (n=3). (E-H) Ectopic expression of Six3 immediately following its induction at E7.5 in Fpn1ffe/KI (F,H, indicated asterisks) when compared with wild-type (E,G) embryos (n=6). (A,B,E,F) Lateral views of embryo with anterior towards the left. (C,D,G,H) Frontal views: G and H show the same embryos as in E and F, respectively.
Secretion of factors from the AVE is required for induction of the most anterior neural tissue (for a review, see Martinez-Barbera and Beddington, 2001; Srinivas, 2006). To determine whether the mispositioned AVE can induce the anterior neural plate, expression of Six3 was examined at early neural plate stages when its expression can first be detected. As shown in Fig. 7, expression of Six3 is restricted to the most anterior aspect of the neural plate in wild-type embryos, whereas, in Fpn1 mutants, in addition to a reduced intensity of Six3 expression in the prospective forebrain, Six3 expression is induced more caudally in mutant embryos. These results indicate that both the properly and improperly localized AVE cells in Fpn1 mutants are capable of inducing anterior neural identity. Although the expression of Six3 in its normal domain is slightly reduced in Fpn1 mutants when compared with wild-type controls at this stage, by E8.5 expression of Six3 in the telencephalon is disproportionately reduced (Fig. 4C,D), suggesting that maintenance of this pre-forebrain is compromised in Fpn1 mutants. Furthermore, the ectopically induced anterior neural tissue failed to be maintained as ectopic Six3 expression was not observed caudally at E8.5 in Fpn1 mutants.
Following the initial induction of the anterior neural tissue by the AVE, additional epiblast-derived signaling centers maintain and refine this pattern (for a review, see Martinez-Barbera and Beddington, 2001). To determine whether deletion of Fpn1 results in cell non-autonomous effects on the specification and or activity of these signaling centers, the expression of molecular markers of the anterior definitive endoderm (ADE; Cerl), anterior neural ridge (ANR; Fgf8) and the anterior mesoendoderm (AME; Shh) were examined at the appropriate stages. As shown in Fig. 8, while expression of Cerl appears equivalent in the AVE of wild-type and mutant embryos at E7.5, expression in the ADE is reduced. Expression of Shh and its downstream target Ptch1 in the AME at E8.25 reveals that this organizing center does not extend to the anterior limit of the neural plate in mutant embryos (Fig. 8C,D and data not shown). This loss of Shh signaling in the forebrain region is consistent with the holoprosencephaly observed in Fpn1 mutants (Fig. 2G,H). Similarly, expression of Fgf8 at E8.5 marks a much smaller ANR in Fpn1 mutants (Fig. 8E,F). Together, these results indicate that in addition to abnormal induction of the anterior neural domain by the AVE, forebrain defects in Fpn1 mutants are compounded by defects in the maintenance and refinement of this already smaller pre-forebrain owing to defects in the ADE, AME and ANR signaling.
Fig. 8.
