

Abstract
Primate models of spinal cord injury differ from rodent models in several respects, including the relative size and functional neuroanatomy of spinal projections. Fundamental differences in scale raise the possibility that retrograde injury signals, and treatments applied at the level of the spinal cord that exhibit efficacy in rodents, may fail to influence neurons at the far greater distances of primate systems. Thus, we examined both local and remote neuronal responses to neurotrophic factor-secreting cell grafts placed within sites of right C7 hemisection lesions in the rhesus macaque. Six months after gene delivery of brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) into C7 lesion sites, we found both local effects of growth factors on axonal growth, and remote effects of growth factors reflected in significant reductions in axotomy-induced atrophy of large pyramidal neurons within the primary motor cortex. Additional examination in a rodent model suggested that BDNF, rather than NT-3, mediated remote protection of corticospinal neurons in the brain. Thus, injured neural systems retain the ability to respond to growth signals over the extended distances of the primate CNS, promoting local axonal growth and preventing lesion-induced neuronal degeneration at a distance. Remote cortical effects of spinally administered growth factors could “prime” the neuron to respond to experimental therapies that promote axonal plasticity or regeneration.
Introduction
Whereas little spontaneous regeneration occurs in the mammalian spinal cord after injury, a variety of experimental approaches can augment the plastic properties of injured neurons and axons (Blesch and Tuszynski, 2009). In particular, mirroring their growth and guidance-related functions during neural development (Davies, 1994), neurotrophic factors administered in rodent models of adult spinal cord injury (SCI) enhance axonal growth into lesion sites (Murray et al., 2002; Blesch and Tuszynski, 2009). However, it is not known whether growth factors can modulate neuronal responses to spinal cord injury over the extended distances of the primate system. That is, as the distance of axotomy from the neuronal soma increases, the retrograde response to injury reportedly diminishes (McQuarrie and Grafstein, 1973; Richardson et al., 1984; Ben-Yaakov and Fainzilber, 2009). This raises the possibility that signals amplifying adaptive responses to injury on the scale of rodent systems may not be equally effective in primate systems. Sites of cervical SCI in rodent models are typically located 3 cm from the cortex, and this distance increases to 10 cm in rhesus monkey cervical lesion models and 25 cm in human cervical segment injury.
To examine the ability of neurotrophic factors to influence both local and remote axonal responses to injury in primate systems, adult rhesus monkeys were subjected to C7 lateral hemisection lesions. This lesion model supports retention of autonomic function and recovery of gross features of locomotion, but results in persistent deficits in use of the hand on the affected side (Courtine et al., 2005a; Jindrich et al., 2005). Coincident with lesion placement, subjects received implants of autologous cell grafts expressing brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) into the injury site. We now report that neurotrophic factors administered to sites of SCI in primates promote regeneration of axons into the local lesion site while ameliorating distant, corticospinal neuronal atrophy.
Materials and Methods
Experimental design, primate study
Previous rodent studies indicate that the neurotrophic factor BDNF elicits the growth of several supraspinal motor projections into lesion sites in the rodent spinal cord, including raphespinal, cerulospinal, and reticulospinal projections (Jin et al., 2002; Lu et al., 2005). Furthermore, when administered to sites of subcortical axotomy, BDNF prevents injured-induced degeneration of corticospinal motor neurons in rodents (Giehl and Tetzlaff, 1996; Lu et al., 2001). Other studies report that NT-3 elicits sprouting of corticospinal axons into host gray matter surrounding sites of SCI, although NT-3 administration will not support corticospinal axonal regeneration into cellular substrates placed into sites of SCI (Schnell et al., 1994; Grill et al., 1997; Chen et al., 2006). NGF (nerve growth factor), however, elicits growth of spinal nociceptive systems (Tuszynski et al., 1996; Romero et al., 2000), an outcome that could contribute to dysfunctional pain after SCI. Thus, for translation to the primate model, we examined the trophic influences of BDNF and NT-3 on axonal and somal responses to injury. The number of experimental subjects and control groups in primate studies is necessarily limited compared with rodent studies; thus, we studied the following groups (additional mechanism was explored in follow-up rodent studies, described below): Group 1 (n = 4) underwent C7 hemisection lesions alone. Group 2 (n = 5) underwent C7 hemisection lesions, immediate placement into the lesion site of autologous fibroblasts transduced to express BDNF and NT-3, and injections of lentiviral vectors expressing BDNF into ventral host gray matter, 2.5 mm caudal to the lesion site. Lentiviral BDNF injections below the lesion site were performed to examine whether growth factor gradients can elicit axonal regeneration beyond the injured zone (Lu et al., 2004; Pearse et al., 2004; Alto et al., 2009; Kadoya et al., 2009). The same subjects received injections of lentiviral vectors expressing NT-3 into the dorsal columns 2.5 mm rostral to the lesion site, to determine whether ascending dorsal column sensory axons potentially regenerating into the cell graft can regenerate beyond the lesion in response to the rostral trophic stimulus (Lu et al., 2004; Taylor et al., 2006). Group 3 (n = 2) underwent C7 hemisection lesions and immediate placement into the lesion site of autologous fibroblasts transduced to express BDNF and NT-3; viral vectors expressing growth factors were not injected into host parenchyma above or below the lesion. Six months later, all animals underwent anterograde tracing of corticospinal projections using different tracers for each hemisphere (see below), together with immunolabeling of raphespinal and cerulospinal axons. Findings from all groups were compared with traced intact monkeys (n = 4).
Detailed methods, primate study
Fifteen adult rhesus primates (Macaca mulatta; mean age, 8 ± 3.8 years; range, 5.5–12 years) were studied. Animals were housed and surgical procedures performed at the California National Primate Research Center. All surgery and animal care procedures adhered to institutional guidelines. The C7 lateral hemisection spinal cord lesion model was designed to avoid discomfort to the subjects while providing reproducible and quantifiable deficits in hand function. Subjects retained normal function of the nonlesioned (left) side of the body, and recovered ability to locomote bipedally on a treadmill after lesions. However, deficits in hand function on the side of the lesion persisted. The primate subjects of this study were involved in extensive analyses of functional capability before and after lesions, and these studies are the subject of continuing experiments. This report focuses on anatomical outcomes.
Cell biopsy and neurotrophin transduction.
For primate studies, autologous skin fibroblasts were genetically modified to produce human NT-3 or BDNF, as previously described using Moloney leukemia virus retroviral vectors (Grill et al., 1997; Lu et al., 2001). Transduced cells were selected by growth in the neomycin analog G418 and the amount of NT-3 or BDNF protein produced from the cells and secreted into the conditioned medium was assayed by two-site BDNF or NT-3 ELISA (Conner and Varon, 1996), as described previously (Lu et al., 2005). In vitro, before grafting, BDNF-transduced fibroblasts secreted 22.3 ± 6.1 ng of BDNF · 106 cells−1 · d−1, and NT-3-transduced fibroblasts secreted 30.8 ± 4.6 ng of NT-3 · 106 cells−1 · d−1.
Production of lentiviral vectors.
