

Abstract
We have implemented a strategy in which a genetically engineered, single-chain protein specifically recognizes cancer cells and is trafficked to a targeted subcellular compartment, such as the nucleus. The recombinant protein termed IL-13.E13K-D2-NLS has a triple functional property: (1) it binds a cancer-associated receptor, interleukin 13 receptor alpha 2 (IL-13Rα2), using modified IL-13 ligand, IL-13.E13K; (2) it exports its C-terminal portion out of the endosomal compartment using Pseudomonas aeruginosa exotoxin A (PE) translocation domain (D2); and (3) it travels to and accumulates in the nucleus guided by the nuclear localization signal (NLS). Here, we have demonstrated that this protein is transported into the brain tumor cells’ nucleus, using 3 different methods of protein conjugation to dyes for the purpose of direct visualization of the protein’s intracellular trafficking. IL-13.E13K-D2-NLS, and not the controls such as IL-13.E13K-D2, IL-13.E13K-NLS, or IL-13.E13K, accumulated in nuclei very efficiently, which increased with the time the cells were exposed to the protein. Also, IL-13.E13K-D2-NLS did not exhibit nuclear transport in cells with low expression levels of IL-13Rα2. Thus, it is possible to recognize cancer cells through their specific receptors and deliver a conjugated protein that travels specifically to the nucleus. Hence, our molecular targeting strategy succeeded in generating a single-chain proteinaceous agent capable of delivering drugs/labels needed to be localized to the cells’ nuclei or potentially any other subcellular compartment, for their optimal efficacy or ability to exert their specific action.
Keywords: cancer, molecular targeting, subcellular compartments, receptors, trafficking, glioblastoma multiforme
Introduction
Molecular targeting of cancer cells is achieved (a) specifically through the use of ligands/antibodies against tumor-associated or tumor-specific receptors, and (b) nonspecifically using plasma membrane permeable agents targeting activated/overexpressed intracellular elements, such as the oncogenes. In the field of nonviral gene therapy of cancer that employs recombinant proteins, we have pioneered the use of proteinaceous vectors for the targeted intracellular transport of proteins/nonproteinaceous compounds.1 Some bacterial toxins, such as Pseudomonas aeruginosa exotoxin A (PE) or diphtheria toxin (DT), possess an ability to exit the endocytic compartment after being internalized in the process of receptor-mediated internalization and being proteolytically activated by a calcium-dependent serine endoprotease, furin.2-7 This “get cleaved and exit endocytic compartment” ability is possible because of the presence of a specialized domain of PE, domain II (abbreviated here as D2).8,9
Previously, we have exploited PE translocation ability to traffic other, non-PE, or repeated-PE peptide sequences into the cell cytosol.1 This was achieved by incorporating non-PE peptides or an additional catalytic domain III of PE within dispensable domain Ib of the toxin. This domain Ib is downstream of both the furin cleavage site and the N-terminal sequence important for initiation/conduct of exiting of the C-terminal portion of the toxin from the endocytic vesicles (includes portions of domain II and domain III of PE toxin).1 We demonstrated for the first time that in this manner, PE can serve as a vector for intracytosolic delivery of various proteins.1 This approach served as a basis for further developments of, for example, intracellular vaccines,10 or prompted the use of DT protein, having very similar properties to PE toxin, for the same purpose of intracytosolic delivery.11
Most anticancer therapeutics have defined targets such as oncogenes, enzymes, or DNA, all of which are localized to distinct intracellular compartments like cytosol, mitochondria, or nuclei. We reasoned that having direct delivery vectors for therapeutics/labels to these subcellular compartments will lead to an increased specificity and efficacy, and less toxicity. To this end, we have designed a true multiple-specificity delivery vehicle targeting the interleukin-13 receptor alpha 2 (IL-13Rα2) for efficient transport to the nuclei of glioblastoma multiforme (GBM) cells. GBM is a high-grade astrocytoma representing the most common form of primary brain tumors. The treatment of patients with GBM is still a major challenge, and the median survival rate is 14.5 months after diagnosis.12 Several factors specific to GBM have been uncovered in recent years.13-16 For example, a tri-molecular signature of GBM has been documented that includes IL-13Rα2, EphA2 receptor, and a fos-related antigen 1 (Fra-1).17 All 3 factors belonging to the signature are suitable for therapeutic targeting of GBM.18 For example, IL-13Rα2 is expressed in >75% of GBM tumor specimens.19,20 Our laboratory has characterized the IL-13Rα2 receptor as a cancer/testis-like antigen.21 IL-13Rα2 is believed to act as a decoy receptor.22 However, it has been shown that the IL-13 ligand binds to the IL-13Rα2 receptor and is internalized through receptor-mediated endocytosis.23,24 Hence, drugs attached to the IL-13 ligand can be internalized and delivered specifically inside the glioma cells. Our laboratory has designed and produced IL-13–based cytotoxins which have shown anti-GBM tumor activity under both in vitro and in vivo conditions.24 Our laboratory has also designed various IL-13 ligand mutants which specifically bind to the IL-13Rα2, the cancer-associated receptor, and which do not bind to the other, physiological receptor for IL-13 that is also shared with homologous cytokine, IL-4.25,26 IL-13.E13K is one of such IL-13 mutants, which has an amino acid residue at position 13 substituted for lysine.27
The nuclear localization signals (NLS) are employed by a large number of important proteins which travel to and from the nucleus. One well-characterized sequence from Simian virus 40 (SV40) large T antigen is a proven efficient NLS.28-31 This particular NLS is a classical monopartite NLS and comprises a short stretch of basic positively charged amino acids, containing several arginine and lysine residues.28 In the nucleus import pathway, the NLS sequence binds to cytosolic proteins known as importins/karyopherins, which recognize and transport NLS-containing proteins to the nuclear pore complex.32
For our nuclear targeting delivery vector, we combined the above-described recognition and transport signals into a single-chain recombinant protein. Thus, our vector consists of IL-13 or the IL-13 mutant, IL-13.E13K, that specifically recognizes IL-13Rα2 followed by domain II (D2) of PE for endosomal translocation, and finally, the SV40 T antigen NLS to guide the remaining portion of PE to the nucleus. We demonstrate here, in a direct way, that molecularly targeted, genetically engineered proteins specifically recognize GBM cancer cells, travel to, and accumulate in these cells’ nuclei.
Results
Production of IL-13.E13K-D2-NLS, IL-13.E13K-D2, IL-13.E13K-NLS, and IL-13.E13K proteins
We aim at developing effective drug/radioactive isotope delivery vehicles to specific intracellular compartments of cancer cells, based preferentially on recombinant proteins. Hence, we have developed here a recombinant protein delivery vehicle to the nuclei of GBM cells. This delivery vehicle recognizes the IL-13Rα2, which is overexpressed on GBM cells. IL-13.E13K-D2-NLS and its control proteins, IL-13.E13K-NLS, IL-13.E13K-D2, as well as IL-13.E13K, which are not expected to either leave the endosomal compartment or reach the nucleus (Fig. 1A), were produced in E. coli and purified using FPLC. IL-13.E13K-D2-NLS was induced in BL21 E. coli cells using IPTG (Fig. 1B). The induced protein was isolated and further processed using a disulphide-shuffling method and purified using FPLC column, as described previously.25,33 The separated protein from the column was >90% pure (Fig. 1C, inset). The controls IL-13.E13K-D2, IL-13.E13K-NLS, and IL-13.E13K recombinant proteins were expressed, processed, and purified in a similar manner (e.g., Suppl. Fig. S1 and not shown).
Figure 1.
