

Abstract
An additivity-based sequence to reactivity algorithm for the interaction of members of the Kazal family of protein inhibitors with six selected serine proteinases is described. Ten consensus variable contact positions in the inhibitor were identified, and the 19 possible variants at each of these positions were expressed. The free energies of interaction of these variants and the wild type were measured. For an additive system, this data set allows for the calculation of all possible sequences, subject to some restrictions. The algorithm was extensively tested. It is exceptionally fast so that all possible sequences can be predicted. The strongest, the most specific possible, and the least specific inhibitors were designed, and an evolutionary problem was solved.
The pioneers of protein chemistry (1, 2) demonstrated that for many proteins the sequence suffices to specify reactivity. This demonstration was equivalent to showing that for such proteins sequence to reactivity algorithms (SRAs) must exist. However, the search for such SRAs was not highly productive and was largely stalled by looking for these SRAs in two steps: sequence to folding and folding to reactivity. Our SRA relies in its first step on recognition of homology. In the second and more difficult step, it relies on additivity.
Additivity is a major predictive principle in chemistry (3). In protein chemistry amino acid residue additivity was studied by various workers as soon as site-specific mutagenesis became tractable. Additivity was applied to various protein reactions, among them protein unfolding, protein–protein and protein–ligand association, enzyme kinetics, and hydrolysis of internal peptide bonds in proteins (4–14). The use of the additivity principle in biochemistry recently was reviewed (15).
Here we report on a successful, 20-year-long (16) effort to determine an additivity-based SRA (Fig. 1) for predicting the equilibrium constants of some serine proteinases with members of the Kazal family (17, 18) of standard mechanism (17), canonical (19) protein inhibitors. This family is named after L. Kazal, discoverer of the first of the pancreatic secretory trypsin inhibitors, PSTI, that are present in all vertebrates. The family has many members. Among them are ovomucoids, abundant proteins in avian egg whites, which consist of three tandem Kazal domains (20). Ovomucoids from closely related species of birds were shown to differ strikingly in their inhibitory specificity (21). Ovomucoid third domains from 153 species of birds were isolated and sequenced (22–24), and their interactions with serine proteinases were studied. The strongest association equilibrium constant, Ka, for a member of this set is 1.4 × 1012 M−1; the weakest measured is 7.1 × 102 M−1. These results underscore the need for an algorithm because identifying a protein as an ovomucoid third domain does not predict that it will or will not be an effective inhibitor of a particular serine proteinase.
Figure 1.

Flow diagram of the SRA construction. Labels within the rectangles are general. Comments are applications to the Kazal family.
Aside from the large range of Ka values among closely related natural variants, additional strong reasons for choosing a standard mechanism, canonical inhibitor family, were the anticipated additivity of individual contact residue contributions (see below) and the availability in our laboratory of techniques for measuring Ka values for enzyme-inhibitor pairs over the 103 M−1 to 1013 M−1 dynamic range with an accuracy of ±20%.
Materials and Methods
Enzymes.
Bovine chymotrypsin Aα (CHYM) and subtilisin Carlsberg (CARL) were purchased from Worthington and Sigma, respectively, and human leukocyte elastase (HLE) was from Elastin Products, St. Louis. Porcine pancreatic elastase (PPE), freed from all chymotrypsin and trypsin activity, was a gift from the late M. Laskowski, Sr. (Roswell Park Memorial Institute, Buffalo, NY). Streptomyces griseus proteinases A and B (SGPA and SGPB) were purified in this laboratory from pronase. These were compared with standards given by D. A. Estell (Genencor International, Palo Alto, CA), L. Smillie (University of Alberta), and J. Travis (University of Georgia, Athens).
Natural Kazal Domains.
Natural ovomucoid third domains were prepared and characterized as described (22–24). Natural ovomucoid first domains were obtained by CNBr cleavage of entire ovomucoid for OMHPA1 (from gray partridge, Perdix perdix) and OMHMP1 (from Himalayan monal pheasant, Lophophorus imperianus), and by thermolysin hydrolysis for OMBWS1 (from black-winged stilt, Himantopus himantopus). They were characterized by amino acid analysis and sequencing. The R44S variant of human pancreatic secretory trypsin inhibitor (HPSTI) and goose pancreatic secretory trypsin inhibitor are described in Table 3.
Table 3.