Lentiviral vectors were constructed as previously described (Taylor et al., 2006). BDNF or NT-3 was expressed from the cytomegalovirus/β-actin hybrid promoter (Niwa et al., 1991). The vector also contained an internal ribosome entry site to coexpress the reporter gene EGFP (enhanced green fluorescent protein). High-titer stocks of lentivirus were prepared by ultracentrifugation, and titers were determined by infection of HEK293T cells, as previously described (Naldini et al., 1996). In addition, vector stocks were assayed for p24 antigen levels using a HIV-1 p24-specific ELISA kit (DuPont). NT-3 vector preparations contained 112–134 mg/ml p24 and 1.5–3 × 108 IU/ml. BDNF vector preparations contained 95–112 mg/ml p24 and 1–5 × 108 IU/ml.
Surgery and growth factor delivery.
Animals were sedated with 1 mg/kg ketamine intramuscularly, and then deeply anesthetized with 1.5–2.5% isoflurane. After midline cervical skin incisions, the caudal half of the C5 dorsal lamina and the entire C6 dorsal lamina were removed, thereby exposing the C7 spinal cord segment. The dura was slit longitudinally along the midline and gently retracted. An iridectomy microknife with blade rotated 90° to the long axis of the spinal cord was mounted on a stereotaxic arm, and positioned 0.5 mm to the left of the spinal cord midline, aiming to completely transect the right lateral hemicord. The blade was lowered into the spinal cord completely from dorsal to ventral surface to place an initial central incision, thereby separating the left and right halves of the cord. This initial cut established the medial-most position of the lesion. Then, under the visual guidance of the operating microscope and using long iridectomy scissors, the lesion of the right hemicord was completed. The lesion cavity was carefully inspected to ensure complete lateral and dorsoventral transection by the surgeon (M. H. Tuszynski). Immediately after the right lateral hemisection, seven subjects received implants of Gelfoam into the lesion cavity filled with autologous fibroblasts genetically modified to secrete BDNF and NT-3. Five of these animals also received a single injection of 5 μl of lentivirus (1 × 108 IU/ml) encoding BDNF, 2.5 mm caudal to the lesion into the right-sided ventral gray matter (1 mm lateral to midline, 2 mm ventral from cord surface), and injection of 5 μl of lentivirus (1 × 108 IU/ml) encoding NT-3, 2.5 mm rostral to the lesion into the dorsal columns at the midline of the spinal cord (1 mm right-lateral to midline, 0.5 mm ventral from cord surface). All injections were performed through pulled glass micropipettes of 60 μm inner diameter, using a Picospritzer (General Valve), over 5 min. The dura was closed with 6-0 vicryl sutures, and the overlying muscle layers and skin incision were closed. Animals underwent postoperative care in the primate hospital facility. Subjects retained bowel, bladder, and autonomic function, and were generally moving freely around their cages by 3–5 d after lesion.
Anterograde corticospinal tracing.
Six months later, the corticospinal tract (CST) was anterogradely labeled as described previously (Rosenzweig et al., 2009). Briefly, 127 injections of lysine-fixable dextran conjugated to Alexa 488 (D488) (10% in PBS 10,000 molecular weight; Invitrogen) were placed into the left primary motor cortex and 127 injections of lysine-fixable dextran conjugated to biotin [biotinylated dextran amine (BDA)] (10% in PBS 10,000 molecular weight; Invitrogen) were placed into the right primary motor cortex (Rosenzweig et al., 2009). One hundred fifty nanoliters were injected per site through a glass micropipette attached to a Picospritzer. The injection sites included areas of the motor cortex that innervate hand, trunk, and foot areas. Subjects survived an additional 6 weeks to allow for tracer transport to the cervical enlargement.
Retrograde corticospinal tracing.
To confirm the size of CST neurons projecting to the cervical enlargement, an additional intact monkey received injections of the retrograde tracer dextran-conjugated Cascade Blue (1 μl of 10% solution; Invitrogen) into C7. Injections were targeted to intermediate and ventral gray matter to target terminals of corticospinal axons, thereby specifically backlabeling corticospinal somata in the motor cortex. Injections were placed 0.7 mm medial to the dorsal root entry zone and at a depth of 2 mm. Transcardial perfusion was performed 6 weeks later, and a series of 1-in-12 coronal sections (40 μm thick) through the motor cortex were immunolabeled using rabbit anti-Cascade Blue (Invitrogen; 1:20,000). The somal area of retrogradely labeled primate corticospinal neurons was measured using the nucleator method (Stereo Investigator), with the nucleolus as reference object (Smith et al., 2004).
Histology and immunocytochemistry.
After 8 months survival, subjects were sedated with ketamine (25 mg/kg, i.m.), and then very deeply anesthetized with Nembutal (30 mg/kg, i.p.), and perfused transcardially for 1 h with a 4% solution of paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, at 4°C followed by 5% sucrose solution. Spinal cords and brains were removed and placed in a glycerin cryoprotectant solution. Brains were stereotaxically blocked in the coronal plane. The entire spinal cord was removed, and the segment containing the lesion site was taken in a 1-cm-long segment that contained the lesion in its center; this was sectioned in the horizontal or tranverse plane. The remainder of the spinal cord was collected in 1 cm blocks that were sectioned in the coronal plane. Sections were cut on a freezing microtome set at 40 μm intervals and collected in cryoprotectant solution as series of one-in-six sections. One-in-six brain sections were thionin-stained for quantification of cortical layer V neuronal number and soma area, and one-in-six spinal cord sections from the horizontal block containing the lesion site were also thionin-stained. An additional 1-in-12 series of spinal cord sections was immunolabeled to examine host axons, using rabbit anti-D488 (Invitrogen; 1:5000; to label anterogradely traced corticospinal axons arising from the left hemisphere), mouse anti-NF70 (Millipore; 1:10; to label total axons), rabbit anti-serotonin (5HT; Immunostar; 1:10,000; to label raphespinal axons), or rabbit anti-tyrosine hydroxylase (TH) (Millipore; 1:1000; to label cerulospinal axons). Free-floating 40 μm horizontal sections through the lesion site were reacted for endogenous peroxidase activity with 0.6% H2O2 in Tris-buffered saline (TBS), pH 7.4, for 30 min at room temperature, and then nonspecific antibody binding was blocked with 5% normal goat serum in TBS plus 0.25% Triton X for 1 h at room temperature. Sections were then incubated overnight at 4°C in primary antibodies. After TBS washes, sections were incubated with goat anti-mouse or rabbit poly-HRP secondary antibody (Millipore) diluted 1:10 in blocking solution. Sections were then reacted with 1.25 mg/ml DAB plus 0.04% NiCl2 plus 0.012% H2O2, mounted on gelatin-subbed slides, dried, dehydrated, and coverslipped with Pro-tek (Thermo Fisher Scientific). TrkB and TrkC receptor immunolabeling was performed in additional intact subjects that underwent anterograde labeling of corticospinal axons with D488 and retrograde labeling of layer V corticospinal neurons with Cascade Blue injections into C7, as noted above. Cortical or spinal cord sections were incubated in streptavidin Alexa 488 plus primary antibody overnight at 4°C (anti-TrkB; 5 μg/ml; Biosensis; anti-TrkC; 4 μg/ml; Santa Cruz Biotechnology). Sections were then incubated in secondary goat anti-rabbit poly-HRP for 1 h at room temperature, followed by 7 min of tyramide-Cy3 (1:100; Invitrogen). Sections were coverslipped with Fluoromount-G (Thermo Fisher Scientific). For spinal cord sections, z-stacks (eight optical planes at 0.5 μm/optical plane) were acquired with an Olympus FV1000 confocal microscope using excitation wavelengths of 488 and 543 nm. Sequential scanning was used to ensure no bleedthrough of the channels during imaging. Z-stacks were flattened and imported into Photoshop CS2 (Adobe). X–Z and Y–Z stacks have been shown to verify colocalization. For motor cortex sections, a single optical plane was imaged and Cascade Blue-labeled CST neurons were pseudocolored green for optimal visualization.