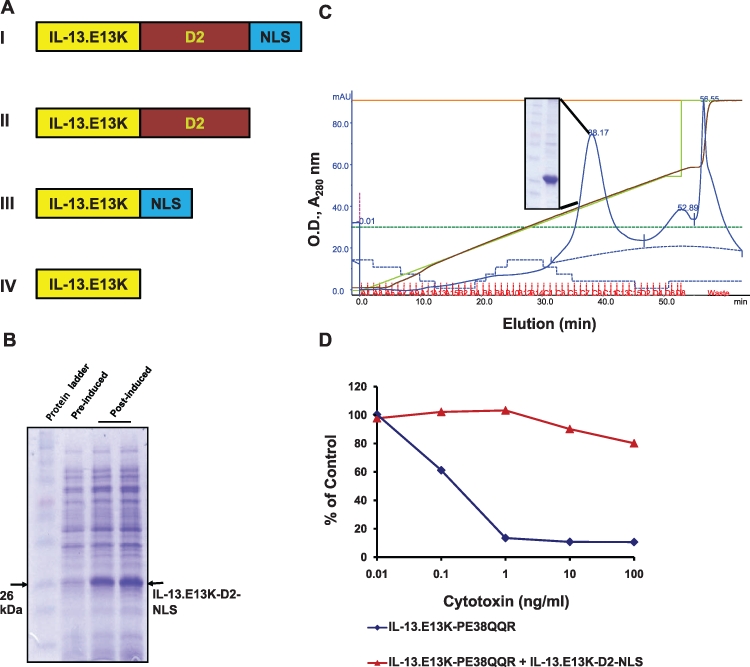
IL-13.E13K-D2-NLS is a nucleus-targeting vector in GBM cells. (A) Schema of single-chain targeted recombinant proteins: (I) IL-13.E13K-D2-NLS composed of building blocks of targeting ligand, endosomal translocation PE domain, and the SV40 T antigen nuclear localization signal sequence; (II) IL-13.E13K-D2 composed of the targeting ligand and the endosomal translocation domain; (III) IL-13.E13K-NLS consisting of the targeting unit and NLS; (IV) IL-13.E13K targeting unit alone. (B) Purification and functional activity of IL-13.E13K-D2-NLS. E. coli BL21 cells were transformed with the recombinant IL-13.E13K-D2-NLS cDNA construct and protein expressed by induction with IPTG. The 12% SDS- PAGE gel shows the pre- and post-IPTG induced cell protein extracts. (C) The expressed protein was further processed and purified on an SP Sepharose column using the FPLC system (inset). (D) The purified protein was subjected to competition assay for the IL-13Rα2 against the IL-13.E13K-PE38QQR toxin. 1 µg/mL of the purified IL-13.E13K-D2-NLS was used to compete against 0.01 to 100 ng/mL of the cytotoxin and the cytoxicity measured using the MTS/PMS assay.
IL-13.E13K-D2-NLS, IL-13.E13K-D2, IL-13.E13K-NLS, and IL-13.E13K compete for IL-13Rα2 on GBM cells
We next wished to confirm that all the purified recombinant proteins bind to the IL-13Rα2 receptor on GBM cells as intended. To this end, we carried out a cell viability assay in which these recombinant proteins bound to the IL-13Rα2 receptor and protected against cytotoxic action of IL-13.E13K-PE38QQR. IL-13.E13K-PE38QQR, as mentioned above, is a recombinant cytotoxin that binds to IL-13Rα2, is internalized, and leads to cell killing through the cleaved active portion of PE, enzymatic domain III. As expected, all recombinant proteins of interest blocked the action of the cytotoxin, resulting in no cell killing: IL13.E13K-D2-NLS (Fig. 1D), IL-13.E13K-D2 (Suppl. Fig. S1C), IL-13.E13K-NLS, and IL-13.E13K (not shown). These results confirm that all the recombinant proteins retain IL-13.E13K ligand binding properties and compete specifically for the IL-13Rα2.
IL-13.E13K-D2-NLS localizes to the nuclei of U-251 MG GBM cells
Next, we wished to monitor the intracellular journey as well as the subcellular localization of our targeted proteins. To this end, we fluorescently labeled these proteins using 3 different approaches/methods. For the first approach, we labeled the carboxyl amino acids of the proteins, so as not to modify the primary amines (lysines) present in the NLS domain of the protein. Thus, we utilized the EDC-Sulfo-NHS and Alexa Fluor 488 labeling techniques. IL-13-D2-NLS and IL-13-D2 were labeled at their carboxylate groups on amino acids with Alexa Fluor 488–hydrazide via EDC (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride) and Sulfo-NHS (N-hydroxysuccinimide) (see Materials and Methods). In the second approach, we directly labeled the primary amines of the proteins, which are present also in the lysines, with the Alexa Fluor 488–TFP reactive dyes. And for the third approach, we carried out an indirect labeling method. Thus, we initially conjugated the proteins at the primary amines with biotin. The biotinylated proteins were detected using HRP-streptavidin and the signals amplified using tyramide signal amplification.
After labeling the proteins using the first conjugation method (i.e., EDC-Sulfo-NHS–Alexa Fluor 488 labeling), we performed cell localization experiments in U-251 MG GBM cells. We observed that the IL-13-D2-NLS effectively localized to the nucleus at 1 hour (Fig. 2A-C), preceded by membrane and cytosolic localization seen at 15 minutes (Fig. 2A). We also performed Z-stack analysis to confirm the localization of the protein inside the nucleus. The Z-stack analysis for the experiment shown in Fig. 2B demonstrated unequivocally the nuclear localization of IL-13.E13K-D2-NLS (Fig. 2C). On the other hand, IL-13-D2 did get internalized into the U-251 MG cells and was found to be primarily in the perinuclear region, but it did not travel into the nucleus at either 15 minutes or 1 hour of the experiment (Fig. 2D and E).
Figure 2.
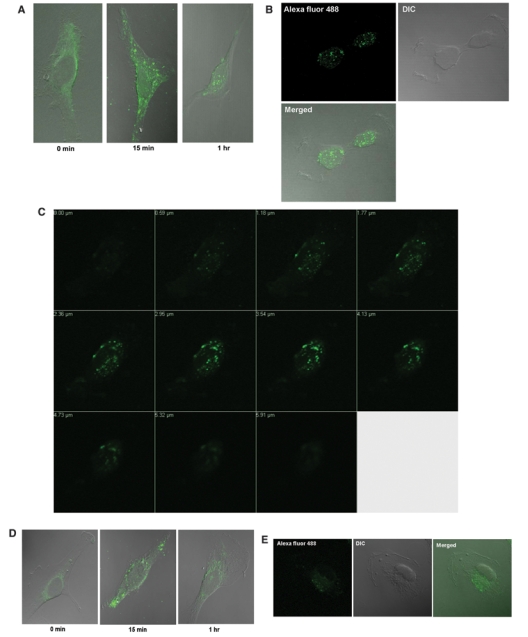
EDC-Sulfo-NHS–Alexa Fluor 488 labeled IL-13-D2-NLS localizes to the nucleus of U-251 MG GBM cells. (A) U-251 MG cells were treated with 500 nM of the EDC-Sulfo-NHS–Alexa Fluor 488 labeled IL-13-D2-NLS and the subcellular localization monitored using Zeiss 510 LSM confocal microscope. (B) As in A, U-251 MG cell at 1 hour. DIC = differential image contrast for depicting the cell morphology. (C) Confocal Z-stack analysis of a U-251 MG cell treated with the labeled IL-13-D2-NLS protein at 1 hour. (D and E) U-251 MG cells were treated with 500 nM of the EDC-Sulfo-NHS–Alexa Fluor 488 labeled IL-13-D2 and the intracellular localization monitored using the Zeiss 510 LSM confocal microscope.