−ΔG° for Kazal domains other than ovomucoid third domains
 |
| Kazal Inhibitors | −ΔG°,
Kcal/mol
|
|||||
|---|---|---|---|---|---|---|
| CHYM | CARL | SGPA | SGPB | HLE | ||
| HSPTI | Measured | 7.6* | 9.4 | 10.2 | 10.2 | 5.8 |
| Predicted | 4.4* | 8.7 | 11.0 | 9.9 | 5.5 | |
| GPSTI | Measured | 6.3 | 5.1 | |||
| Predicted | 6.4 | 6.5 | ||||
| OMBWS1 | Measured | 8.8† | 10.1 | 9.7 | 6.2 | |
| Predicted | 5.6† | 10.7 | 10.0 | 7.3 | ||
| OMHPA1 | Measured | 6.4 | 6.1 | 5.9 | ||
| Predicted | 6.0 | 7.4 | 5.7 | |||
| OMHMP1 | Measured | 7.2 | 6.9 | 6.1 | ||
| Predicted | 6.2 | 6.9 | 6.0 | |||
Primary structures of the Kazal inhibitors used in this table are given. The consensus contact residue set is shaded yellow. Residues in red differ from those in OMTKY3, the ones in black do not. The residues in gray are insertions and extensions compared to OMTKY3. HPSTI is the R44S variant of human pancreatic secretory trypsin inhibitor (45), and GPSTI is goose pancreatic secretory trypsin inhibitor (46). OMBWS1, OMHPA1 and OMHMP1 are ovomucoid first domains from black-winged stilt (Himantopus himantopus), grey partridge (Perdix perdix) and Himalayan monal pheasant (Lophophorus imperianus), respectively. Limitations of material and very low predicted values prevented us from making the 13 missing measurements. The nonadditive (designated by *) and partially additive (designated by
) cases are emphasized.
Recombinant Turkey Ovomucoid Third Domain (OMTKY3) Variants.
These were expressed in the periplasmic space of Escherichia coli as fusion proteins with protein A domains. Their isolation, purification, and extensive characterization was described (25) for the P1 variants.
Ka Determination.
The extensive modification of the procedure of Green and Work (26) is described (25). The conditions were 21 ± 2°C, pH 8.30, ionic strength 0.10 M. The measurement range was 103 M−1 to 1013 M−1, the accuracy ±20%.
Results and Discussion
Selection of Targets and the Wild Type.
Among the natural third domains we sequenced most are effective inhibitors of some chymotrypsins, elastases, and subtilisins. Only a few inhibit trypsins and Glu-specific SGP (27). None are effective inhibitors of furin and proprotein convertases. Therefore, we selected six enzymes: CHYM, PPE, CARL, SGPA, SGPB, and HLE. All are among the best studied of serine proteinases. Three are bacterial and three are mammalian. They all have hydrophobic S1 pockets that range from small (PPE) to very large (CHYM). All of them are strongly inhibited by OMTKY3 (28) as are many other enzymes. In addition, OMTKY3 very nearly represents the most probable sequence among the 153 species we examined (24). Therefore, it was chosen as the wild type.
Selection of Contact Positions.
X-ray crystallography of complexes of OMTKY3, of many of its variants, and of some PSTI variants with SGPB, CHYM and HLE showed that a consensus set of 12 contact positions in the inhibitors is in contact with the enzymes (29–34) (Fig. 2). When sequences of ovomucoid third domains from 153 species are compared, the seven most variable positions in the 51 residue domains all lie in the consensus contact residue set (24). Analysis of Kas of most of these variants indicates that changes among the residues in the consensus contact set frequently cause very large changes in Ka. In sharp contrast, changes in noncontact residues often do not affect Ka values beyond experimental error. The few clear Ka changes caused by noncontact residues are all small. Therefore, it appears that the 12 residue consensus set suffices to determine changes in Ka relative to OMTKY3. However, further reduction is possible. Most of the hypervariable contact residues are also highly exposed but two of the contact residues, P3 Cys-16 and P15′ Asn-33, serve clear structural roles and show no (P3) or very little variation (P15′) from sequence to sequence. This strong conservation at P3 and P15′ persists in all of the 471 known sequences (see data at http://www.chem.purdue.edu/LASKOWSKI) of Kazal inhibitors. Therefore, we designated the remaining 10-residue set (Figs. 1 and 2) as the variable consensus contact set.
Figure 2.

The covalent structure (22) of OMTKY3. The bars indicate disulfide bridges. The arrow points to the reactive site peptide bond between P1 and P1′ residues (47). The 12 colored residues comprise the consensus contact residue set (24, 29–31). Of these 12, the two in green are structural and accept very few mutations in evolution. In contrast, the remaining 10 are hypervariable (22, 48). Changing one of the white (noncontact) residues for another has little effect on ΔG°, whereas changing one of the colored ones often has a very large effect.
Expression of Variants, Measurement of ΔG°.
Because additivity will be assumed, to have a general SRA we need to have the wild type (OMTKY3) and 19 possible coded variants at each of the 10 variable contact positions. Happily, all 191 variants could be expressed and were soluble and stable. The 191 × 6 = 1,146 equilibrium constants for the interaction of each variant with our six selected enzymes were measured. The values for P1 variants were already reported (25). The remainder will be published separately and may be requested now from the corresponding author. Again happily, all of the equilibrium constants fell into the 103 M−1 to 1013 M−1 dynamic range of measurements. The lowest was 4.8 × 103 M−1 and the highest was 8.2 × 1012 M−1. This was not a foregone conclusion. In a set of seven eglin C variants at P1, the Leu-45 variant (wild type) interacts with SGPA and SGPB too strongly for direct measurement whereas the Asp-45 and Glu-45 variants interact with PPE too weakly (35). For each of the six selected enzymes, for variants at each of the 10 variable consensus contact positions, i, there are 19 association equilibrium constants Ka(X). These were converted to ΔG°(X) = −RT ln Ka (X) values. These in turn were converted at each of the 10 variable consensus contact positions to ΔΔG° (XTKY i X) = ΔG° (X) − ΔG° OMTKY3 values.