Axon density within neurotrophin-producing grafts and the lesion site was quantified using NIH Image software, as previously described (Tuszynski et al., 2002; Lu et al., 2005). Total numbers of 5HT- and TH-labeled axons within the lesion site were quantified in four randomly selected fields (0.1 mm2) per subject, from two separate sections located at least 240 μm apart. The total number of pixels (neurofilament label, NF) or total number of axons (serotonin label, 5HT; tyrosine hydroxylase label, TH) per square millimeter was calculated to obtain compare axon density within the lesion site among groups.
Stereology of layer V cortical neurons.
Stereology of layer V cortical neurons was conducted using Stereo Investigator software (MicroBrightField) on a Nissl-stained series of one-in-six sections through the motor cortex. Live images were received from an Olympus Optical OLY-200 video camera mounted on an Olympus Optical BX60 microscope fitted with a Ludl 99S000 XYZ motorized stage. Area 4, found on the banks of the central sulcus, extends from a rostral boundary demarcated by the appearance of the superior precentral dimple, to a caudal boundary formed by the convergence of the central sulcus to midline (see Fig. 5G). Within area 4, layer V is characterized by a dense population of large pyramidal neurons between less dense layers IV and VI that are mostly devoid of pyramidal neurons (Murray and Coulter, 1981; Dum and Strick, 1991; Nudo et al., 1995). After defining the boundaries of layer V on the computer graphic display of each section, the Stereo Investigator software placed within each laminar boundary a set of optical fractionator frames (50 × 50 μm) in a pseudorandom fashion corresponding to a sampling frame of 10%. The optical fractionator relies on a systematic uniform sampling scheme to generate estimates of cell number unbiased by tissue processing or assumptions regarding neuronal size and shape. Within each optical dissector, cells were counted only if the nucleolus reached focus within the fractionator sample frame, which was set at the middle 75% of total tissue thickness for each section. Using these methods, a minimum of 200 large (>15 μm) pyramidal neurons per subject were counted using the nucleolus as a reference object, to assess effects of axotomy on large-diameter cortical neurons; the majority of these neurons project to the spinal cord (Murray and Coulter, 1981; Dum and Strick, 1991; Nudo et al., 1995). Separately, to assess effects of spinal cord lesions on the total population of layer V cortical neurons, all neurons of diameter >8 μm were measured (Smith et al., 1999, 2004). As above, neuronal number was quantified using the optical fractionator (West et al., 1996; Long et al., 1999; Smith et al., 1999, 2004) and neuronal size was quantified using the nucleator (Gundersen, 1988). Cells were included in counts if they met the following criteria to exclude non-neuronal cells (Pakkenberg and Gundersen, 1997; Simic et al., 1997): (1) cell diameter of >8 μm, (2) typical pyramidal-shaped neuronal morphology, and (3) clearly visible nucleus and nucleolus within the stereology inclusion boundaries. The distance between sections used for quantification and the size and spacing between counting frames were set to achieve a coefficient of error <5% (Gundersen and Jensen, 1987). In area 4, every 48th section was used for analysis (12 sections per primate). A total of 200–300 neurons per animal was quantified for determination of somal area using the nucleator method. Analyses of somal area distributions were performed in Matlab (The MathWorks).
Figure 5.
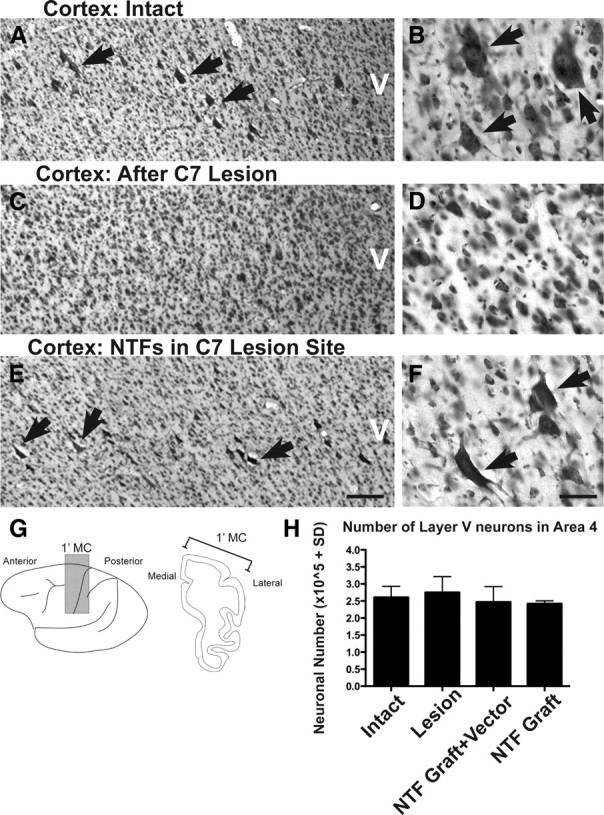
Cortical responses to injury and spinally administered neurotrophins. A, C, E, Representative thionin-stained coronal section from an intact monkey (A), lesion-only monkey (C), and lesion plus neurotrophin (NTF)-treated monkey (E). The arrows indicate large pyramidal neurons located within layer V. B, D, F, Higher magnification demonstrates a reduction in large layer V neurons after C7 hemisection that is prevented by growth factor administration to the spinal cord. G, H, Stereologic quantification of thionin-stained layer V total neuron number within the primary motor cortex (dark shading; G) indicates that total neuronal number in motor cortex is preserved after C7 lesions in all groups. Error bars indicate SEM. Scale bar: E (for A, C, E), 250 μm; F (for B, D, F), 25 μm.
To assess the proportion of projecting corticospinal neurons that survive 8 months after C7 spinal cord injury, we also quantified the total number of medullary axons in neurofilament-labeled sections of the medulla. Three 40-μm-thick sections, spaced 240 μm apart, per subject were labeled for neurofilament within the medullary pyramids. Three images per section were acquired at 20× magnification using an Optronic Microfire A/R digital camera and imported into NIH ImageJ. HRP reaction product was thresholded and presented as the mean pixel density obtained from four primates per group (intact and lesion-only). Total axon number per square millimeter was quantified per section and averaged over three sections per subject to yield mean axon density.
Based in part on findings of the analysis in the preceding paragraph, coronal sections through area 4 of the motor cortex were examined in five additional subjects that underwent C7 hemisection lesions (without experimental treatment) followed by 7 d (n = 1) or 14 d (n = 4) survival, to detect apoptotic nuclei. The 40-μm-thick coronal sections through area 4 were stained for terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) using the in situ cell death detection POD kit (Roche Applied Science), according to manufacturer's instructions, and counterstained with thionin. The boundaries of area 4, layer V, were determined as described above for neuronal counts, and 500–600 thionin-stained large (>13 μm diameter) pyramidal neurons per animal were analyzed for the presence of TUNEL reaction product within the nucleus. No cells (neurons or glia) detectably expressed the TUNEL label, however, and quantitative analysis was therefore not performed.
Electron microscopy.