The above experiments strongly suggested an ability of IL-13-D2-NLS, but not IL-13-D2, to localize to U-251 GBM cells’ nuclei. However, in order to obtain even higher-resolution pictures and confirm the nuclear localization using a different conjugation method, we carried out a direct labeling of IL-13.E13K-D2-NLS and IL-13.E13K-D2 with the Alexa Fluor 488 dye. In this approach, Alexa Fluor 488 tetrafluorophenyl (TFP) reactive dye molecules attach directly to the primary amines of the amino acids of the proteins, forming stable protein-dye conjugates. The conjugate of the proteins were visualized using either SDS-PAGE (Fig. 3A) or fluorescence signals (Fig. 3B). The Typhoon scan showed that only protein-dye conjugates emitted fluorescence signals, but not the unconjugated proteins (Fig. 3B). The labeled IL-13.E13K-D2-NLS was observed as a doublet on the gel, which can be attributed to differential labeling levels of the protein and hence different molecular weight bands. We next repeated the localization experiments in U-251 MG cells and confirmed what we had observed earlier: IL-13.E13K-D2-NLS localized to the nuclei at 1 hour (Fig. 3C and D), whereas IL-13.E13K-D2 protein never trafficked into the nucleus (Fig. 3E).
Figure 3.
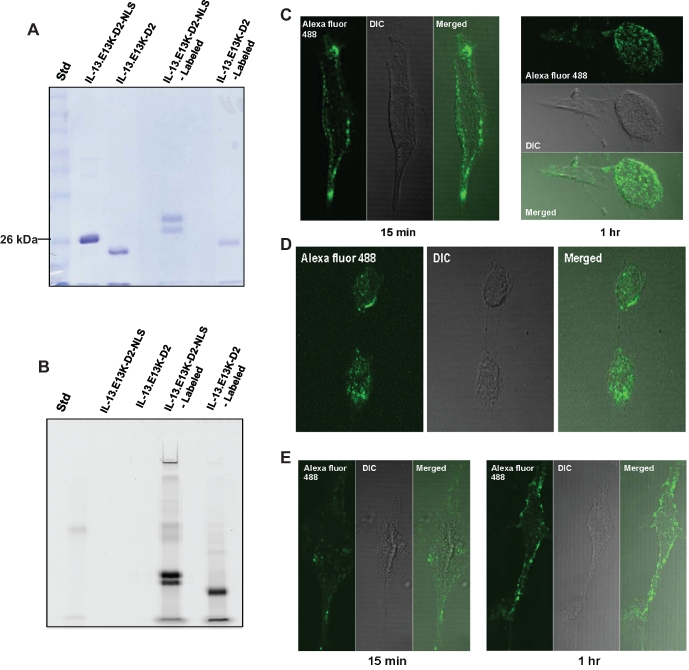
IL-13.E13K-D2-NLS travels to and very efficiently accumulates in the nuclei of GBM cells, IL-13Rα2 overexpressors. (A) Conjugation of IL-13.E13K-D2-NLS and IL-13.E13K-D2 to Alexa Fluor 488. The primary amines in the IL-13.E13K-D2-NLS and the IL-13.E13K-D2 were conjugated directly to the tetrafluorophenyl (TFP)-reactive Alexa Fluor 488 dye. The labeled proteins were run on 12% SDS-PAGE gel and stained with Coomassie blue. (B) As in A, but the blot scanned using Typhoon 9210 to detect fluorescent proteins. (C and D) Alexa Fluor 488 labeled IL-13.E13K-D2-NLS localizes to the nucleus of U-251 MG GBM cells. The intracellular localization of IL-13.E13K-D2-NLS labeled with Alexa Fluor 488 in U-251 MG cells was examined using confocal microscope. (E) As in C and D, but the protein used was control IL-13.E13K-D2. The data are representative of 3 independent experiments.
In order to examine whether yet another visualization method would document the same nuclei-localization ability of our recombinant constructs, we decided to use a signal-amplification method via tyramide molecules. We initially labeled our proteins using biotin-XX sulfosuccinimidyl ester (biotin-XX, SSE), which reacts very efficiently with the primary amines of the proteins, forming stable protein-biotin conjugates. The biotinylated proteins were analyzed using SDS-PAGE/Western blot, and the protein-biotin conjugates were detected using streptavidin-HRP. The Western blot indicated that both the proteins had been biotinylated (Fig. 4A). The number of biotins on each of these proteins was quantified by performing FluoReporter Biotin Quantitation Assay based on the standard curve in Figure 4B. Using a quadratic fit equation, the IL-13.E13K-D2-NLS and IL-13.E13K-D2 had similar levels of labeling (DOL) of 13.87 and 14.45 when labeled at a protein:dye molar ratio of 1:4 and 1:8, respectively. Next, these biotinylated proteins were tested in the neutralization of cell killing in a cytotoxicity assay, as shown in Figure 4C. Both biotinylated IL-13.E13K-D2-NLS and IL-13.E13K-D2 (BL) proteins blocked the action of IL-13Rα2–specific cytotoxin-mediated U-251 MG cell killing (Fig. 4C). The cell localization experiment was then conducted, and the proteins were detected using HRP-streptavidin tyramide signal amplification. We found that in the case of IL-13.E13K-D2-NLS, the protein was bound to the cell membrane with some cytosolic localization at 5 minutes (Fig. 5A). At 4 hours, cells nuclei started to demonstrate IL-13.E13K-D2-NLS localization, whereas almost all the cells had a significant portion of the protein inside their nuclei at 8 and 24 hours (Fig. 5A). The Z-stack analyses of the 24-hour experimental time point shown in Figure 5B clearly demonstrated that the IL-13.E13K-D2-NLS traveled and accumulated to the nucleus. In the case of IL-13.E13K-D2, the protein was mostly bound to the cell membrane with some molecules undergoing internalization at 5 minutes, while at 8 and 24 hours the protein was predominantly internalized and localized in the perinuclear region of cells (Fig. 5C). The Z-stack analyses of the 24-hour experimental time point however failed to demonstrate any significant migration of IL-13.E13K-D2 protein to the nucleus (Fig. 5D).
Figure 4.
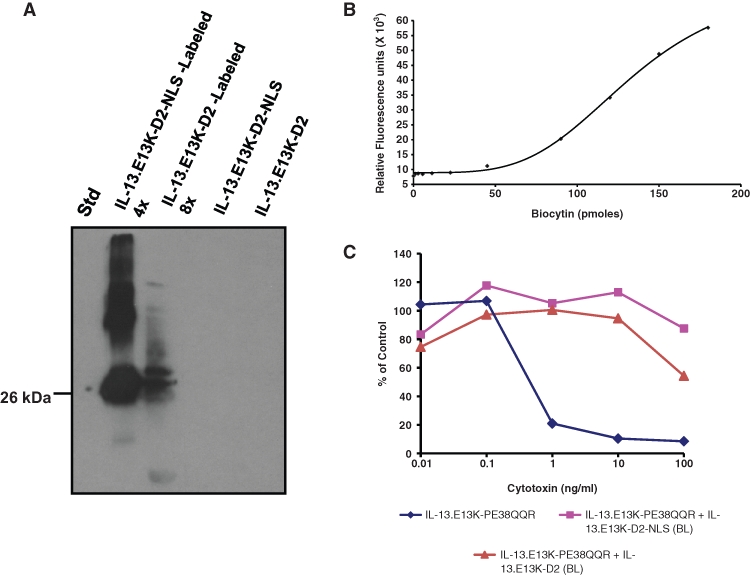
Biotin-labeled IL-13.E13K-D2-NLS retains binding to IL-13Rα2 on GBM cells. (A) Western blot for the biotin-labeled IL-13.E13K-D2-NLS and IL-13.E13K-D2 probed with streptavidin-HRP. (B) Standard curve for the bio-fluoreporter assay for quantification of the biotin molecules on the labeled proteins. Biocytin was used as the standard and the fluorescence was measured using the SpectraMax 340 plate reader. (C) Neutralization of the cytotoxicity assay for the biotin-labeled (BL) IL-13.E13K-D2-NLS and the IL-13.E13K-D2. 1 µM each of the biotin-labeled proteins was competed against the IL-13.E13K-PE38QQR cytotoxin.