The SRA.
If a variant differs from OMTKY3 only by changes among the 51 − 12 = 39 noncontact positions, we state that its predicted ΔG° is simply the value for OMTKY3. If a substitution is among the 10 variable consensus residues, we add to ΔG°TKY the single ΔΔG° (XTKY i X) term. For more changes in contact, we sum the ΔΔG° (XTKY i X) terms
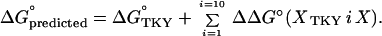 |
1 |
The simple additivity of the ΔΔG° (XTKY i X) is the major approximation involved in this work. It is its use that makes our SRA so simple. There are some restrictions on the application of Eq. 1 to predict ΔG° for the association of any Kazal domain with any of the six enzymes we selected. Many Kazal family members are present in multidomain proteins, such as ovomucoid (20). Here we predict only for single Kazal domains, such as PSTI or for isolated domains of multidomain proteins such as turkey ovomucoid. The decision not to vary the green residues in Fig. 2 (P3 Cys-16 and P15′ Asn-33) requires that these must be present in all of the inhibitors we predict. A group of Kazal inhibitors, called nonclassical (36), have at P5 a disulfide-bridged Cys. We have no information on disulfide-bridged Cys, only on reduced Cys. The SRA is based on an assumption that the variable consensus contact set of residues is additive. This additivity is equivalent to assuming that the interactions, if any, among the contact residues present in the free inhibitors do not change appreciably on formation of complexes with enzymes. This assumption is not valid for the P2 Thr-17–P1′ Glu-19 residue pair (Fig. 2). The side chains of these two residues are hydrogen-bonded to each other in the free inhibitor (37). The hydrogen bond shortens on complex formation with SGPB (29) and CHYM (30) but not with HLE (31). The same shortening is seen when the structure of variant 3 of PSTI (38) (this variant has P2 Thr-17 and P1′ Glu-19) is compared in free inhibitor and in complex with chymotrypsinogen (34). A few tests based on ΔΔG° value indicate major P2–P1′ pair nonadditivity. We therefore restrict our predictions to the 39 P2–P1′ pairs we measured rather than to all possible 400 pairs. A simple wording of the restriction is that either P2 Thr or P1′ Glu (or both) must be present. The elimination of this restriction would widen the scope of SRA to more Kazal inhibitors, and in other protein systems there will be patches of nonadditivity in generally additive situations. For avian ovomucoid third domains where the P2 Thr–P1′ Glu is very common, the P2–P1′ restriction allows one to deal with substantial majority of the domains. For Kazal inhibitors at large where this is not common, the current SRA encompasses a significant minority (about 30%) of sequenced domains.
Testing the SRA.
To use Eq. 1 to obtain ΔG°predicted, we need only the sequence of the Kazal domain of interest. Therefore, there is no need to have the protein. However, to test the SRA, we need to have the protein to obtain its ΔG°measured for interaction with the enzymes.
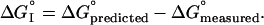 |
2 |
The subscript I on ΔG°I was designed (7) to indicate interactions and therefore to be a measure of nonadditivity. In our system, the nonadditivity component of this term is a measure of the change in interactions on complex formation. However, ΔG°I also contains two other terms. One arises from the changes in noncontact residues (white circles in Fig. 2), which are neglected in Eq. 1. The other arises from errors in measurement of ΔG° values as ΔG°measured (directly) and more importantly ΔG°predicted (indirectly) involve such measurements.
The test set of proteins for an SRA can be either designed or natural. In our case, the choice was easy. We already had a set of ΔG° values for the interaction of the six enzymes we study with 92 different, natural ovomucoid third domains. The contact residues in these domains are hypervariable (24). These domains were gathered to serve as a primary data source for the construction of the SRA (4, 39). As we became facile with the production of recombinant OMTKY3 variants in the 1990s (25), the acquisition of natural variants ceased. The SRA described above is based solely on the data on 190 single variants of OMTKY3 in the contact region (Fig. 2) and on the OMTKY3 itself. The set of the natural variant data were thus available as a test set. The raw test set contains somewhat less (443) than 92 × 6 = 552 ΔG° values as limitations in the amounts of available material did not allow for the determination of some binding constants for weak interactions. The 443 ΔG° values were sieved for sequences that meet the current SRA restrictions and 398 remained. Predictions were made for all of them (Eq. 1) and the ΔG°I values (Eq. 2) were calculated. The average absolute value of ΔG°I is 510 cal/mol. This is small compared with the range of 13.5 Kcal/mol in ΔG°. It is also small compared with the expectations of inhibitor designers. Surprisingly, this value is significantly smaller than the 900 cal/mol for the set of 17 P1 variants of OMTKY3 interacting with SGPB (40) based on the determined three-dimensional structures of the relevant complexes. Our predictions here are based on the SRA and the sequence alone.