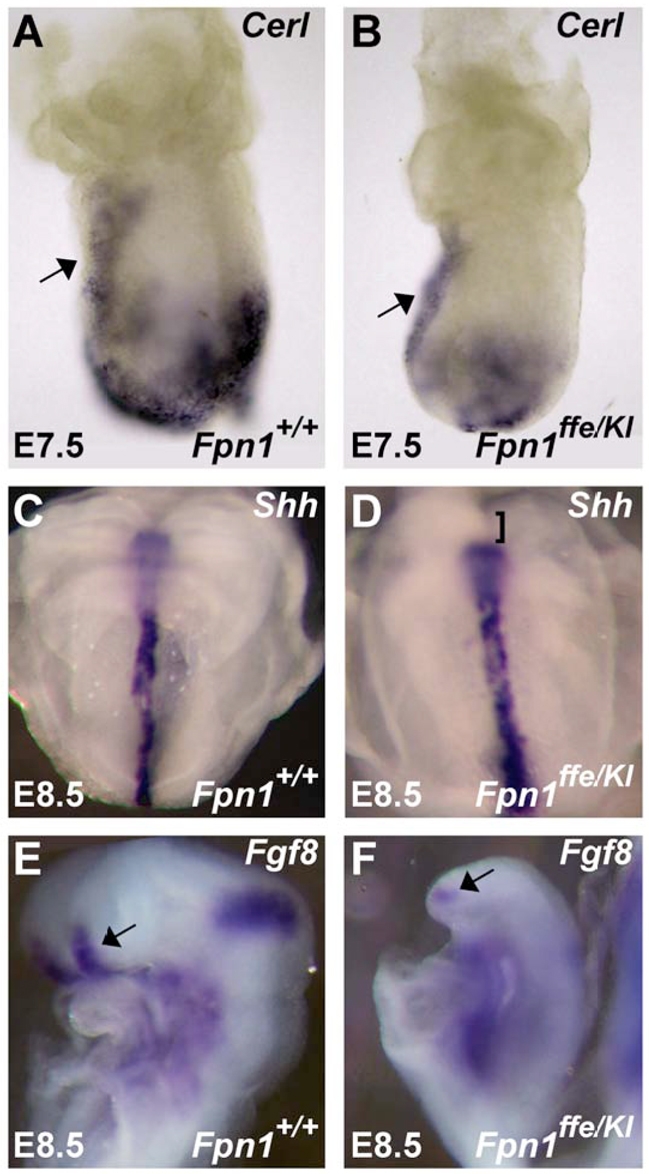
Fpn1ffe/KI mutants exhibit defects in multiple signaling centers within the epiblast that maintain and refine the anterior forebrain. (A,B) Expression of Cerl is reduced in the anterior definitive endoderm (ADE) but not the AVE (arrow) of Fpn1ffe/KI mutants at E7.5 (n=3). (C,D) Expression of Shh does not extend to the anterior limit of the neural plate in Fpn1ffe/KI mutant (bracket; D) at E8.5 when compared with wild-type embryo (C). (E,F) Fgf8 expression in the anterior neural ridge (ANR) is also reduced at E8.5 in Fpn1ffe/KI mutants (arrows). (A,B,E,F) Lateral views of embryos with anterior towards the left. (C,D) Frontal view.
Forebrain defects in Fpn1 mutants are partially due to iron deficiency
Although the cell-autonomous effect of Fpn1 loss on AVE function may be due to the accumulation of iron in the VE that leads to their altered migration, we reasoned that the cell non-autonomous effect of Fpn1 loss on ADE, AME and ANR function and the resulting failure to maintain the anterior neural tissue was probably due to a reduction in iron transported to these cells. To test the hypothesis that iron deficiency can result in loss of anterior neural character, wild-type embryos were cultured from E7.5 to E8.5 in the presence or absence of an iron chelator (200 μM; 1,2-Dimethyl-3-hydroxy-4-pyridone) and subjected to in situ hybridization analysis to examine expression of the prospective telencephalic marker Six3. As shown in Fig. 9, culture of wild-type embryos in water (vehicle control) did not affect expression of Six3 in the telencephalon. Strikingly, culture of wild-type embryos in the presence of the iron chelator during this crucial period phenocopies the forebrain truncation observed in Fpn1ffe/ffe mutants. These results suggest that forebrain truncations in Fpn1 mutants are due, at least in part, to the reduced transport of iron across the VE to the developing embryo.
Fig. 9.