Figure 5.
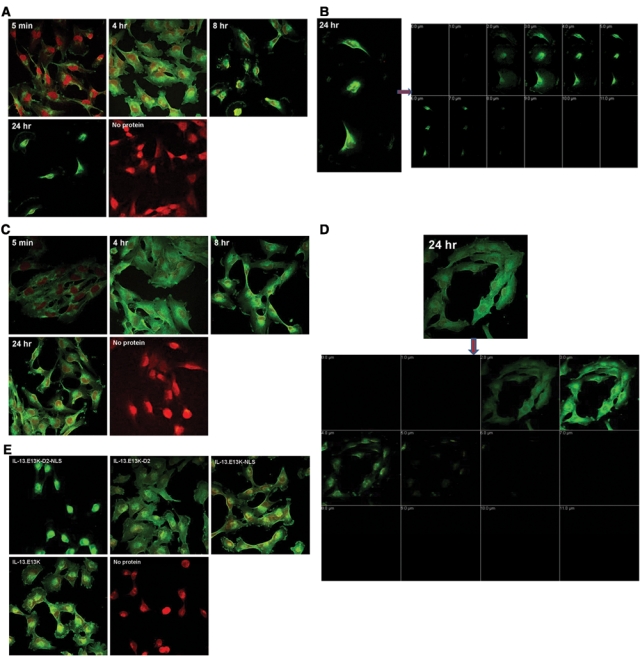
Biotin-labeled IL-13.E13K-D2-NLS retains nuclear localization ability in GBM cells. (A) Biotin-labeled IL-13.E13K-D2-NLS (1 µM) was added to U-251 MG cells for different time points and the biotin signal was amplified by tyramide–Alexa Fluor 488 labeling technique. Subcellular localization of the proteins was monitored using confocal microscope. (B) The Z-stack confocal analysis of IL-13.E13K-D2-NLS nuclear accumulation. (C and D) The biotin-labeled IL-13.E13K-D2 does not localize to the nuclei of U-251 MG GBM cells. The localization experiment was carried out as for IL-13.E13K-D2-NLS. Cells not exposed to the labeled protein were used as controls. The Z-stack analysis is that of the experiment at 24 hours. The pictures are representative of 3 independent experiments. (E) Localization experiment for IL-13.E13K-D2-NLS, IL-13.E13K-D2, IL-13.E13K-NLS and the IL-13.E13K proteins in U-251 MG cells. At 24 hours, only the IL-13.E13K-D2-NLS was prominently localized in the nucleus whereas the other three proteins had major perinuclear, but no nuclear localization. The data are representative of 2 independent experiments.
We have also carried out experiments in which we have labeled the proteins with different molar ratios of the biotin. The protein:dye ratios used were 1:12, 1:8, and 1:4. When both IL-13.E13K-D2-NLS and IL-13.E13K-D2 proteins were labeled at a 1:12 ratio, we observed similar localization patterns for these proteins as described above, except we did not observe any nuclear localization at 4 hours (not shown). This is suggestive of higher occupancy of lysine sites in the NLS by the dye, thus influencing the signal’s functional capability.
We also carried out cell localization experiments with another control protein, IL-13.E13K-NLS, which is devoid of domain II of PE and should not be able to undergo endosomal translocation and subsequent nuclear transport; it should behave like the IL-13.E13K ligand alone. We thus labeled IL-13.E13K-NLS and IL-13.E13K proteins with the biotin. The SDS-PAGE/Western blot of the IL-13.E13K-D2-NLS, IL-13.E13K-D2, IL-13.E13K-NLS, and IL-13.E13K proteins conjugated with biotin and probed with streptavidin-HRP indicated that all the proteins were similarly labeled (Suppl. Fig. S2A) and also that all the biotin-conjugated proteins bound to the IL-13Rα2 on GBM cells (Suppl. Fig. S2B). We then used all the conjugated proteins concomitantly in an experiment using U-251 MG cells (Fig. 5E). As expected from the previous experiments, IL-13.E13K-D2-NLS traveled to the cells’ nuclei, while IL-13.E13K-D2 did not. Both IL-13.E13K-NLS and IL-13.E13K ligand alone behaved like IL-13.E13K-D2, since they did demonstrate perinuclear localization but no nuclear transport whatsoever at 24 hours of experiment in GBM cells (Fig. 5E). Comparatively the same results were obtained at 5-minute, 4-hour, and 8-hour time points to that observed at 24 hours.
IL-13.E13K-D2-NLS localizes to the nucleus of G48a GBM cells
We repeated the nuclear trafficking of IL-13.E13K-D2-NLS experiments in another GBM cell line, G48a,13 which overexpresses IL-13Rα2. We obtained corresponding results to the U-251 MG cells. Again, almost all the cells had the IL-13.E13K-D2-NLS protein inside their nuclei not only at 8 and 24 hours, but already at 4 hours of the experiment (Fig. 6A). Again, at 5 minutes, we observed mainly plasma membrane binding with partial internalization of the protein (Fig. 6A). The Z-stack analysis for the 24-hour experiment directly documented the nuclear localization of IL-13.E13K-D2-NLS (Fig. 6B). Also, similar results were observed for the IL-13.E13K-D2, IL-13.E13K-NLS, and IL-13.E13K proteins in G48 cells as in U-251 MG cells (Fig. 6C). IL-13.E13K-D2 as well as IL-13.E13K-NLS and IL-13.E13K were not found to have the ability to localize to nuclei at any of the time points and exhibited mainly membrane/cytosolic/perinuclear localization (Fig. 6C, and data not shown).
Figure 6.
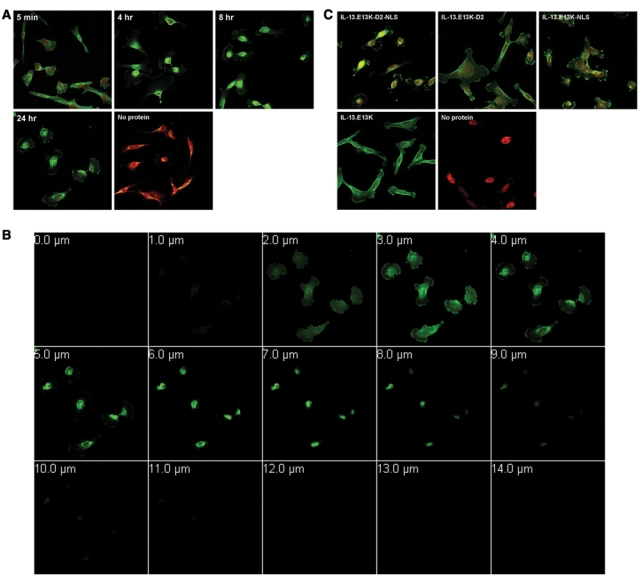
Biotin-labeled IL-13.E13K-D2-NLS localizes to the nuclei of G48a GBM cells. (A) Cellular localization experiments carried out in another high IL-13Rα2 expressor, G48a cells. Wells having no labeled proteins were used as controls. (B) Z-stack analysis of G48a cells treated with IL-13.E13K-D2-NLS at 24 hours. (C) Cellular localization experiments for the IL-13.E13K-D2-NLS, IL-13.E13K-D2, IL-13.E13K-NLS, and the IL-13.E13K were carried out in G48a cells concomitantly and their trafficking documented using confocal microscope. Cells with no labeled proteins were used as controls. The data are representative of 2 independent experiments.