Eqs. 1 and 2 lend themselves to error analysis. From the fits obtained and the reproducibility of the about 3,000 ΔG° values determined in our laboratory, we conclude that the average standard deviation, σ, is ΔG° ± 100 cal/mol. This finding corresponds to log Ka ± 0.075 and Ka ± 20%. Note that this is an average value. At both the upper Ka = 1013 M−1 and lower Ka = 103 M−1, σ is much greater. σ is significantly smaller for OMTKY3, which was measured very frequently and served as a control in various experiments. However, in view of the difficulties in estimating individual σ values, we consider only a constant one. We make the calculations at 2σ level.
In Eq. 2, both ΔG°measured and ΔG°predicted have experimental errors. For ΔG°measured, it is σ. For ΔG°predicted, the error depends on k, the number of variable consensus contact residues in OMTKY3 that are replaced by others in the predicted variant. Eq. 2 can be rewritten as:
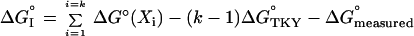 |
3 |
because each ΔΔG° (XTKY i X) term is the difference between ΔG° (Xi) and ΔG°TKY. Of the terms of interest to us here, the ΔG° (Xi) terms and the ΔG°measured terms are independent variables. On the other hand, when several ΔG°TKY terms occur, these are not. For the entire system
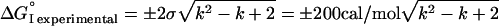 |
4 |
We divided the entire 398-member ΔG° measured set on the basis of the number of contact region substitutions. Cases where ΔG°I is within the limits of Eq. 4 are called additive, outside the limits but within twice the limits partially additive, and outside that nonadditive (Table 1). Obviously larger absolute errors were allowed at k = 6 than for k = 0 (changes not in contact). However, after that correction is made, small deviations from additivity will not show at all at k = 0 and 1 as there is no additivity there. Such corrections are expected to be much smaller for k = 2 where there is only one potential pairwise nonadditivity than at k = 6 where there are many pairwise, triple, quadruple, pentuple, and one hexuple potential nonadditivities.
Table 1.
Predictive success for ovomucoid third domains differing from OMTKY3 by k contact residues
| Number of consensus contact residue
changes
|
Total | % | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |||
| Additive | 11 | 67 | 78 | 42 | 28 | 23 | 3 | 252 | 63 |
| Partially additive | 0 | 12 | 23 | 25 | 16 | 26 | 4 | 106 | 27 |
| Nonadditive | 0 | 5 | 12 | 11 | 3 | 7 | 2 | 40 | 10 |
| Total | 11 | 84 | 113 | 78 | 47 | 56 | 9 | 398 | 100 |
The 398 ΔG°measured set also was divided according to the enzyme (Table 2). Clearly, CARL is the worst. It is the only member of the subtilin family of enzymes in our set. Also there is no three-dimensional structure of OMTKY3 or any Kazal inhibitor in complex with CARL, even though there are many structures of CARL in complex with other standard mechanism (17), canonical (19) protein inhibitors. Thus, some of its contact residues may not be in the consensus set. SGPA is the best. We are unable to account for its difference from SGPB but we use SGPA for calculation of the distribution of ΔG° values for all possible contact region sequences.
Table 2.
Predictive success for ovomucoid third domains by enzyme
| CHYM | PPE | CARL | SGPA | SGPB | HLE | |
|---|---|---|---|---|---|---|
| Additive | 45 | 44 | 29 | 56 | 45 | 31 |
| Partially additive | 17 | 16 | 24 | 14 | 20 | 13 |
| Nonadditive | 7 | 8 | 14 | 1 | 6 | 8 |
| Total | 69 | 68 | 67 | 71 | 71 | 52 |
The test above was made on avian ovomucoid third domains that are similar to OMTKY3. For these, the largest k value is 6. It will be more interesting to make a comparably large test with other members of the Kazal family. However, only a small set was available. Table 3 summarizes what we obtained after sieving this set for our restrictions, which in this case were more bothersome than for avian ovomucoid third domain. The two top sequences are for PSTIs, the bottom three for avian ovomucoid first domains. It should be noted that domains in many multidomain Kazal inhibitors could be readily divided into a type and b type domains (41). Ovomucoid first and second domains are a type whereas the third domains are b type. Therefore, ovomucoid first and third domains while homologous are very different proteins. For HPSTI and OMHMP1 k = 8; for the three other comparisons k = 9, the largest number allowed by our sieve. Of the 17 cases where comparisons could be made, 15 are additive, one is partially additive and one is nonadditive, a somewhat similar distribution as in ovomucoid third domains. On this limited number of tests, we conclude that SRA holds for all Kazal inhibitors provided that the restrictions are met.