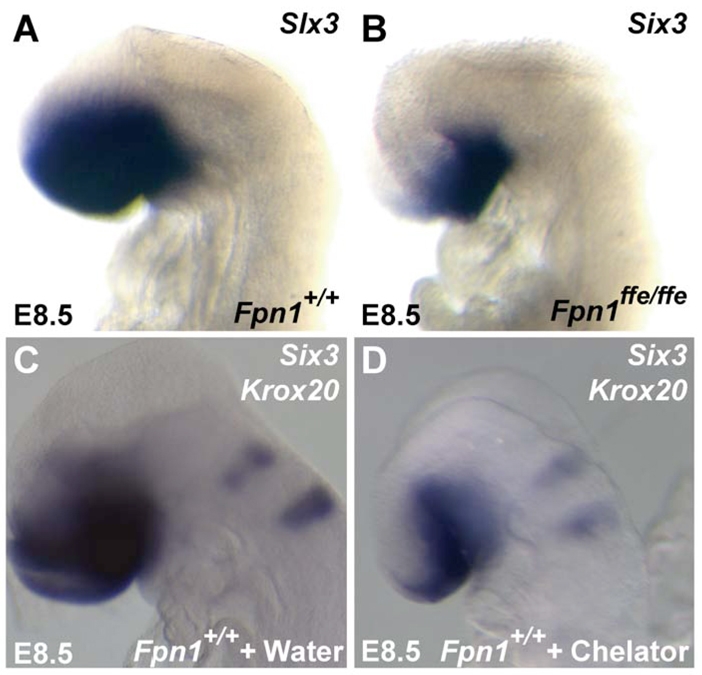
Wild-type embryos cultured in iron chelators phenocopies the forebrain defects of Fpn1 mutants. (A,B) Six3 expression in wild-type (A) and Fpn1ffe/ffe mutant (B) embryos at E8.5. (C,D) Six3 and Krox20 expression in wild-type embryos cultured in the presence of water (vehicle control; C) or 200 μM of the iron chelator 1,2-dimethyl-3-hydroxy-4-pyridone (D). Four out of five iron chelator-treated embryos show reduced Six3 expression compared with those treated with vehicle control.
DISCUSSION
Here we analyzed the developmental pathology leading to NTDs in embryos that are mutant for the iron exporter Fpn1. We have created an allelic series of Fpn1 mutations that reveal graded developmental defects depending on the residual activity of the iron transport activity. A null mutation in Fpn1 results in embryonic lethality before E7.5 (Donovan et al., 2005). A hypomorphic mutation in Fpn1 (Fpn1ffe) results in NTDs consisting primarily of exencephaly and forebrain truncations. Transheterozygous Fpn1 mutants (Fpn1ffe/KI) exhibit more severe NTDs that consisted of exencephaly, craniorachischisis and more drastic forebrain truncations. The expression pattern of Fpn1 during neurulation suggests that it is required in the VE, an observation that was confirmed by conditional deletion of Fpn1 in the VE lineage, resulting in neural tube and forebrain defects. Fpn1 function in the VE is essential for proper morphogenesis of the AVE, as the AVE is diffusely localized in the anterior region of mutant embryos. Consequently, anterior neural character, as determined by Six3 expression at E7.5, is induced to a slightly lesser degree in its normal domain but also ectopically in more caudal regions of the embryo. However, this anterior neural tissue is not maintained, as Six3 expression by E8.5 is disproportionately reduced. Our data suggest that failure to maintain anterior neural identity is due to reduction of a number of signaling centers, including the ADE, AME and ANR. Finally, the failure to maintain anterior neural tissue identity is due to loss of iron transport activity of Fpn1 from the VE to the embryo, as anterior truncations are phenocopied by culture of wild-type embryos in iron chelators during a crucial period (E7.5 to E8.5) of forebrain maintenance.