IL-13.E13K-D2-NLS does not localize to the nucleus of LN229 GBM cells
We carried out identical experiments with biotin-labeled IL-13.E13K-D2-NLS in LN229 cells, very low expressors of IL-13Rα2.34 We observed that the protein displayed weak binding to the cell surface with some internalization, but we did not observe any nuclear localization for the IL-13.E13K-D2-NLS protein at any of the experimental time points (Fig. 7A) similarly to the 3 control proteins used in our assays (Fig. 7C). The Z-stack analysis for the IL-13.E13K-D2-NLS protein localization in the LN229 cells at 24 hours depicted a low internalization activity and no nuclear localization for the protein in these cells (Fig. 7B).
Figure 7.
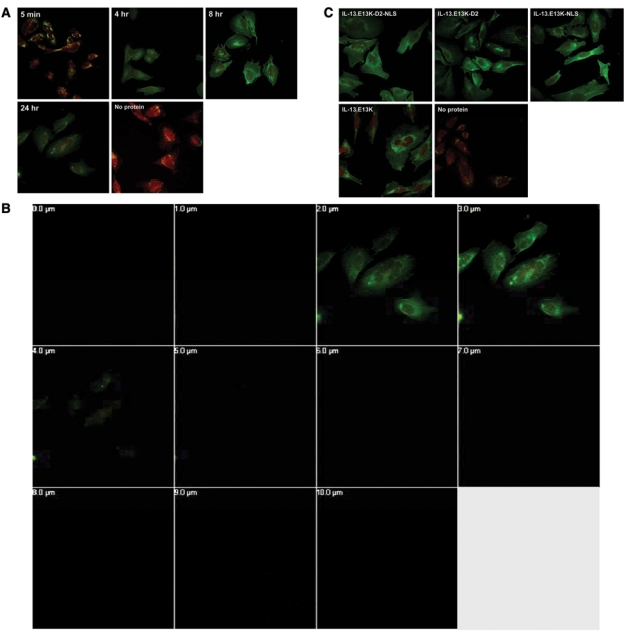
Biotin-labeled IL-13.E13K-D2-NLS does not localize to the nuclei of LN229 cells. (A) Cellular localization experiments carried out in low IL-13Rα2 expressing cells, LN229 GBM cells. (B) Z-stack analysis of the 24-hour treatment of LN229 cells with IL-13.E13K-D2-NLS. (C) LN229 cells exposed to IL-13.E13K-D2-NLS, IL-13.E13K-D2, IL-13.E13K-NLS, and the IL-13.E13K concomitantly. Wells treated with no labeled proteins were used as controls. The data are representative of 2 independent experiments.
Discussion
We have attempted to construct a universal proteinaceous module that would recognize cancer cells specifically and be able to travel via intracellular routes specifically to the cells’ nuclei. We have achieved this goal by targeting IL-13Rα2 in GBM cells with a single-chain protein composed of a modified receptor ligand, modified specialized cytosol translocation bacterial toxin domain D2 of PE, and a nuclear localization signal from the SV40 T antigen to form the IL-13.E13K-D2-NLS delivery vector. We have shown in direct tracking/visualization experiments that this vector binds to plasma membranes of GBM cells, enters endocytic compartment, concentrates in the perinuclear region, and then enters nuclei in a time-dependent manner. IL-13.E13K-D2-NLS persistently remains and accumulates in the nuclei up to at least 24 hours. Control proteins without either the NLS or the D2 of PE, and the IL-13.E13K ligand, could follow all of these steps with the exception of the last one (i.e., the efficient entry into the nucleus). Thus, we have directly demonstrated the journey of a designer protein–based vector from the cell surface to the nucleus of GBM cells.
To ascertain the direct demonstration of the phenomenon of IL-13.E13K-D2-NLS traveling from the membrane of cells to their nuclei, we have used 3 different methods of protein labeling with tractable dyes. First, we conjugated the proteins at the carboxyl groups of amino acids with Alexa Fluor 488 using EDC-Sulfo-NHS labeling technique and were able to demonstrate that NLS-guided vector traveled readily to the GBM cells’ nuclei, but the signals obtained were relatively weak and the procedure cumbersome as well as not readily reproducible. Therefore, we moved to the approach of directly conjugating proteins at primary amines with Alexa Fluor 488 and again observed analogous nuclear localization of the protein. We observed similar cell localization for recombinant proteins containing both wild-type IL-13 and mutant IL-13.E13K domains. The signals improved, but we strove to work out a system in which intracellular journey of IL-13.E13K-D2-NLS could be even more easily demonstrated. We then tried attaching proteins at primary amines with biotin and subsequently used HRP-streptavidin and tyramide–Alexa Fluor 488 signal amplification as a potentially more optimal technique. We repeated the intracellular localization experiments and found readily detectable nuclear localization of IL-13.E13K-D2-NLS and cytoplasmic/perinuclear localization of IL-13.E13K-D2. Also, the control proteins such as IL-13.E13K-NLS and IL-13.E13K demonstrated internalization but no nuclear localization. Very few cells had IL-13.E13K-NLS and IL-13.E13K sporadically in the nucleus. This could be due to the large number of these protein molecules in the vicinity of these cells’ nuclei. Another reason can be attributed to the metabolism of cancer cells and the increased number of nuclear import factors such as importin/karyopherins in malignant cells.35 With the last conjugation method, in which we biotinylated the proteins and then used the tyramide amplification method for detection, we also learned indirectly the importance of the lysine amino acids used for the conjugation and contained within the NLS, since lowering the number of attached biotins had a favorable effect on IL-13.E13K-D2-NLS accumulation to nuclei. These experiments strongly suggest the possibility of attaching various labels/drugs/dyes to a protein such as IL-13.E13K-D2-NLS in expectation of efficient delivery to the nuclei.
We have originally found that it is possible to target cancer cells and deliver peptides/proteins into the cell cytosol using modified PE.1 This finding was used in trying to accomplish the same goal with DT derivatives, which proved to be successful as well.11 In a recent report, a MAb was linked to the NLS peptide conjugated to an Auger electron emitter.36 The construct was able to recognize cells and kill them, although no direct demonstration of nuclear delivery was provided. It is also difficult to explain why the NLS peptides leave the antibody and the endocytotic compartment in order to travel to nuclei. Another group has engineered a recombinant protein similarly to our current design.37 This protein targets a nonspecifically overexpressed EGFR and, when conjugated to an α-emitter, kills EGFR-positive GBM cells. The construct was shown to internalize, but no direct evidence for the journey to the nucleus was provided. Besides, α-emitters are not necessarily needed to be in the nucleus in order to kill cells, since their cytotoxic radiation has a range of several layers of cells. Therefore, it cannot be excluded that only internalization might be required for such conjugates to work.