Why do Kazal inhibitors work so well in the SRA? (i) The association is nearly lock and key rather than induced fit. (ii) The putative P1 residue always inserts into the S1 cavity of the enzyme, however deleterious this is to local binding. (iii) Compared with other protein reactions the ΔΔG° terms observed here are very large. (iv) In contrast to antigen–antibody interactions the main chain–main chain contributes importantly to the interaction energy (42). (v) In contrast to nuclease–nuclease protein inhibitor system, the side-chain interactions are largely hydrophobic and not electrostatic (10). (vi) The inhibitor is globally convex and local interactions are mainly of the inhibitor's side chains inserting into the enzyme's pockets. (vii) A method of measuring Kas accurately and over a wide dynamic range is available. Many of us think that it is the last of these that is most important for success.
It seems highly likely that the SRAs can be developed for several other families of standard mechanism canonical protein inhibitors of serine proteinases. However, what is most intriguing is whether additivity-based SRAs can be developed for some other protein–ligand and protein–protein interactions. We are hopeful.
Applications of the Additivity-Based SRA.
Eq. 1 is very simple. Aside from the SRA data that are already available, it requires only the sequence of the variable consensus contact set. Therefore, the value of ΔG° can be predicted for proteins whose sequences are known only from DNA sequencing and that were never isolated or expressed. We made many such predictions. Predictions also can be made for postulated nodal sequences in phylogenetic trees or for sequences constructed to meet some design objectives (see example below). Because the additivity-based SRA is so very fast many potential designs can be screened before settling on the ones we want to express. But most surprisingly we can calculate ΔG° values for all possible sequences of Kazal inhibitors interacting with a stated enzyme.
For the demonstration, we chose SGPA as it is the most additive. However, studies with the five other enzymes gave similar results. Subject to our restrictions (see above) there are 39 × 208 ≅ 1012 possible sequences that affect the value of ΔG°. All of these were calculated on a personal computer in a few days. There was not enough memory to store them all but their distribution could be compiled and is given in Fig. 3. Even though it appears continuous it is not. There is a largest (weakest possible inhibitor) ΔG° value on the left side (not shown) and smallest (strongest possible inhibitor) shown on the right side. This value could have been obtained from the calculation but additivity provides an even simpler way (39). A sequence composed of best residues at each of the 10 positions is the best possible sequence. Such sequences and their Kas are listed for all six enzymes in Table 4. They are all very strong. All are larger than the 1013 M−1 upper limit of the measurement range. Additivity allows for simple calculation of the weakest possible Ka (largest ΔG°) but this is not likely to be useful. Additivity also allows for the calculation of the average ΔG° (−5.13 kcal/mol for SGPA).
Figure 3.

Distribution of predicted association free energies with SGPA (in blue) of all possible Kazal sequences subject to restrictions stated in the text. Predicted free energies of association of all known subject to restriction ovomucoid third domains with SGPA are shown in red. A white bar indicates the 103 M−1 to 1013 M−1 Ka range of direct measurements. Black arrow marks ΔG°min, the strongest possible association with SGPA, Ka = 8.0 × 1015 M−1. Similar sets of data were obtained for the other five enzymes. They are excluded for brevity.
Table 4.
Kazal sequences predicted to produce the greatest possible Kas
| Sequences | Predicted
Ka, M−1
|
||||||
|---|---|---|---|---|---|---|---|
| CHYM | PPE | CARL | SGPA | SGPB | HLE | ||
| P6 P1 P14′P18′ | |||||||
| CHYM | QCWCTYEYR… . AN..P | 4.0 × 1017 | 2.3 × 104 | 1.3 × 109 | 6.0 × 1011 | 1.1 × 1010 | 3.2 × 106 |
| PPE | TEYCTAEYM… . PN..V | 3.1 × 106 | 5.1 × 1013 | 2.0 × 1010 | 1.3 × 1012 | 8.4 × 1010 | 1.5 × 1010 |
| CARL | WDFCYCEWD… . GN..Q | 3.9 × 103 | 8.6 × 106 | 1.2 × 1017 | 5.6 × 109 | 9.6 × 108 | 1.7 × 107 |
| SGPA | SYYCTCEYT… . GN..V | 4.4 × 108 | 1.3 × 1011 | 9.7 × 1010 | 8.0 × 1015 | 9.8 × 1013 | 1.3 × 1010 |
| SGPB | SDYCTCIYK… . GN..V | 3.2 × 108 | 9.8 × 109 | 1.8 × 1013 | 3.7 × 1014 | 2.8 × 1015 | 6.0 × 108 |
| HLE | RLWCTIEYF… . GN..D | 4.6 × 108 | 7.7 × 109 | 1.9 × 106 | 5.9 × 109 | 2.4 × 108 | 2.4 × 1016 |
Based on our SRA, these sequences in a Kazal inhibitor scaffold are predicted to produce greatest possible Kas (in bold) for each enzyme at pH 8.3, 21°C. Only the residues in the consensus contact region (see Fig. 2) are shown. C denotes half cystine, C denotes cysteine.