For electron microscopy, 40-μm-thick transverse sections from subjects in groups 1 and 2 were postfixed in 4% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, for 4 d at 4°C (these subjects had previously been transcardially perfused with 4% PFA, as described above). The sections were next rinsed in PBS, postfixed in 1% osmium tetroxide, dehydrated in graded ethanol, and embedded in Durcopan ACM (Electron Microscopy Sciences). Semithin sections were cut at ∼1 μm thickness and stained with toluidine blue for light microscopy. Ultrathin sectioning of embedded tissue blocks containing the lesion or graft site was performed using an RMC Products PowerTome X ultramicrotome (Boeckeler Instruments). Ultrathin sections (60–70 nm) were serially collected on Formvar-coated copper one-hole grids, counterstained with uranyl acetate and lead citrate, and examined in a Tecnai G2 Spirit TWIN transmission electron microscope (FEI Company) equipped with a Gatan Orius SC 1000B digital camera (Gatan). Approximately two to five ultrathin sections were examined per animal for graft morphology evaluations.
Statistics.
Multiple-group comparisons were made using ANOVA followed by post hoc Fisher's; two-way comparisons were made using Student's two-tailed t test. Significance was defined as p < 0.05. Results are reported as mean ± SEM, aside from neuronal stereological data, which by convention are presented as mean ± SD. The examiner was blinded to group identity for all quantification procedures listed above.
Experimental design, rodent study
As presented below in Results, analysis of primate data indicated a remote effect of growth factor administration to the injured spinal cord on corticospinal neuronal somal atrophy over the extended distances of the primate spinal cord. To gain insight into the specific requirement for BDNF or NT-3 for trophic protection in this injured system, additional rodent experiments were performed. Female Fisher 344 rats (125–150 g) underwent C5 laminectomy and retrograde labeling of identified corticospinal motor cortex neurons using injections of cholera toxin B subunit (CTB) into the spinal cord. Two weeks later, the same subjects underwent dorsal column lesions to transect the majority corticospinal projection, followed by grafting of syngeneic fibroblasts expressing either BDNF, NT-3, or both growth factors into the lesion site. The following groups were examined: group 1, sham (laminectomy only; N = 6); group 2, lesion only (N = 6); group 3, lesion plus green fluorescent protein (GFP)-expressing fibroblast grafts placed in lesion site (N = 6); group 4, lesion plus BDNF-secreting fibroblast grafts placed in lesion site (N = 6); group 5, lesion plus NT-3-secreting fibroblast grafts placed in lesion site (N = 6); group 6, lesion plus fibroblast grafts secreting both BDNF and NT-3 in lesion site (N = 6). Subjects survived 6 weeks after the lesion, and then underwent quantification of identified corticospinal neurons in the cortex.
Detailed methods, rodent study
Surgical procedures.
All procedures adhered to Institutional Animal Care and Use Committee guidelines and animals had ad libitum access to food and water throughout the study. Adult Fisher 344 female rats (125–150 g) were anesthetized with a mixture of ketamine (25 mg/ml), xylazine (1.3 g/ml), and acepromazine (0.25 mg/ml). To specifically label corticospinal motor neurons in the motor cortex, 2 μl of a 1% solution of CTB (List Biological Laboratories) was injected into the dorsal columns at C5. Injections were made at the spinal midline at depths of 0.7 and 1.2 mm from the dorsal surface of the spinal cord. Two weeks later, rats were reanesthetized and a bilateral C4 dorsal column spinal cord lesion was made with a wire knife (David Kopf Instruments), as previously described (Weidner et al., 2001). Briefly, the wire knife was lowered 1.1 mm into the spinal cord, extruded to create a 2.5 mm arc, and raised to completely transect the dorsal component of the rat corticospinal tract. Immediately after the lesion, syngeneic rat fibroblasts genetically modified to express BDNF, NT-3, or the reporter gene GFP, were injected directly into the lesion site at the spinal cord midline. Two microliters of cells (100,000 cells/μl) were injected slowly using a Picospritzer penetrating 0.5 and 1.0 mm from the dorsal spinal cord surface. Before in vivo grafting, NT-3-transduced cells secreted 47 ng of NT-3 · 106 cells−1 · d−1, and BDNF-transduced cells secreted 36 ng of BDNF · 106 cells−1 · d−1.
Perfusion, histology, and immunolabeling.
Six weeks later, rats were deeply anesthetized with ketamine and xylazine and perfused transcardially with 4% paraformaldehyde in 0.1 m phosphate buffer. Brains were removed and postfixed in the same fixative overnight, and then transferred to 30% sucrose for 2–3 d. Brains were sectioned on a freezing microtome set at 40 μm through the rostrocaudal extent of the motor cortex. A series of 1-in-12 sections was immunolabeling for CTB. Nonspecific antibody binding was blocked in 5% normal goat serum in TBS plus 0.25% Triton X (TBS-T) for 1 h. Sections were incubated in mouse anti-CTB (0.25 μg/ml; Biodesign International) in the same solution overnight at 4°C, washed in TBS, and incubated in goat anti-mouse poly-HRP (1:10; Millipore) in 3% normal goat serum in TBS for 1 h at room temperature. Sections were then reacted with 1.25 mg/ml DAB plus 0.04% NiCl2 plus 0.012% H2O2, mounted on gelatin-subbed slides, dried, dehydrated, and coverslipped with Pro-tek (Thermo Fisher Scientific). A one-in-six series of sections through the lesion site were Nissl stained to assess lesion extent and graft survival. Axonal responses to BDNF and NT-3 in the spinal cord of the rodent have been reported previously (Lu et al., 2005); in the present study, we confirmed previous patterns of neurotrophic responsiveness to BDNF and NT-3 in the lesion site using labels for 5HT, TH, and neurofilament.
Stereological quantification of rat corticospinal neurons.
Stereology of light-level CTB-labeled corticospinal neurons in the rat was conducted as described previously in the primate study. The rat motor cortex was defined in the rostrocaudal direction based on a +3 to −1.5 mm range relative to bregma, and the mediolateral boundaries were determined by the position of CTB-labeled neurons. Every 12th section (spaced 480 μm apart) was used for analysis (9–10 sections/subject). After defining the boundaries of layer V on the computer graphic display of each section, the Stereo Investigator software placed within each laminar boundary a set of optical fractionator frames (50 × 50 μm) in a pseudorandom fashion corresponding to a sampling frame of 5%. Within each optical dissector, cells were counted only if the nucleus reached focus within the fractionator sample frame, which was set at the middle 75% of total tissue thickness for each section. Using these methods, a minimum of 200 CTB-labeled neurons per subject was counted, and total number of CST neurons was calculated for each animal. In addition, somal area was determined using the nucleator and the median somal area for each subject calculated. Data are presented as the average neuronal number or somal area per group.
Statistics.
Statistical analysis was performed as in primate studies: Multiple-group comparisons were made using ANOVA followed by post hoc Fisher's, with a significance criterion of p < 0.05. Results are presented as mean ± SEM, and stereological data as mean ± SD.
Results
Primate studies
All subjects survived the experimental procedures. Lesions resulted in deficits of fine, but not gross, features of locomotion and persistent deficits in right hand use (Courtine et al., 2005b; Jindrich et al., 2005). Functional outcomes after growth factor administration are a subject of ongoing experiments in this model and are not further addressed here.