IL-13.E13K-D2-NLS might be best suited for conjugation with radiopharmaceuticals such as Auger electrons, chemotherapeutics, and photosensitizers. Auger electrons are low-energy electrons, similar to β– decay, that are emitted when another electron from a high-energy transition state moves to a low-energy inner-vacancy shell, transferring the differential energy to these electrons.38 Therefore, Auger electron therapy is very useful for specific tumor cell killing because of its very low bystander effects.39 Chemotherapeutics such as standard doxorubicin was used successfully in delivering to brain tumors by liposomes,40 but directing it specifically to nuclei of the targeted cells could promise both higher efficacy and safety through a novel dual-targeted approach. Also, delivering topotecan, a DNA topoisomerase 1 inhibitor, to the cells’ nuclei will be more effective. Photosensitizers, used in photodynamic therapy, would also be an ideal conjugating therapeutic because they generate singlet oxygen species which are active only around a 40-nm radius of action and have to been shown to be more efficacious if localized to the nucleus.41,42
In summary, we have generated a multiple-specificity vector for nuclear delivery targeting the IL-13Rα2–positive cancer cells, which can be further used for radio labeling and/or conjugation to various labels/chemotherapeutics/genetic elements, offering potentially more effective and safer treatment options for GBM and other cancers.
Materials and Methods
Cell culture
Human GBM cell lines U-251 MG and LN229 were obtained from American Type Culture Collection (Manassas, VA). U-251 MG cells were maintained in DMEM (Lonza, Walkersville, MD) supplemented with 1x nonessential amino acids (Invitrogen, Carlsbad, CA) and 10% FCS (Hyclone, Logan, UT). LN229 cells were grown in DMEM supplemented with 10% FCS. G48a cells were grown and maintained in RPMI 1640 (Lonza) supplemented with glucose, adjusted to 4 gm/L of media and 10% FCS.13
Cloning, production, and purification of targeted proteins
A duplex primer cloning strategy was employed wherein SV40 T-antigen NLS 5′ and 3′ sequence primers were synthesized (Invitrogen) and made into duplex DNA (containing Xho1/BamH1 ends) by incubating the primers in favorable annealing conditions. The annealed duplex was then subcloned into the IL-13-D2 containing plasmid using Xho1/BamH1 at the 3′ end to produce IL-13-D2-NLS. The IL-13-D2 plasmid was engineered by subcloning it from a previously generated IL-13-D2-PE38QQR plasmid.24 The IL-13 mutant recombinant constructs were made by replacing the wild-type IL-13 sequence from the parent plasmid with the mutant IL-13.E13K sequence.25 The NH2-terminal end of NLS domain was joined to the COOH terminal of IL-13.E13K domain using the HindIII site to form the IL-13.E13K-NLS plasmid. Also, all of these recombinant constructs were transformed in DH5α E. coli cells for amplification. All the constructs were sequenced at the DNA sequencing laboratory of the Comprehensive Cancer Center at Wake Forest University and analyzed for their in-frame DNA sequence using an automated sequence analyzer prior to protein expression.
Also, the IL-13/IL-13.E13K-D2-NLS and other control DNA constructs have been created in a manner such that they enable the expression of these proteins under the IPTG-inducible T7 promoter in BL21 (λDE3) E. coli protein expression system, as previously described.43 In brief, the recombinant constructs were transformed in BL21 cells and the cells were grown in Luria-broth media supplemented with 100 µg/mL of ampicillin at 37°C shaker. When the A600 of the bacterial culture media reached around 1.4, the recombinant protein expression in the cells was induced by addition of 1 nmol/L of IPTG and allowed to incubate for a further 90 minutes. The expressed proteins in the inclusion bodies were then denatured using 7 M guanidine (MP Biomedicals, Salon, OH) and 1,4-dithiothreitol (Sigma, St. Louis, MO). The reduced protein was then renatured in a buffer containing arginine/L-glutathione oxidase (Sigma). The protein was further dialyzed and purified by SP Sepharose ion-exchange liquid chromatography (GE Healthcare, Piscataway, NJ) using a fast protein liquid chromatography system (GE Healthcare). The purified proteins were subsequently run on SDS-PAGE gels to identify the purity of the isolated proteins. All of the proteins obtained were >90% pure.
Colorimetric MTS/PMS cell viability assay
1 × 103 U-251 MG cells were plated per well in quadruplicates for each concentration to be tested. IL-13.E13K-PE38QQR is an IL-13Rα2-based cytotoxin against GBM.24 After 24 hours of incubation at 37°C for the cells to attach, a fixed concentration (i.e., 1 µM) of the IL13.E13K-D2-NLS and other purified proteins as well as biotin-labeled (BL) proteins were added and incubated at 37°C for 1 hour. After 1-hour incubation, increasing concentrations of the IL-13.E13K-PE38QQR ranging from 0.1 to 100 ng/mL was added, and the plate was incubated for 48 hours. Cells treated with cyclohexamide and just the cytotoxin were used as controls. After 48 hours, cell viability was measured using the MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]/PMS [phenazine methosulfate] dye (Promega, Madison, WI) per the manufacturer’s instructions. The absorbance from the assay was measured at 490 nm using the plate reader SpectraMax 340 PC (Molecular Devices, Sunnyvale, CA), and data were plotted as percentage of control versus concentration of the toxin used.
IL-13-D2-NLS and IL-13-D2 labeling with EDC-Sulpho-NHS and Alexa Fluor 488
Purified IL-13-D2-NLS and the IL-13-D2 proteins were labeled at their carboxylate amino acids via EDC (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride). EDC reacts with a carboxyl group on an amino acid of the protein and forms an amine reactive O-acylisourea intermediate that swiftly reacts with an amino group to form an amide bond and release the isourea by-product. The intermediate is unstable in aqueous solutions and therefore, 2-step conjugation procedures require N-hydroxysuccinimide (Sulfo-NHS) stabilization. Sulfo-NHS reacts with the O-acylisourea intermediate and stabilizes it. Next, Alexa Fluor 488–hydrazide was added, which replaced the Sulfo-NHS and formed a stable amide bond on the carboxyl groups of the protein to form labeled protein conjugates.
The proteins were initially dissolved in the activation buffer (0.05 M MES, 0.5 M NaCl, pH 6) at the concentration of 1 mg/mL using buffer-exchange columns. Later, 2 mM EDC (Thermo Scientific, Waltham, MA) and 5 mM Sulfo-NHS (Thermo Scientific) were added to the proteins and allowed to react for 15 minutes at RT. Subsequently, 0.14 µL of 2-mercaptoethanol was added to quench the unreacted EDC. The protein-EDC-Sulfo-NHS conjugates were then dissolved in the coupling buffer (0.1 M sodium phosphate, 0.15 M NaCl, pH 7.5) using buffer exchange columns (Pierce, Rockford, IL). Next, Alexa Fluor 488–hydrazide (dissolved in the coupling buffer) (Invitrogen) was added at 25 molar concentration excess to the proteins and incubated in the dark at RT for 30 minutes. After incubation, 10 mM hydroxylamine (Thermo Scientific, Rockford, IL) was added to quench the excess fluor. The excess unreacted hydrazide fluor was removed using Pierce protein desalting columns.
Localization studies on IL-13Rα2 positive U-251 MG cells using Alexa Fluor 488 EDC-Sulfo-NHS labeled proteins
25,000 U-251 MG GBM cells were plated on coverslip/per well in a 24-well plate. The wells were plated in duplicates for each time point. The cells were allowed to adhere to the coverslips for 24 hours, after which 500 nM each of the Alexa Fluor 488 EDC-Sulpho-NHS labeled proteins were added to the U-251 MG cells for 15 minutes and 4 hours. After the incubations, the cells were fixed with acetone (pre-chilled at –20°F) for 10 minutes and washed with PBS 4x times. The coverslips were then mounted on the slides using the gel mount (Biomeda, Foster City, CA) and observed with LSM 510 Zeiss Confocal Microscope (Cellular Imaging Core, Comprehensive Cancer Center, Wake Forest University) and the images processed using Zeiss LSM Image Browser (version 4.2).