The designs in Table 4 are not only very strong but generally rather specific for only one of the six enzymes. There are a few exceptions. The strongest inhibitor for SGPB is less than 10-fold stronger for SGPB than SGPA. It is possible to sacrifice some of the strength for specificity (39). Table 5 lists the results. The inhibitors are now very specific and still moderately strong. The opposite notion (not shown) is of interest. Defense against parasites is a postulated function of many proteinase inhibitors. As there are very many possible parasites, least specific inhibitors may well be nature's goal.
Table 5.
The sequences of most specific possible Kazal inhibitors for the six enzyme sets
| Sequences | Predicted
Ka, M−1
|
||||||
|---|---|---|---|---|---|---|---|
| CHYM | PPE | CARL | SGPA | SGPB | HLE | ||
| P6 P1 P14′P18′ | |||||||
| CHYM | DSGCTWEIR… . AN..S | 3.2 × 1013 | 1.6 × 100 | 3.4 × 105 | 4.6 × 104 | 1.4 × 104 | 1.2 × 101 |
| PPE | KPQCGAEHW… . PN..M | 3.5 × 100 | 3.9 × 108 | 3.5 × 102 | 6.9 × 102 | 5.6 × 101 | 6.7 × 103 |
| CARL | WDLCYEEWD… . FN..E | 1.1 × 10−2 | 8.1 × 10−1 | 9.8 × 1013 | 1.4 × 102 | 7.1 × 100 | 1.3 × 101 |
| SGPA | PWKCPFEFS… . GN..V | 1.3 × 103 | 1.7 × 10−1 | 1.9 × 101 | 2.7 × 109 | 2.1 × 105 | 2.9 × 102 |
| SGPB | SPPCTKKSL… . GN..N | 1.7 × 10−1 | 3.7 × 10−2 | 8.1 × 10−1 | 1.6 × 105 | 1.3 × 108 | 1.5 × 100 |
| HLE | RLRCVIEYL… . GN..D | 2.4 × 104 | 1.6 × 104 | 7.1 × 101 | 1.2 × 105 | 7.5 × 103 | 8.5 × 1014 |
Based on our SRA, these are the sequences of the consensus contact residue set (see Fig. 2) that are predicted to produce the most specific inhibitor at pH 8.3, 21°C, for each of the six enzymes.
It is of interest to compare the distribution of all possible variants to that of a large set of natural variants. The predicted values (predicted because the lowest values on the left are too weak to measure) for all species of birds, whose ovomucoid third domains were sequenced and comply with our restrictions (140/153) are shown in red (Fig. 3). It is seen that the variance of this distribution is narrower than that of all possible values and that they are much stronger than the average of all possible Kazal inhibitors. Except for the few very weak ones they fall in the measurable range. Nature appears to evolve very strong but not strongest possible inhibitors.
The comparison of the distributions allows us to answer an interesting question in evolution. The variable consensus residue sets of Kazal inhibitors are hypervariable, and yet the residues in this set exert very large effects on ΔG° values. This is an apparent contradiction. According to the widely accepted neutral mutation theory (43), the structural and functional residues in proteins are conserved. It is the surface residues that are not functionally important that rapidly fix mutations. If inhibition of serine proteinases were not the function of ovomucoids, there would be no paradox (44). But the superposition of the two distributions clearly implies that the residues in the consensus contact set are selected for strong inhibition and one of the functions of ovomucoids is the inhibition of serine proteinases.
Having the complete distribution of reactivity parameters for all possible variants of a protein was highly useful. We anticipate that the readers will come up with many better uses of such data and that such uses will drive others to attempt to develop additivity-based SRAs for the systems they study.
Acknowledgments
The Purdue predecessors on this project (M. Baillargeon, W. C. Bogard, C. W. Chi, R. Duran, M. Empie, D. A. Estell, W. R. Finkenstadt, H. F. Hixson, D. F. Kowalski, T. R. Leary, J. Lebowitz, J. Luthy, C. March, J. Mattis, R. E. McKee, J. McKie, C. Niekamp, K. Ozawa, M. Praissman, J. Schrode, R. W. Sealock, and K. A. Wilson) established the standard mechanism, replaced residues near the reactive site, and characterized ovomucoid. Early work on structures of avian ovomucoid third domains by W. Bode and R. Huber was of great importance. M. Laskowski, Sr., L. Smillie, J. Travis, W. Bachovchin, and N. Yoshida donated serine proteinases. M. C. Laskowski helped with error analysis, and M. J. Laskowski assisted with the title. The National Institutes of Health supported this research at Purdue for the first 18 (of 20) years.