Diffuse induction and improper maintenance of the forebrain in Fpn1 mutants
The analysis of multiple mouse mutants with forebrain truncations indicate that this defect can result from a number of different mechanisms that fall into two general groups. The first group of mutants fail to induce the anterior neural domain because of defects in the AVE, whereas in the second group of mutants the forebrain is induced but not maintained (for a review, see Martinez-Barbera and Beddington, 2001). Within the second group, a failure to maintain the forebrain can be due to defects in the ADE, AME or ANR. Our data suggest that forebrain truncations in Fpn1 mutants are due to disruptions in all of these mechanisms. As schematized in Fig. 10, the AVE in Fpn1 mutants is formed and induces the pre-forebrain domain; however, the AVE in mutant embryos is diffusely localized within the anterior region of the embryo. The presence of scattered AVE cells results in diffuse and ectopic induction of anterior neural character. Despite induction of only a slightly weaker anterior neural domain, by E8.5, Six3 expression is very much reduced, indicating that Fpn1 function is also required for maintenance of the anterior neural domain. The failure to maintain anterior neural character appears to be due to reductions in multiple signaling centers, including the ADE, AME and ANR. Specifically, the failure to maintain Six3 expression is due in part to defects in the ADE, as Cerl expression in the ADE is reduced compared with control embryos. Furthermore, the AME does not extend to the normal anterior limit in Fpn1 mutants, as shown by the lack of expression of Shh in the most anterior portion of the neural plate. This reduction of AME signaling is likely to be responsible for the mild holoprosencephaly observed in Fpn1 mutant embryos, as Shh signaling from the AME is required for splitting the forebrain into the bilateral cerebral hemispheres (for a review, see McMahon et al., 2003). In addition, Fgf8 expression in the ANR is also reduced in Fpn1 mutants at E8.5, which probably also contributes to the reduction of forebrain markers, particularly Foxg1 in Fpn1 mutants. As Fpn1 is not expressed in the embryo proper throughout neurulation and deletion of Fpn1 in the embryonic lineage does not result in NTDs, the requirement for Fpn1 in the establishment of these signaling centers is probably secondary to the requirement for Fpn1 in transporting iron across the VE to the developing embryo. Importantly, the failure to maintain anterior neural identity is probably due to the iron deficiency that is predicted to result from the loss of Fpn1 activity. This is supported by the observation that in Fpn1 mutants forebrain truncations can be phenocopied by culturing wild-type embryos in the presence of iron chelators from E7.5 to E8.5, precisely when the anterior neural tissue fails to be maintained in Fpn1 mutants.
Effect of Fpn1 mutation on iron homeostasis in the early embryo
Previous studies of the expression pattern of Fpn1 and characterization of knockout mouse models have demonstrated a crucial role for Fpn1 in absorption of iron across the intestinal epithelium, the placenta and VE (Donovan et al., 2000; Donovan et al., 2005). Fpn1 protein is expressed on the basal surface of intestinal epithelium, where it is required for the transport of dietary iron absorbed by these cells into the bloodstream. For example, conditional deletion of Fpn1 in the intestine resulted in iron overload in the duodenal enterocytes, owing to the failure to release iron into the blood stream in the absence of Fpn1 (Donovan et al., 2005). As in the intestinal enterocytes, iron is taken up by the VE from the maternal environment by a number of iron transporters and is exported from these cells to the embryo by the Fpn1 protein (Fig. 10). Consequently, similar to duodenal enterocytes, mutation of Fpn1 in the VE should also result in the accumulation of iron in these cells. However, iron levels in the VE are below the detection limit of traditional histological staining protocols such as Prussian Blue, as we have not been able to detect staining using this technique in the VE (data not shown). Elevated iron levels in the VE could result in the creation of reactive oxygen species and oxidative stress, potentially disrupting the activity of organizing centers within the VE. In support of this model, we observed defects in morphogenesis of the AVE such that although the specification and signaling from the AVE in Fpn1 mutants appears normal, the AVE is diffusely localized to the anterior region of the embryo. This results in diffuse and ectopic induction of anterior neural markers a day later in development.
Another predicted consequence of reduction of Fpn1 exporter activity in the VE would be a lack of iron transported to the developing embryo resulting in iron deficiency (Fig. 10). Our data indicate that iron deficiency also plays a role in forebrain defects in Fpn1 mutants as culture of wild-type embryos between E7.5 and E8.5 can phenocopy these forebrain defects. Meanwhile, signaling centers within the epiblast that maintain anterior forebrain character are affected in a non-cell autonomous fashion. These include the AME, ADE and ANR. Together, these data fit a model (Fig. 10) where both iron overload in the AVE and iron deficiency in the embryo proper contribute to the forebrain defects observed in Fpn1 mutants.