Direct labeling of IL-13.E13K-D2-NLS and IL-13.E13K-D2 with Alexa Fluor labels
The proteins were directly labeled with Alexa Fluor 488 dye using the Alexa Fluor 488 Microscale Protein Labeling Kit from Invitrogen (Carlsbad, CA) per the manufacturer’s instructions. A molar ratio of 25 of the dye to the protein was used to label both the proteins. The proteins were run on 12% SDS-PAGE. The gel was scanned using Typhoon 9210 (Amersham Pharmacia Biotech) for fluorescence signals and later stained using Coomassie blue dye.
Localization studies on IL-13Rα2 positive U-251 MG cells for Alexa Fluor 488 directly labeled proteins
25,000 U-251 MG GBM cells were plated on coverslips per well in a 24-well plate. The wells were plated in duplicates for each time point. After 24 hours for allowing the cells to adhere and attach to the plate, 1 µM/well of Alexa Fluor directly labeled proteins were added to the U-251 MG cells for 15 minutes and 4 hours. After the incubations, the cells were fixed with 5% paraformaldehyde (Ted Pella, Redding, CA) for 15 minutes at 37°C and washed with 1X PBS (3 times). The cells were then permeabilized with 0.1% Triton-X-100/0.2% BSA-PBS for 10 minutes at RT. After permeabilization, the cells were washed 3 times with 1X PBS. Subsequently, Topro-3 iodide (Invitrogen) was added at a concentration of 1:1000 dilution of the 1 mM stock to stain the cell nuclei. The coverslips were then mounted on the slides using the gel mount (Biomeda) and observed with LSM 510 Zeiss Confocal Microscope (Cellular Imaging Core, Comprehensive Cancer Center, Wake Forest University) and the images processed using Zeiss LSM Image Browser (version 4.2).
Direct labeling of IL-13.E13K-D2-NLS and IL-13.E13K-D2 with biotin and tyramide signal amplification system
The Biotin-XX Microscale Protein Labeling Kit (Invitrogen) was used to label the proteins per the manufacturer’s instructions. A different molar ratio of 12, 8, or 4 biotin-dye to the proteins was used. The biotin-labeled proteins were run on a gel and a Western blot carried out using streptavidin-HRP (Pierce, Rockford, IL) to detect for biotin-labeled proteins. The number of biotin molecules attached to the proteins was determined by the FluoReporter Biotin Quantitation Assay Kit (Invitrogen) per the manufacturer’s guidelines. The fluorescent signals were measured using the plate reader SpectraMax 340 PC (Molecular Devices) and data were plotted as concentration of the standard biocytin in pmoles versus relative fluorescence units.
Localization studies on IL-13Rα2 positive U-251 MG, G48a, and IL-13Rα2 low expressors-LN229 cell with biotin-conjugated proteins
12,500 U-251 MG or G48a or LN229 GBM cells were plated on coverslips per well in a 24-well plate. The wells were plated in duplicates for each time point. After 24 hours, 1 µM/well of biotin-labeled proteins was added onto the cells for 15 minutes, 4, 8, and 24 hours. After the incubations, the cells were fixed with 4% paraformaldehyde (Ted Pella, Redding, CA) for 15 minutes at 37°C and washed with PBS 4x times. The cells were then permeabilized with 0.1% Triton-X-100/0.2% BSA-PBS for 10 minutes at RT. After permeabilization, the cells were washed 3 times with 1X PBS. After washings, Tyramide Signal Amplification Kit (Invitrogen) using Alexa Fluor 488 dyes and HRP-streptavidin was carried out per the manufacturer’s instructions. Topro-3 iodide (Invitrogen) was added at a concentration of 1:1000 dilution of the 1 mM stock to stain the cell nuclei. After the tyramide staining, wells were washed and mounted with gel mount (Biomeda) and observed with LSM 510 Zeiss confocal microscope (Cellular Imaging Core, Comprehensive Cancer Center, Wake Forest University) and the images processed using Zeiss LSM Image Browser (version 4.2).
Immunoblotting
500 ng/well of each of the recombinant biotin conjugated proteins were loaded onto a 12% SDS-PAGE gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Pierce, Rockford, IL). Blots were blocked with 5% milk-phosphate buffered saline (PBS) for 1 hour at room temperature (RT). Biotin-proteins were detected using streptavidin conjugated with horseradish peroxidase (Thermo Fisher Scientific, Rockford, IL) diluted 1:16000 in blocking buffer. Detection was performed using the ECL plus Western Blotting Detection System (Amersham Biosciences, UK). Membranes were exposed to autoradiographic film Kodak Biomax XR. Films were scanned at 600x dpi and images compiled using Jasc Paint Shop Pro v 6.0.
Supplementary Material
Footnotes
Dr. Waldemar Debinski is a consulting scientific advisor and a shareholder in Targepeutics, Inc. This work was not supported by any industrial entity. Hetal Pandya and Denise Gibo report no potential conflicts of interest.
This study was supported by a grant from the NIH/NCI R01 CA74145.
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. Debinski W, Siegall CB, FitzGerald D, Pastan I. Substitution of foreign protein sequences into a chimeric toxin composed of transforming growth factor alpha and Pseudomonas exotoxin. Mol Cell Biol. 1991. March;11(3):1751-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Debinski W, Pastan I. Monovalent immunotoxin containing truncated form of Pseudomonas exotoxin as potent antitumor agent. Cancer Res. 1992. October 1;52(19):5379-85 [PubMed] [Google Scholar]
- 3. Debinski W, Obiri NI, Pastan I, Puri RK. A novel chimeric protein composed of interleukin 13 and Pseudomonas exotoxin is highly cytotoxic to human carcinoma cells expressing receptors for interleukin 13 and interleukin 4. J Biol Chem. 1995. July 14;270(28):16775-80 [DOI] [PubMed] [Google Scholar]
- 4. Chiron MF, Fryling CM, FitzGerald D. Furin-mediated cleavage of Pseudomonas exotoxin-derived chimeric toxins. J Biol Chem. 1997. December 12;272(50):31707-11 [DOI] [PubMed] [Google Scholar]
- 5. Inocencio NM, Moehring JM, Moehring TJ. Furin activates Pseudomonas exotoxin A by specific cleavage in vivo and in vitro. J Biol Chem. 1994. December 16;269(50):31831-5 [PubMed] [Google Scholar]
- 6. Moehring JM, Inocencio NM, Robertson BJ, Moehring TJ. Expression of mouse furin in a Chinese hamster cell resistant to Pseudomonas exotoxin A and viruses complements the genetic lesion. J Biol Chem. 1993. February 5;268(4):2590-4 [PubMed] [Google Scholar]
- 7. Ogata M, Chaudhary VK, Pastan I, FitzGerald DJ. Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J Biol Chem. 1990. November 25;265(33):20678-85 [PubMed] [Google Scholar]
- 8. Jinno Y, Ogata M, Chaudhary VK, et al. Domain II mutants of Pseudomonas exotoxin deficient in translocation. J Biol Chem. 1989. September 25;264(27):15953-9 [PubMed] [Google Scholar]
- 9. Siegall CB, Ogata M, Pastan I, FitzGerald DJ. Analysis of sequences in domain II of Pseudomonas exotoxin A which mediate translocation. Biochemistry. 1991. July 23;30(29):7154-9 [DOI] [PubMed] [Google Scholar]