Abbreviations
- SRA
sequence to reactivity algorithm
- PSTI
pancreatic secretory trypsin inhibitor
- CHYM
bovine chymotrypsin Aα
- CARL
subtilisin Carlsberg
- HLE
human leukocyte elastase
- PPE
porcine pancreatic elastase
- SGP
Streptomyces griseus proteinase
- HPSTI
human pancreatic secretory trypsin inhibitor
- OMTKY3
turkey ovomucoid third domain
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.031581398.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.031581398
References
- 1.Anfinsen C B. Science. 1973;96:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Merrifield B. Science. 1986;232:341–347. doi: 10.1126/science.3961484. [DOI] [PubMed] [Google Scholar]
- 3.Benson S W. Thermochemical Kinetics. New York: Wiley; 1968. [Google Scholar]
- 4.Laskowski M, Jr, Tashiro M, Empie M W, Park S J, Kato I, Ardelt W, Wieczorek M. In: Proteinase Inhibitors. Katunuma N, Umezawa H, Holzer H, editors. Tokyo: Springer; 1983. pp. 55–68. [Google Scholar]
- 5.Horovitz A, Rigbi M. J Theor Biol. 1985;116:149–159. doi: 10.1016/s0022-5193(85)80135-1. [DOI] [PubMed] [Google Scholar]
- 6.Alber T. Annu Rev Biochem. 1989;58:765–798. doi: 10.1146/annurev.bi.58.070189.004001. [DOI] [PubMed] [Google Scholar]
- 7.Wells J A. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 8.Serrano L, Horovitz A, Avron B, Bycroft M, Fersht A R. Biochemistry. 1990;29:9343–9352. doi: 10.1021/bi00492a006. [DOI] [PubMed] [Google Scholar]
- 9.Horovitz A, Serrano L, Fersht A R. J Mol Biol. 1991;219:5–9. doi: 10.1016/0022-2836(91)90852-w. [DOI] [PubMed] [Google Scholar]
- 10.Schreiber G, Fersht A R. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 11.Blaber M, Baase W A, Gassner N, Matthews B W. J Mol Biol. 1995;246:317–330. doi: 10.1006/jmbi.1994.0087. [DOI] [PubMed] [Google Scholar]
- 12.Ardelt W, Laskowski M., Jr J Mol Biol. 1991;220:1041–1053. doi: 10.1016/0022-2836(91)90370-l. [DOI] [PubMed] [Google Scholar]
- 13.Sandberg W S, Terwilliger T C. Proc Natl Acad Sci USA. 1996;90:10753–10757. doi: 10.1073/pnas.93.20.10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajpal A, Kirsch J F. Proteins. 2000;40:49–57. [PubMed] [Google Scholar]
- 15.Dill K A. J Biol Chem. 1997;272:701–704. doi: 10.1074/jbc.272.2.701. [DOI] [PubMed] [Google Scholar]
- 16.Laskowski M., Jr Biochem Pharmacol. 1980;29:2089–2094. doi: 10.1016/0006-2952(80)90177-x. [DOI] [PubMed] [Google Scholar]
- 17.Laskowski M, Jr, Kato I. Annu Rev Biochem. 1980;49:593–626. doi: 10.1146/annurev.bi.49.070180.003113. [DOI] [PubMed] [Google Scholar]
- 18.Laskowski M, Jr, Qasim M A. Biochim Biophys Acta. 2000;7:324–337. doi: 10.1016/s0167-4838(99)00284-8. [DOI] [PubMed] [Google Scholar]
- 19.Bode W, Huber R. Eur J Biochem. 1992;204:433–451. doi: 10.1111/j.1432-1033.1992.tb16654.x. [DOI] [PubMed] [Google Scholar]
- 20.Kato I, Schrode J, Kohr W J, Laskowski M., Jr Biochemistry. 1987;26:193–201. doi: 10.1021/bi00375a027. [DOI] [PubMed] [Google Scholar]
- 21.Rhodes M B, Bennet N, Feeney R E. J Biol Chem. 1960;235:1686–1693. [Google Scholar]
- 22.Laskowski M, Jr, Kato I, Ardelt W, Cook J, Denton A, Empie M W, Kohr W J, Park S J, Parks K, Schatzley B L, et al. Biochemistry. 1987;26:202–221. doi: 10.1021/bi00375a028. [DOI] [PubMed] [Google Scholar]
- 23.Laskowski M, Jr, Apostol I, Ardelt W, Cook J, Giletto A, Kelly C A, Lu W, Park S J, Qasim M A, Whatley H E, et al. J Protein Chem. 1990;9:715–725. doi: 10.1007/BF01024766. [DOI] [PubMed] [Google Scholar]
- 24.Apostol I, Giletto A, Komiyama T, Zhang W, Laskowski M., Jr J Protein Chem. 1993;12:419–433. doi: 10.1007/BF01025042. [DOI] [PubMed] [Google Scholar]