Lesions completeness and graft survival
The main component of the primate corticospinal tract is located in the dorsolateral white matter, and this projection was completely transected in all subjects (Fig. 1). Cell grafts survived in the lesion cavity for at least 8 months after transplantation and integrated well into host tissue (Fig. 1). Host cells spontaneously penetrated the lesion site in control, nongrafted animals, and many of these cells exhibited morphological features of fibroblasts and Schwann cells (Fig. 1). Semithin sections stained with DAB plus gold chloride for myelin demonstrated extensive penetration of myelinated axons into neurotrophic factor-secreting grafts, whereas few myelinated axons were present in the cellular matrix occupying the lesion site in nongrafted animals (Fig. 2). Ultrastructural examination confirmed the presence of fibroblasts and Schwann cells in the lesion site of both nongrafted control and grafted subjects (Fig. 2), although the number of both cell types occupying the lesion site was greater in grafted lesion cavities. Furthermore, collagen was present in both grafted and nongrafted lesion sites (Fig. 2), providing an extracellular matrix that could potentially support axonal growth. Indeed, both myelinated and unmyelinated axons were detected in the lesion sites of both control and grafted subjects, although the density of penetrating axons and Schwann cells was greater in grafted subjects (Fig. 2).
Figure 1.
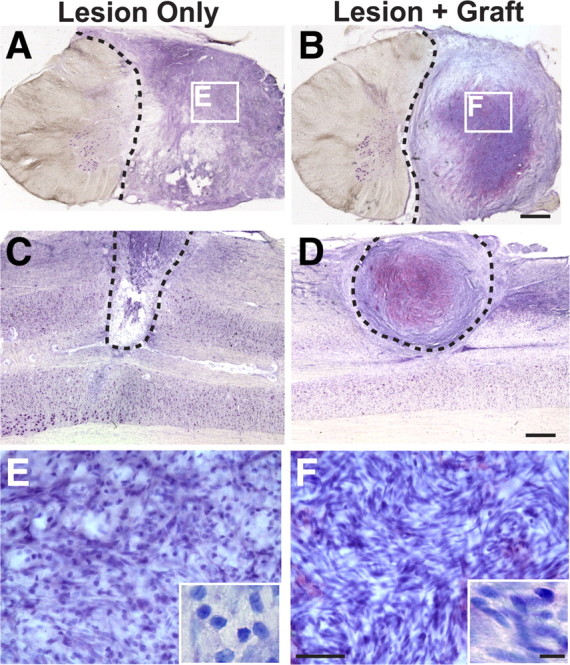
Lesion/graft site after primate C7 hemisection. Thionin-stained transverse (A, B) and horizontal (C, D) sections through monkey spinal cord lesion site 8 months after C7 lateral hemisection (the dashed line indicates lesion border). In both nongrafted subjects (A, C) and subjects grafted with autologous fibroblasts genetically modified to secrete BDNF and NT-3 (B, D), cells fill the lesion site. Cells are both rounded and spindle-shaped, typical of fibroblast, Schwann cells, and leptomeningeal cell morphology (Fig. 2). Higher magnification insets in E and F reveal greater density cell within grafted subjects. Scale bars: B (for A, B), D (for C, D), 1 mm; F (for E, F), 50 μm; inset, 10 μm.
Figure 2.
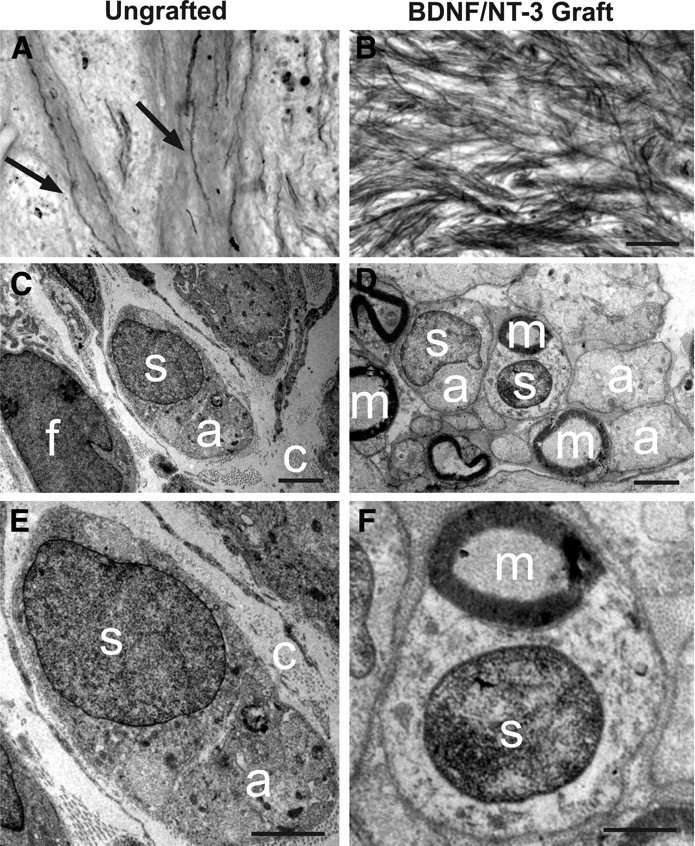
Myelin stain and ultrastructure of lesion/graft site. A, Myelin stain of the lesion site within ungrafted subject indicates occasional penetration of myelinated axons (arrows) into spontaneous cell matrix that forms in injury zone. B, In contrast, myelinated axons extensively penetrate BDNF/NT-3-secreting fibroblast graft placed in lesion site. C, Ultrastructure of lesion site in ungrafted subject demonstrates cellular influx consisting of both fibroblasts (f) and occasional ensheathing Schwann cells (s). Bundles of unmyelinated axons (a) are observed within cytoplasm of associated Schwann cell in ungrafted lesion cavity; nuclear–axonal juxtaposition is typical of Schwann cell morphology. Ungrafted subjects also exhibit abundant extracellular collagen fibrils (c), providing a substrate for axonal penetration into control lesion site. D, Subjects that received grafts of BDNF/NT-3-secreting fibroblasts into the lesion cavity generally exhibit a greater density of both cells and axons. Abundant Schwann cells are present and are associated with many myelinated axons (m). E, Higher magnification of nongrafted lesion site, demonstrating association of Schwann cell with unmyelinated axon, and extracellular collagen fibrils. F, Higher magnification of BDNF/NT-3 grafted subject within lesion site, demonstrating association of Schwann cell with myelinated axon. Scale bars: B (for A, B), 100 μm; C, D, 2 μm; E, F, 1 μm.
Axonal responses to growth factors in the spinal cord lesion site
Axons extensively penetrated BDNF/NT-3-secreting cell grafts in the lesion cavity, as revealed by neurofilament (NF), serotonin (5HT), and tyrosine hydroxylase (TH) labeling (Fig. 3). Significantly greater numbers of axons of each of these phenotypes were detected in BDNF/NT-3-secreting grafts compared with control lesioned animals (p < 0.05 in all cases) (Fig. 3). However, corticospinal axons did not penetrate either control lesion sites or grafts of BDNF/NT-3-secreting cells (Fig. 4). Notably, many corticospinal axons remained adjacent to the lesion site in both control and BDNF/NT-3 grafted subjects 8 months after injury (Fig. 4). Thus, lesioned primate raphespinal and cerulospinal axons exhibit trophic responses to a combination of BDNF and NT-3 expressed in a lesion site, consistent with reports in rodent models (Lu et al., 2005). However, corticospinal axons did not regenerate into lesion sites containing the neurotrophic factors BDNF and NT-3, a finding that is also consistent with rodent studies (Grill et al., 1997).