The Fpn1ffe mouse model provides a unique opportunity to study the effects of iron deficiency on early neural development
Previous studies of mouse models of iron homeostasis have failed to provide insight into the requirement for iron during early development of the neural tube as these mutations either provide too severe or too mild defects in iron uptake. For example, Fpn1-null mice exhibit embryonic lethality before neural tube closure (Donovan et al., 2005), illustrating the essential role for Fpn1 in the delivery of iron to the embryo through the VE. In contrast to the crucial requirement for Fpn1 in transport of iron from the VE to the embryo, multiple different proteins can uptake iron into the VE. Mutant mouse models of iron uptake proteins do not exhibit defects in neurulation nor does iron deficiency manifest until later in embryogenesis when significantly more iron is needed for growth of the fetus. For example, in most cells, transferrin-bound iron from the bloodstream enters the cell by binding to transferrin receptor. Transferrin receptor mutants either exhibit no embryonic phenotypes (Trfr2−/−) or do not exhibit defects in neural development until after 10.5 (Trfr−/−), when metabolic needs increase (Levy, 1999; Fleming, 2002). The multi-ligand endocytic receptor complex that includes cubilin (Cubn) and megalin (Lrp2) is expressed in the VE and mediates endocytosis of a number of ligands, including folic acid, Vitamin B12, cholesterol and lipids, in addition to transferrin bound to iron (for a review, see Kozyraki and Gofflot, 2007). Interestingly, megalin-null mutant embryos exhibit exencephaly and forebrain defects reminiscent of those exhibited by Fpn1ffe/KI mutants (Spoelgen et al., 2005; Willnow et al., 1996). However, the fact that megalin mediates transfer of numerous nutrients important for neural tube closure and that conditional ablation of the gene encoding megalin only in the epiblast lineage also results in NTDs (Spoelgen et al., 2005) suggests a possible different role from that of Fpn1. As the Fpn1ffe mutation incompletely impairs the ability of Fpn1 to transport iron across the VE, this hypomorphic allele provides a unique opportunity to examine the effects of iron deficiency in Fpn1ffe/ffe and Fpn1ffe/KI mutants within a crucial range where embryogenesis can proceed but neural tube closure and forebrain patterning are affected. Furthermore, we have found that the degree of malformation is directly proportional to the amount of functional Fpn1 expressed and theoretically the amount of iron transported across the VE. For example, neural tube closure and anterior neural patterning are more profoundly affected in Fpn1ffe/KI mutants compared with Fpn1ffe/ffe mutants. Furthermore, our analysis of the mechanism of neural pattering defects in this mouse model has provided valuable insight into the developmental processes most sensitive to iron deficiency.
The analysis of Fpn1 mutants in zebrafish has also failed to demonstrate a role for Fpn1 in early embryogenesis. Two loss-of-function fpn1 (slc40a1 – Zebrafish Information Network) mutations have been identified in the zebrafish anemia mutant weissherbst (weh) (Donovan et al., 2000). In zebrafish, fpn1 is expressed in the YS and is required for the transport of iron from the maternally derived yolk stores to the embryonic circulation. Consequently, weh mutants develop hypochromic anemia but fail to develop defects earlier in embryogenesis. These interspecies differences in the severity of the phenotype may be due to differences in maternal contribution between zebrafish and mouse. The timing of when the embryo needs to completely rely on the mutant zygotic product would depend on the stability of maternally deposited proteins and transcripts. In the mouse, maternal contribution of transcripts and proteins is minimal and following implantation essentially non-existent. Therefore, it is possible that the differences in the phenotypes of Fpn1 mutants in zebrafish versus the mouse are due to differences in maternal contribution. These species-specific differences in maternal contribution could explain the early embryonic lethality in Fpn1 null mutants and profound patterning defects in Fpn1ffe/KI versus the lack of early developmental defects in zebrafish weh mutants.
Iron deficiency and NTDs?