- 10. London SD, Schmaljohn AL, Dalrymple JM, Rice CM. Infectious enveloped RNA virus antigenic chimeras. Proc Natl Acad Sci USA. 1992. January 1;89(1):207-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stenmark H, Moskaug JO, Madshus IH, Sandvig K, Olsnes S. Peptides fused to the amino-terminal end of diphtheria toxin are translocated to the cytosol. J Cell Biol. 1991. June;113(5):1025-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stupp R, Dietrich PY, Ostermann KS, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002. March 1;20(5):1375-82 [DOI] [PubMed] [Google Scholar]
- 13. Debinski W, Gibo DM. Fos-related antigen 1 modulates malignant features of glioma cells. Mol.Cancer Res. 2005. April;3(4):237-49 [DOI] [PubMed] [Google Scholar]
- 14. Mintz A, Gibo DM, Slagle-Webb B, Christensen ND, Debinski W. IL-13Ralpha2 is a glioma-restricted receptor for interleukin-13. Neoplasia. 2002. September;4(5):388-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mintz A, Gibo DM, Madhankumar AB, Cladel NM, Christensen ND, Debinski W. Protein- and DNA-based active immunotherapy targeting interleukin-13 receptor alpha2. Cancer Biother Radiopharm. 2008. October;23(5):581-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005. October;3(10):541-51 [DOI] [PubMed] [Google Scholar]
- 17. Wykosky J, Gibo DM, Stanton C, Debinski W. Interleukin-13 receptor alpha 2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clin Cancer Res. 2008. January 1;14(1):199-208 [DOI] [PubMed] [Google Scholar]
- 18. Debinski W. Drug cocktails for effective treatment of glioblastoma multiforme. Expert Rev Neurother. 2008. April;8(4):515-7 [DOI] [PubMed] [Google Scholar]
- 19. Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. 1999. May;5(5):985-90 [PubMed] [Google Scholar]
- 20. Saikali S, Avril T, Collet B, et al. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J Neurooncol. 2007. January;81(2):139-48 [DOI] [PubMed] [Google Scholar]
- 21. Debinski W, Gibo DM. Molecular expression analysis of restrictive receptor for interleukin 13, a brain tumor-associated cancer/testis antigen. Mol Med. 2000. May;6(5):440-9 [PMC free article] [PubMed] [Google Scholar]
- 22. Bernard J, Treton D, Vermot-Desroches C, et al. Expression of interleukin 13 receptor in glioma and renal cell carcinoma: IL13 Ralpha2 as a decoy receptor for IL13 1. Lab Invest. 2001. September; 81(9):1223-31 [DOI] [PubMed] [Google Scholar]
- 23. Kawakami K, Taguchi J, Murata T, Puri RK. The interleukin-13 receptor alpha2 chain: an essential component for binding and internalization but not for interleukin-13-induced signal transduction through the STAT6 pathway. Blood. 2001. May 1;97(9):2673-9 [DOI] [PubMed] [Google Scholar]
- 24. Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin Cancer Res. 1995. November;1(11):1253-8 [PubMed] [Google Scholar]
- 25. Madhankumar AB, Mintz A, Debinski W. Interleukin 13 mutants of enhanced avidity toward the glioma-associated receptor, IL13Ralpha2. Neoplasia. 2004. January;6(1):15-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thompson JP, Debinski W. Mutants of interleukin 13 with altered reactivity toward interleukin 13 receptors. J Biol Chem. 1999. October 15;274(42):29944-50 [DOI] [PubMed] [Google Scholar]
- 27. Debinski W, Gibo DM, Obiri NI, Kealiher A, Puri RK. Novel anti-brain tumor cytotoxins specific for cancer cells. Nat Biotechnol. 1998. May;16(5):449-53 [DOI] [PubMed] [Google Scholar]
- 28. Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984. December;39(3 Pt 2):499-509 [DOI] [PubMed] [Google Scholar]
- 29. Hubner S, Xiao CY, Jans DA. The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T-antigen nuclear localization sequence by importin. J Biol Chem. 1997. July 4;272(27):17191-5 [DOI] [PubMed] [Google Scholar]
- 30. Rihs HP, Peters R. Nuclear transport kinetics depend on phosphorylation-site-containing sequences flanking the karyophilic signal of the Simian virus 40 T-antigen. EMBO J. 1989. May;8(5):1479-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rihs HP, Jans DA, Fan H, Peters R. The rate of nuclear cytoplasmic protein transport is determined by the casein kinase II site flanking the nuclear localization sequence of the SV40 T-antigen. EMBO J. 1991. March;10(3):633-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiao CY, Hubner S, Jans DA. SV40 large tumor antigen nuclear import is regulated by the double-stranded DNA-dependent protein kinase site (serine 120) flanking the nuclear localization sequence. J Biol Chem. 1997. August 29;272(35):22191-8 [DOI] [PubMed] [Google Scholar]
- 33. Mintz A, Gibo DM, Madhankumar AB, Debinski W. Molecular targeting with recombinant cytotoxins of interleukin-13 receptor alpha2-expressing glioma. J Neurooncol. 2003. August;64(1-2):117-23 [DOI] [PubMed] [Google Scholar]
- 34. Kawakami K, Kawakami M, Liu Q, Puri RK. Combined effects of radiation and interleukin-13 receptor-targeted cytotoxin on glioblastoma cell lines. Int J Radiat Oncol Biol Phys. 2005. September 1;63(1):230-7 [DOI] [PubMed] [Google Scholar]
- 35. Jans DA, Xiao CY, Lam MH. Nuclear targeting signal recognition: a key control point in nuclear transport? Bioessays. 2000. June;22(6):532-44 [DOI] [PubMed] [Google Scholar]
- 36. Costantini DL, Chan C, Cai Z, Vallis KA, Reilly RM. (111)In-labeled trastuzumab (Herceptin) modified with nuclear localization sequences (NLS): an Auger electron-emitting radiotherapeutic agent for HER2/neu-amplified breast cancer. J Nucl Med. 2007. August;48(8):1357-68 [DOI] [PubMed] [Google Scholar]
- 37. Rosenkranz AA, Vaidyanathan G, Pozzi OR, Lunin VG, Zalutsky MR, Sobolev AS. Engineered modular recombinant transporters: application of new platform for targeted radiotherapeutic agents to alpha-particle emitting 211 At. Int J Radiat Oncol Biol Phys. 2008. September 1;72(1):193-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buchegger F, Perillo-Adamer F, Dupertuis YM, Delaloye AB. Auger radiation targeted into DNA: a therapy perspective. Eur J Nucl Med Mol Imaging. 2006. November;33(11):1352-63 [DOI] [PubMed] [Google Scholar]
- 39. Goddu SM, Rao DV, Howell RW. Multicellular dosimetry for micrometastases: dependence of self-dose versus cross-dose to cell nuclei on type and energy of radiation and subcellular distribution of radionuclides. J Nucl Med. 1994. March;35(3):521-30 [PubMed] [Google Scholar]
- 40. Madhankumar AB, Slagle-Webb B, Wang X, et al. Efficacy of interleukin-13 receptor-targeted liposomal doxorubicin in the intracranial brain tumor model. Mol Cancer Ther. 2009. March;8(3):648-54 [DOI] [PubMed] [Google Scholar]
- 41. Liang H, Shin DS, Lee YE, et al. Subcellular phototoxicity of 5- aminolaevulinic acid (ALA). Lasers Surg Med. 1998;22(1):14-24 [DOI] [PubMed] [Google Scholar]
- 42. Takemura T, Ohta N, Nakajima S, Sakata I. Critical importance of the triplet lifetime of photosensitizer in photodynamic therapy of tumor. Photochem Photobiol. 1989. September;50(3):339-44 [DOI] [PubMed] [Google Scholar]
- 43. Madhankumar AB, Mintz A, Debinski W. Alanine-scanning mutagenesis of alpha-helix D segment of interleukin-13 reveals new functionally important residues of the cytokine. J Biol Chem. 2002. November 8;277(45):43194-205 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.