Figure 3.

Local and supraspinal axons regenerate into neurotrophin-secreting cell grafts in the lesioned primate spinal cord. A, Neurofilament-labeled axons extend into the cellular matrix spontaneously filling the lesion in a nongrafted injury site, 8 months after injury. B, C, Significantly greater numbers of neurofilament-labeled axons penetrate the lesion site in subjects that received grafts of autologous fibroblasts genetically modified to secrete BDNF and NT-3 (B), quantified in C (*p < 0.05). D–F, Raphespinal axons grow into control lesion site (D) but exhibit a 2.5-fold increase in growth into BDNF/NT-3-secreting grafts (E), quantified in F. G–I, Cerulospinal axons labeled by TH exhibit little growth into control lesion site (G), but growth is significantly increased in the presence of growth factors (H), quantified in I. Error bars indicate SEM. Scale bars: B (for A, B), E (for D, E), H (for G, H), 25 μm.
Figure 4.
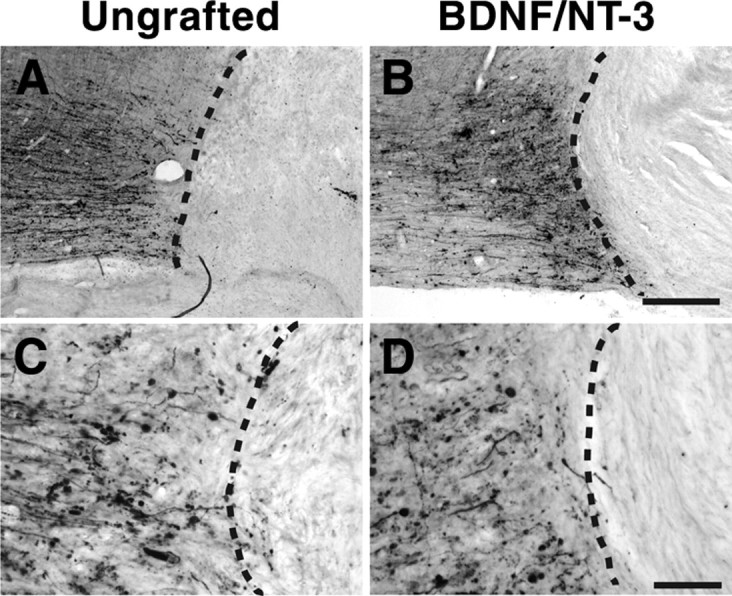
Neurotrophin-secreting grafts do not promote CST regeneration. Shown are photomicrographs of horizontal spinal cord sections: rostral, left; caudal, right (dashed line indicates lesion interface). A–D, Transected CST axons anterogradely labeled with dextran-488 do not enter the cellular matrix of either ungrafted lesion site (A, C) or lesion site containing fibroblasts that secrete BDNF and NT-3 (B, D). Scale bars: B (for A, B), 250 μm; D (for C, D), 100 μm.
Corticospinal motor neurons undergo atrophy, but not death, after primate spinal cord injury
There is uncertainty regarding the fate of corticospinal motor neurons after spinal cord injury (Hains et al., 2003; Wannier et al., 2005), a point of some relevance to future efforts to promote regeneration and functional recovery at prolonged delays after SCI (Kadoya et al., 2009). To investigate the fate of corticospinal motor neurons after primate SCI, thionin-stained sections of the primary motor cortex (area 4) were examined using stereological methods. Stereological quantification of all layer V neurons, using a lower size limit of 8 μm, revealed that total neuronal number was preserved after C7 lateral hemisection in lesioned control subjects 8 months after SCI compared with intact animals (Fig. 5). Confirming the preservation of neuronal number, quantification of axon number in the medullary pyramids did not reveal differences between intact and the lesioned control group examined 8 months after SCI (Fig. 6). Furthermore, assessment of apoptotic cell markers in the motor cortex at time points of 7 and 14 d after injury yielded no evidence of apoptosis, although positive labeling was detected in control material subjected to DNase treatment (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results suggest that SCI in the adult primate does not lead to detectable neuronal loss in the motor cortex 8 months after transection.
Figure 6.

Axons are retained within the medullary pyramids after SCI. A, B, To further confirm that CST neurons and their projections are retained 8 months after C7 hemisection, axon numbers were quantified in the medullary pyramids of intact (A) and lesioned (B) subjects (that did not receive BDNF/NT-3 grafts), using NF70 immunolabeling. C, Quantification of NF70 labeling density reveals no significant difference between intact or lesioned monkeys (p = 0.4). Error bars indicate SEM. Scale bar: B (for A, B), 25 μm.
Neurotrophic factors act across the extended distance of the adult primate CNS to prevent motor cortex neuronal atrophy
After axotomy, neurons can adopt a fate of death, sustained atrophy or full morphological recovery. To examine cell fate in the motor cortex 8 months after primate C7 spinal cord injury, we quantified cortical neuron somal size using stereological methods. In intact monkeys, large pyramidal neurons (500 μm2) are present exclusively within layer V (Fig. 5) and constitute 10–20% of large neurons in this region (Humphrey et al., 1978). We confirmed that this size criterion reliably identifies corticospinal neurons by measuring somal area in corticospinal motor neurons that were retrogradely labeled in one primate using injections of dextran conjugated-Cascade Blue into the spinal cord (Fig. 7): all retrogradely labeled corticospinal motor neurons in the primary motor cortex of the primate exceeded the 500 μm2 threshold. Subsequent quantification of large-diameter neurons (>500 μm2) in four intact subjects revealed that neurons of this size constitute 21% of the neuronal population in layer V motor cortex (Fig. 7). Subjects that underwent C7 lateral hemisection lesions exhibited a 77% reduction in the number of large-diameter neurons in the motor cortex 8 months after injury, compared with intact monkeys (ANOVA, p < 0.01; post hoc Fisher's, p < 0.001) (Fig. 7). Notably, BDNF/NT-3 growth factor expression in the lesion site, plus BDNF/NT-3 lentiviral gene delivery in the perilesion region, resulted in a loss of only 28% of large-diameter neurons compared with intact animals, representing a significant improvement compared with lesioned controls (post hoc Fisher's, p < 0.05) (Fig. 7). Two subjects received grafts of BDNF/NT-3-expressing cells in the lesion site, without additional perilesion vector injections, and these animals exhibited a similar degree of cell rescue: large-diameter neurons were reduced by only 21% compared with intact animals (Fig. 7). To determine whether the effect was limited to the largest neurons, we also quantified somal area of all layer V neurons and divided these into bins representing the largest 20%, the middle 20%, and the smallest 20% of the overall neuronal population. Notably, neurons in each size group became atrophic after C7 hemisection, and spinal growth factor administration partially ameliorated the atrophy of the largest 20% of neurons, and completely prevented atrophy of the middle 20% and smallest 20% of layer V neurons (Fig. 7). Thus, although growth factor gene delivery did not detectably support corticospinal axonal regeneration into the injured spinal cord, growth factor expression in the lesion site substantially and significantly ameliorated corticospinal neuronal atrophy in the cortex over the extended distances of the primate nervous system. The preservation of corticospinal somata could have implications for priming these neurons to regenerate (see Discussion).
Figure 7.