Our data suggest that iron deficiency is at least partially responsible for the forebrain truncations observed in Fpn1 mutants; however, whether defects in neural tube closure are also due to iron deficiency remains to be determined. Data in humans suggest that this hypothesis is likely. Of significance, women of childbearing age are disproportionately affected with iron deficiency. Some retrospective epidemiological studies have suggested a link between iron deficiency and the increased risks for a NTD-affected pregnancy (Felkner et al., 2005; Groenen et al., 2004). Furthermore, clinical trials suggest that peri-conception vitamin supplementation that includes iron in addition to folic acid can lower the risk of NTDs further than folic acid supplementation alone (for a review, see Czeizel, 2009). These data suggest that although folic acid is an essential nutrient for preventing NTDs, other nutrients such as iron may also play an important role. In humans, neural tube closure occurs during the third and fourth weeks of gestation, before many women are aware of the pregnancy. Our data suggest that the VE may play an essential role in ‘loading’ the embryo with iron to allow for successful neural tube closure, implying that supplementation with both folic acid and iron prior to conception may reduce the incidence of NTD-affected pregnancies even further.
Acknowledgements
We are grateful to Drs Anna-Katerina Hadjantonakis and Nancy Andrews for providing the Ttr-Cre and Fpn1flox alleles, respectively. The Fpn1ffe allele was generated in the Sloan-Kettering Institute Mouse Mutagenesis Project in collaboration with Kathryn Anderson (U01-HD43478) and Gordon Fischell. We thank Kathryn Anderson, Anamaris Colberg-Poley, Jerry Kaplan and Ivana De Domenico for helpful discussions. We also thank Gerald Rosenthal, Lori Bulwith and Andrew Pollock for technical assistance. This work was partially supported by a Basal O'Conner Award from the March of Dimes, by a Young Investigator Award from the Spina Bifida Association and by R01-HD058629 to I.E.Z. L.A.N. is a HHMI investigator. Deposited in PMC for release after 6 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- Andrews N. C. (1999). Disorders of iron metabolism. New Engl. J. Med. 341, 1986-1995 [DOI] [PubMed] [Google Scholar]
- Ang S. L., Conlon R. A., Jin O., Rossant J. (1994). Positive and negative signals from mesoderm regulate the expression of mouse Otx2 in ectoderm explants. Development 120, 2979-2989 [DOI] [PubMed] [Google Scholar]
- Barnes J. D., Crosby J. L., Jones C. M., Wright C. V., Hogan B. L. (1994). Embryonic expression of Lim-1, the mouse homolog of Xenopus Xlim-1, suggests a role in lateral mesoderm differentiation and neurogenesis. Dev. Biol. 161, 168-178 [DOI] [PubMed] [Google Scholar]
- Belaoussoff M., Farrington S. M., Baron M. H. (1998). Hematopoietic induction and respecification of A-P identity by visceral endoderm signaling in the mouse embryo. Development 125, 5009-5018 [DOI] [PubMed] [Google Scholar]
- Belo J. A., Bouwmeester T., Leyns L., Kertesz N., Gallo M., Follettie M., De Robertis E. M. (1997). Cerberus-like is a secreted factor with neutralizing activity expressed in the anterior primitive endoderm of the mouse gastrula. Mech. Dev. 68, 45-57 [DOI] [PubMed] [Google Scholar]
- Bielinska M., Narita N., Wilson D. B. (1999). Distinct roles for visceral endoderm during embryonic mouse development. Int. J. Dev. Biol. 43, 183-205 [PubMed] [Google Scholar]
- Blom H. J., Shaw G. M., den Heijer M., Finnell R. H. (2006). Neural tube defects and folate: case far from closed. Nat. Rev. Neurosci. 7, 724-731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet P., Chazaud C., Oulad-Abdelghani M., Dolle P., Chambon P. (1995). Sequence and expression pattern of the Stra7 (Gbx-2) homeobox-containing gene induced by retinoic acid in P19 embryonal carcinoma cells. Dev. Dyn. 204, 372-382 [DOI] [PubMed] [Google Scholar]