- 25.Lu W, Apostol I, Qasim M A, Warne N, Wynn R, Zhang W L, Anderson S, Chiang Y W, Ogin E, Rothberg I, et al. J Mol Biol. 1997;266:441–461. doi: 10.1006/jmbi.1996.0781. [DOI] [PubMed] [Google Scholar]
- 26.Green N M, Work E. Biochem J. 1953;54:347–352. doi: 10.1042/bj0540347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komiyama T, Bigler T L, Yoshida N, Noda K, Laskowski M., Jr J Biol Chem. 1991;266:10727–10730. [PubMed] [Google Scholar]
- 28.Ardelt W, Laskowski M., Jr Biochemistry. 1985;24:5313–5320. doi: 10.1021/bi00341a007. [DOI] [PubMed] [Google Scholar]
- 29.Read R J, Fujinaga M, Sielecki A R, James M N G. Biochemistry. 1983;22:4420–4433. doi: 10.1021/bi00288a012. [DOI] [PubMed] [Google Scholar]
- 30.Fujinaga M, Sielecki A R, Read R J, Ardelt W, Laskowski M, Jr, James M N G. J Mol Biol. 1987;195:397–418. doi: 10.1016/0022-2836(87)90659-0. [DOI] [PubMed] [Google Scholar]
- 31.Bode W, Wei A Z, Huber R, Meyer E, Travis J, Neumann S. EMBO J. 1986;5:2453–2458. doi: 10.1002/j.1460-2075.1986.tb04521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang K, Lu W, Anderson S, Laskowski M, Jr, James M N G. Protein Sci. 1995;4:1985–1997. doi: 10.1002/pro.5560041004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bateman K S, Anderson S, Lu W, Qasim M A, Laskowski M, Jr, James M N G. Protein Sci. 2000;9:83–94. doi: 10.1110/ps.9.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hecht J J, Szardenings M, Collins J, Schomburg D. J Mol Biol. 1991;220:711–722. doi: 10.1016/0022-2836(91)90112-j. [DOI] [PubMed] [Google Scholar]
- 35.Qasim M A, Ganz P J, Saunders C W, Bateman K S, James M N G, Laskowski M., Jr Biochemistry. 1997;36:1598–1607. doi: 10.1021/bi9620870. [DOI] [PubMed] [Google Scholar]
- 36.Pszenny V, Angel S O, Duschak V G, Paulino M, Ledesma B, Yabo M I, Guarnera E, Ruiz A M, Bontempi E J. Mol Biochem Parasitol. 2000;2:241–249. doi: 10.1016/s0166-6851(00)00202-4. [DOI] [PubMed] [Google Scholar]
- 37.Bode W, Epp O, Huber R, Laskowski M, Jr, Ardelt W. Eur J Biochem. 1985;147:387–395. doi: 10.1111/j.1432-1033.1985.tb08762.x. [DOI] [PubMed] [Google Scholar]
- 38.Hecht H J, Szardenings M, Collins J, Schomburg D. J Mol Biol. 1992;225:1095–1103. doi: 10.1016/0022-2836(92)90107-u. [DOI] [PubMed] [Google Scholar]
- 39.Laskowski M, Jr, Park S J, Tashiro M, Wynn R. In: Protein Recognition of Immobilized Ligands: UCLA Symposia on Molecular and Cellular Biology, New Series. Hutchens T W, editor. New York: Liss; 1989. pp. 149–168. [Google Scholar]
- 40.Fujinaga M, Huang K, Bateman K S, James M N G. J Mol Biol. 1998;284:1683–1694. doi: 10.1006/jmbi.1998.2277. [DOI] [PubMed] [Google Scholar]
- 41.Scott M J, Huckaby C S, Kato I, Kohr W J, Laskowski M, Jr, Tsai M J, O'Malley B W. J Biol Chem. 1987;262:5899–5907. [PubMed] [Google Scholar]
- 42.Jackson R M. Protein Sci. 1999;8:603–613. doi: 10.1110/ps.8.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimura M. The Neutral Theory of Molecular Evolution. Cambridge, U.K.: Cambridge Univ. Press; 1983. [Google Scholar]
- 44.Graur D, Li W H. J Mol Evol. 1989;28:131–135. doi: 10.1007/BF02143504. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y Z, Ikei S, Yamaguchi Y, Sameshima H, Sugita H, Morivasu M, Ogawa M. J Int Med Res. 1996;24:59–68. doi: 10.1177/030006059602400108. [DOI] [PubMed] [Google Scholar]
- 46.Wilimowska-Pelc A, Stachowiak D, Gladysz M, Olichwier Z, Polanowski A. Acta Biochim Polon. 1996;43:489–496. [PubMed] [Google Scholar]
- 47.Schechter I, Berger A. Papain Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 48.Laskowski M, Jr, Kato I, Kohr W J, Park S J, Tashiro M, Whatley H E. Cold Spring Harbor Symp Quant Biol. 1988;52:545–553. doi: 10.1101/sqb.1987.052.01.062. [DOI] [PubMed] [Google Scholar]