Spinal growth factor treatment in primates ameliorates cortical neuronal atrophy: stereological assessment. A, Retrograde labeling with CTB indicates that a majority of identified layer V corticospinal neurons exhibit a somal area of >500 μm2 (see text). B, Frequency distribution of all thionin-stained neurons within layer V. The number of neurons within 100-μm2-sized bins is uniformly reduced after spinal cord injury (red line), and these numbers are restored by BDNF/NT-3 treatment (blue line) compared with intact animals (black line). C, C7 lesions induce a significant reduction in the proportion of large cortical motor neurons (>500 μm2), and treatment with BDNF/NT-3 substantially prevents this neuronal atrophy (NTF graft plus vector: BDNF/NT-3-secreting cell grafts in lesion cavity plus lenti-BDNF and lenti-NT-3 vector injections in host spinal cord parenchyma; NTF graft: BDNF/NT-3-secreting graft in lesion site; ANOVA, p < 0.001; post hoc Fisher's, *p < 0.001). Furthermore, neurotrophin treatment prevents atrophy of a range of layer V somal sizes. D–F, Somal sizes were divided into largest 20% (D), middle 20% (E), and smallest 20% (F). The size of layer V neurons after lesion only were significantly smaller compared with intact monkeys. BDNF/NT-3 treatment prevented the atrophy of both the middle 20% and the smallest 20% and partially prevented atrophy of the largest 20% of neurons. *p < 0.05, intact versus lesion and NTF graft plus vector. Error bars indicate SEM. Scale bar: A, 25 μm.
Primate corticospinal motor neurons and axons express TrkB and TrkC
To access whether primate CST neuronal cell bodies and spinal cord axons express TrkB (the high-affinity receptor for BDNF) and/or TrkC (the high-affinity receptor for NT-3), we performed immunolabeling on motor cortex sections containing Cascade Blue retrogradely labeled CST neurons and on spinal cord sections containing BDA anterogradely labeled CST axons. TrkB and TrkC immunoreactivity was detected in both corticospinal axons in the cervical spinal cord, and in corticospinal motor neuronal somata in the motor cortex (Fig. 8). Specificity of Trk receptor antibodies was confirmed by coimmunolabeling monkey dorsal root ganglion (DRG) with neurofilament 200 (Nf200), a marker of large-diameter DRG neurons (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Colocalization of Nf200 with TrkC but not TrkB was observed, as expected (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 8.
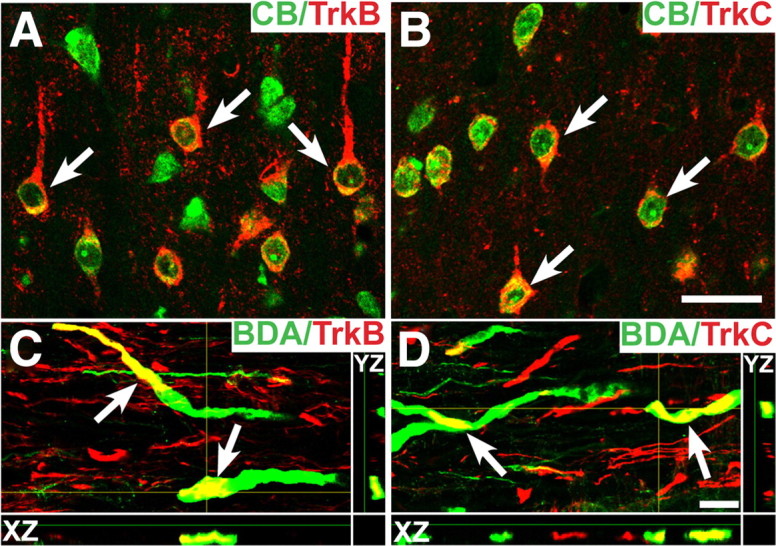
Primate TrkB and TrkC immunolabeling. A, TrkB and Cascade Blue (CB) double labeling showing expression of TrkB in intact corticospinal motor neuron cell somata. Corticospinal neurons are backlabeled after injection of CB into the spinal cord. The arrows indicate cells containing both TrkB and CB. B, TrkC and CB double labeling also shows expression of TrkC in retrogradely identified corticospinal motor neurons. C, Confocal z-stacks demonstrate that TrkB is also detected on corticospinal axons in the cervical spinal cord, double-labeled with BDA (arrows). D, TrkC is also present on corticospinal axons (arrows). The yellow crosshairs indicate the position of a single x–z and y–z plane to confirm colocalization. Scale bars: B (for A, B), 25 μm; D (for C, D), 5 μm.
Rodent studies
Primate studies did not identify whether BDNF or NT-3 conferred trophic protection to cortical motor neurons. To identify the specific trophic factor responsible for remote protection of corticospinal neuronal somata after SCI, we examined neurotrophic effects of BDNF, NT-3, and both growth factors in a rat model. Neurotrophic effects were specifically assessed in corticospinal motor neurons by retrogradely labeling this spinally projecting population with CTB injections at the C3 spinal cord segment 1 week before placing a C5 corticospinal dorsal column lesions. As observed in the primate model, rodents exhibited a significant shrinkage of corticospinal motor neurons 6 weeks after spinal cord lesions, but cell loss was not observed using stereological quantification (Fig. 9). Identified corticospinal motor neurons exhibited a mean 20% reduction in cell size compared with intact animals (ANOVA, p < 0.05; post hoc Fisher's, p < 0.01) (Fig. 9), a value similar to that observed in primates, and treatment with BDNF or BDNF/NT-3 prevented this atrophy (p < 0.05, post hoc Fisher's comparing BDNF treated to lesion control, and BDNF/NT-3 treated to lesion control). NT-3 administration to the spinal cord lesion site did not prevent retrograde corticospinal neuronal atrophy in the motor cortex. These findings suggest that BDNF, but not NT-3, exerts trophic actions on cortical neurons when administered to the remote spinal cord site of axonal transection.
Figure 9.

Rodent study: spinally administered BDNF prevents cortical neuronal atrophy. A–F, Representative photomicrographs of corticospinal neurons retrogradely labeled with Cascade Blue within the rat motor cortex, after cervical corticospinal dorsal column lesion. Neuronal atrophy is evident after lesion-alone, lesion with GFP graft, or lesion with NT-3 graft. Atrophy is ameliorated by BDNF graft and BDNF plus NT-3 grafts to spinal cord. G, Stereological quantification confirms that corticospinal somal size exhibits a 20% reduction after C5 spinal cord dorsal column lesion; this atrophy is prevented by BDNF-secreting and BDNF/NT-3 grafts, but not NT-3 or control GFP grafts. H, Stereological quantification of CTB-labeled CST neuronal number indicates no cell loss after injury in any group, consistent with findings of primate study. *p < 0.05. Error bars indicate SEM. Scale bar: F (for A–F), 25 μm.
Discussion
This study provides evidence that neurotrophic factors can act over the extended distances of the primate CNS to significantly ameliorate injury-related cellular degeneration. The distance of the corticospinal cell body from axotomy in the cervical spinal cord of a rhesus monkey (∼10 cm) is ∼40% that of the human (∼25 cm) and 3.3-fold greater than the rat (∼3 cm). It is also notable that, although neurotrophic factors delivered to the tips of injured corticospinal axons influenced the remote cell body response to injury, they did not detectably influence the behavior of the injured axons themselves, as assessed by measures of regeneration. These findings indicate that retrograde injury signals are clearly transported to the remote somata of the injured primate corticospinal projection and that the responses can be modified by local actions of growth factors on injured axons, without affecting the growth of these axons per se at the lesion site. Yet the same growth factors clearly elicit the regeneration of other axonal systems into the spinal cord lesion site, including raphaespinal and cerulospinal axons, consistent with previous findings in rodent models (Lu et al., 2005). Therefore, growth factors act both locally and distantly to influence the responses of axons to injury over the expanded scale of the primate spinal cord, yet responses differ among specific neuronal populations.