- Crossley P. H., Martin G. R. (1995). The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development 121, 439-451 [DOI] [PubMed] [Google Scholar]
- Czeizel A. E. (2009). Periconceptional folic acid and multivitamin supplementation for the prevention of neural tube defects and other congenital abnormalities. Birth Defects Res. A Clin. Mol. Teratol. 85, 260-268 [DOI] [PubMed] [Google Scholar]
- De Domenico I., McVey Ward D., Kaplan J. (2008). Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 9, 72-81 [DOI] [PubMed] [Google Scholar]
- Detrait E. R., George T. M., Etchevers H. C., Gilbert J. R., Vekemans M., Speer M. C. (2005). Human neural tube defects: developmental biology, epidemiology, and genetics. Neurotoxicol. Teratol. 27, 515-524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S. J., Moynihan J., Paw B. H., Drejer A., Barut B., Zapata A., et al. (2000). Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776-781 [DOI] [PubMed] [Google Scholar]
- Donovan A., Lima C. A., Pinkus J. L., Pinkus G. S., Zon L. I., Robine S., Andrews N. C. (2005). The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1, 191-200 [DOI] [PubMed] [Google Scholar]
- Echelard Y., Epstein D. J., St-Jacques B., Shen L., Mohler J., McMahon J. A., McMahon A. P. (1993). Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75, 1417-1430 [DOI] [PubMed] [Google Scholar]
- Eichholzer M., Tonz O., Zimmermann R. (2006). Folic acid: a public-health challenge. Lancet 367, 1352-1361 [DOI] [PubMed] [Google Scholar]
- Felkner M. M., Suarez L., Brender J., Scaife B., Hendricks K. (2005). Iron status indicators in women with prior neural tube defect-affected pregnancies. Matern. Child Health J. 9, 421-428 [DOI] [PubMed] [Google Scholar]
- Fleming R. E., Ahmann J. R., Migas M. C., Waheed A., Koeffler H. P., Kawabata H., Britton R. S., Bacon B. R., Sly W. S. (2002). Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. USA 99, 10653-10658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindley J. C., Davidson D. R., Hill R. E. (1995). The role of Pax-6 in eye and nasal development. Development 121, 1433-1442 [DOI] [PubMed] [Google Scholar]
- Groenen P. M., van Rooij I. A., Peer P. G., Ocke M. C., Zielhuis G. A., Steegers-Theunissen R. P. (2004). Low maternal dietary intakes of iron, magnesium, and niacin are associated with spina bifida in the offspring. J. Nutr. 134, 1516-1522 [DOI] [PubMed] [Google Scholar]
- Hallonet M., Kaestner K. H., Martin-Parras L., Sasaki H., Betz U. A., Ang S. L. (2002). Maintenance of the specification of the anterior definitive endoderm and forebrain depends on the axial mesendoderm: a study using HNF3beta/Foxa2 conditional mutants. Dev. Biol. 243, 20-33 [DOI] [PubMed] [Google Scholar]
- Hayashi S., Lewis P., Pevny L., McMahon A. P. (2002). Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 119, S97-S101 [DOI] [PubMed] [Google Scholar]
- Kozyraki R., Gofflot F. (2007). Multiligand endocytosis and congenital defects: roles of cubilin, megalin and amnionless. Curr. Pharm. Des. 13, 3038-3046 [DOI] [PubMed] [Google Scholar]
- Kwon G. S., Hadjantonakis A. K. (2009). Transthyretin mouse transgenes direct RFP expression or Cre-mediated recombination throughout the visceral endoderm. Genesis 47, 447-455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon G. S., Viotti M., Hadjantonakis A. K. (2008). The endoderm of the mouse embryo arises by dynamic widespread intercalation of embryonic and extraembryonic lineages. Dev. Cell 15, 509-520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy J. E., Jin O., Fujiwara Y., Kuo F., Andrews N. C. (1999). Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 21, 396-399 [DOI] [PubMed] [Google Scholar]
- Maretto S., Cordenonsi M., Dupont S., Braghetta P., Broccoli V., Hassan A. B., Volpin D., Bressan G. M., Piccolo S. (2003). Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA 100, 3299-3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Barbera J. P., Beddington R. S. (2001). Getting your head around Hex and Hesx1: forebrain formation in mouse. Int. J. Dev. Biol. 45, 327-336 [PubMed] [Google Scholar]
- Martinez Barbera J. P., Clements M., Thomas P., Rodriguez T., Meloy D., Kioussis D., Beddington R. S. (2000). The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development 127, 2433-2445 [DOI] [PubMed] [Google Scholar]
- McMahon A. P., Ingham P. W., Tabin C. J. (2003). Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 53, 1-114 [DOI] [PubMed] [Google Scholar]
- Nagy A., Gertsenstein M., Vintersten K., Behringer R. R. (2003). Manipulating the Mouse Embryo- a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Oliver G., Mailhos A., Wehr R., Copeland N. G., Jenkins N. A., Gruss P. (1995). Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development 121, 4045-4055 [DOI] [PubMed] [Google Scholar]
- Parr B. A., Shea M. J., Vassileva G., McMahon A. P. (1993). Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development 119, 247-261 [DOI] [PubMed] [Google Scholar]
- Rubenstein J. L., Shimamura K., Martinez S., Puelles L. (1998). Regionalization of the prosencephalic neural plate. Annu. Rev. Neurosci. 21, 445-477 [DOI] [PubMed] [Google Scholar]
- Shimamura K., Rubenstein J. L. (1997). Inductive interactions direct early regionalization of the mouse forebrain. Development 124, 2709-2718 [DOI] [PubMed] [Google Scholar]
- Spoelgen R., Hammes A., Anzenberger U., Zechner D., Andersen O. M., Jerchow B., Willnow T. E. (2005). LRP2/megalin is required for patterning of the ventral telencephalon. Development 132, 405-414 [DOI] [PubMed] [Google Scholar]
- Srinivas S. (2006). The anterior visceral endoderm-turning heads. Genesis 44, 565-572 [DOI] [PubMed] [Google Scholar]
- Tallquist M. D., Soriano P. (2000). Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26, 113-115 [DOI] [PubMed] [Google Scholar]
- Tao W., Lai E. (1992). Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron 8, 957-966 [DOI] [PubMed] [Google Scholar]
- Tomihara-Newberger C., Haub O., Lee H. G., Soares V., Manova K., Lacy E. (1998). The amn gene product is required in extraembryonic tissues for the generation of middle primitive streak derivatives. Dev. Biol. 204, 34-54 [DOI] [PubMed] [Google Scholar]
- Wilkinson D. G., Bhatt S., Chavrier P., Bravo R., Charnay P. (1989). Segment-specific expression of a zinc-finger gene in the developing nervous system of the mouse. Nature 337, 461-464 [DOI] [PubMed] [Google Scholar]
- Willnow T. E., Hilpert J., Armstrong S. A., Rohlmann A., Hammer R. E., Burns D. K., Herz J. (1996). Defective forebrain development in mice lacking gp330/megalin. Proc. Natl. Acad. Sci. USA 93, 8460-8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohn I. E., Sarkar A. A. (2008). Modeling neural tube defects in the mouse. Curr. Top. Dev. Biol. 84, 1-35 [DOI] [PubMed] [Google Scholar]
- Zohn I. E., Anderson K. V., Niswander L. (2005). Using genomewide mutagenesis screens to identify the genes required for neural tube closure in the mouse. Birth Defects Res. A Clin. Mol. Teratol. 73, 583-590 [DOI] [PubMed] [Google Scholar]
- Zohn I. E., Li Y., Skolnik E. Y., Anderson K. V., Han J., Niswander L. (2006). p38 and a p38-interacting protein are critical for downregulation of E-cadherin during mouse gastrulation. Cell 125, 957-969 [DOI] [PubMed] [Google Scholar]
- Zohn I. E., De Domenico I., Pollock A., Ward D. M., Goodman J. F., Liang X., Sanchez A. J., Niswander L., Kaplan J. (2007). The flatiron mutation in mouse ferroportin acts as a dominant negative to cause ferroportin disease. Blood 109, 4174-4180 [DOI] [PMC free article] [PubMed] [Google Scholar]