Corticospinal motor neurons in the primate cortex were likely protected by BDNF administration to the spinal cord rather than NT-3, because BDNF but not NT-3 reduced cortical neuronal atrophy in the lesioned rat spinal cord. Primate and rat corticospinal axons express detectable levels of both TrkB and TrkC, yet only BDNF was neuroprotective. This observation highlights the ability of neurotrophic factors to independently influence cellular mechanisms in distinct neuronal compartments, most likely via differential activation of either local signal transduction mechanisms in axons, or binding and transport of growth factor/receptor complexes to influence nuclear transcriptional mechanisms (Miller and Kaplan, 2001; Ginty and Segal, 2002). Although we have previously reported that rat corticospinal axons do not express detectable levels of TrkB (Lu et al., 2001), the present study used a different antibody and labeling method to successfully detect the receptor. It remains possible that BDNF stimulates release of an intermediary molecule in the local environment of the injured axon that in turn reduces the retrograde response of corticospinal neurons in the cortex, rather than directly acting solely on TrkB-expressing corticospinal axons at the lesion site. However, it is unlikely that a factor secreted by the grafted fibroblasts, independent of BDNF expression, influenced the cortical response to injury because the parallel rodent experiment did not detect a protective effect of control fibroblast grafts.
Despite effects of local growth factor secretion on distant corticospinal atrophy in the cortex, corticospinal axons did not regenerate into cell grafts placed in the lesion cavity. These findings are consistent with previous observations in rats (Grill et al., 1997), yet stand in marked contrast to regeneration of raphaespinal and cerulospinal axons into BDNF/NT-3-secreting grafts. Previously, NT-3 has been reported in rat models to promote local sprouting of corticospinal axons after spinal cord injury, but only within spared host gray matter adjacent to the lesion site (Schnell et al., 1994; Grill et al., 1997; Chen et al., 2006). In the present primate model, host gray matter was not spared on the side of the lesion, and NT-3 was not expressed in host gray or white matter in regions containing corticospinal axons; thus, we could not assess whether primate corticospinal axons sprout in response to NT-3. Clearly, however, corticospinal axons in both primate and rodent models fail to regenerate into a lesion site expressing either BDNF or NT-3. Indeed, eliciting regeneration of injured corticospinal axons has been extraordinarily challenging in experimental models of SCI in any species. Yet this system is critical for motor control in higher primates, including humans, and to date only modification of gene expression in the cell somata has proven successful in eliciting even relatively modest regeneration from this system (Hollis et al., 2009). We developed this primate model in part to directly address the need to identify approaches for enhancing plasticity and regeneration of corticospinal systems in primates, and this effort is ongoing.
Findings of this study also indicate that corticospinal neurons survive spinal axotomy for prolonged time periods but remain in a chronically atrophic state. Lesioned corticospinal motor neurons do not die, as revealed by the following: (1) persistent cell numbers in layer V of motor cortex, (2) persistence of labeled corticospinal axons in the medullary pyramids over extended time periods, and (3) an absence of activation of apoptotic markers in the motor cortex within 2 weeks of spinal cord injury. Corticospinal cell death occurs as a function of distance of transection from the cell body, with more proximal lesions (at the level of the internal capsule) causing cell death (Giehl and Tetzlaff, 1996), whereas more distant spinal cord lesions do not result in detectable cell loss. Axon collaterals above a spinal lesion may be sufficient to sustain corticospinal survival. Indeed, corticospinal axons in primates exhibit remarkably extensive collateralization and decussation on reaching the spinal cord (Rosenzweig et al., 2009).
In summary, our results demonstrate that growth factors can signal over the extended scale of the primate CNS, exerting both remote actions on corticospinal systems and local effects on noncorticospinal axons to modify responses to injury. In particular, the prevention of corticospinal neuronal atrophy may maintain these neurons in a state that may be more conducive to elicit plasticity or regeneration using additional experimental manipulations, a subject of ongoing study at the present time.
Footnotes
This work was supported by National Institutes of Health Grant NS42291, the Veterans Administration, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and the Bernard and Anne Spitzer Charitable Trust. We thank Maya Culbertson for technical assistance.
References
- Alto LT, Havton LA, Conner JM, Hollis Ii ER, Blesch A, Tuszynski MH. Chemotropic guidance facilitates axonal regeneration and synapse formation after spinal cord injury. Nat Neurosci. 2009;12:1106–1113. doi: 10.1038/nn.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yaakov K, Fainzilber M. Retrograde Injury Signaling in Lesioned Axons. Results Probl Cell Differ. 2009;48:327–338. doi: 10.1007/400_2009_14. [DOI] [PubMed] [Google Scholar]
- Blesch A, Tuszynski MH. Spinal cord injury: plasticity, regeneration and the challenge of translational drug development. Trends Neurosci. 2009;32:41–47. doi: 10.1016/j.tins.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Chen Q, Zhou L, Shine HD. Expression of neurotrophin-3 promotes axonal plasticity in the acute but not chronic injured spinal cord. J Neurotrauma. 2006;23:1254–1260. doi: 10.1089/neu.2006.23.1254. [DOI] [PubMed] [Google Scholar]
- Conner JM, Varon S. Characterization of antibodies to nerve growth factor: assay-dependent variability in the cross-reactivity with other neurotrophins. J Neurosci Methods. 1996;65:93–99. doi: 10.1016/0165-0270(95)00151-4. [DOI] [PubMed] [Google Scholar]
- Courtine G, Roy RR, Raven J, Hodgson J, McKay H, Yang H, Zhong H, Tuszynski MH, Edgerton VR. Performance of locomotion and foot grasping following a unilateral thoracic corticospinal tract lesion in monkeys (Macaca mulatta) Brain. 2005a;128:2338–2358. doi: 10.1093/brain/awh604. [DOI] [PubMed] [Google Scholar]
- Courtine G, Jindrich DL, McKay HL, Roy R, Havton LA, Zhong H, Liu JJ, Mnatsakanyan A, Bernot TJ, Betts S, Yang H, Tuszynski MH, Edgerton VR. Effects of complete cervical hemisection on performance of locomotion and fine motor activity in Rhesus monkeys. Soc Neurosci Abstr. 2005b;31:784–8. [Google Scholar]
- Davies AM. The role of neurotrophins in the developing nervous system. J Neurobiol. 1994;25:1334–1348. doi: 10.1002/neu.480251103. [DOI] [PubMed] [Google Scholar]
- Dum RP, Strick PL. The origin of corticospinal projections from the premotor areas in the frontal lobe. J Neurosci. 1991;11:667–689. doi: 10.1523/JNEUROSCI.11-03-00667.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giehl KM, Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur J Neurosci. 1996;8:1167–1175. doi: 10.1111/j.1460-9568.1996.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol. 2002;12:268–274. doi: 10.1016/s0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- Grill R, Murai K, Blesch A, Gage FH, Tuszynski MH. Cellular delivery of neurotrophin-3 promotes corticospinal axonal growth and partial functional recovery after spinal cord injury. J Neurosci. 1997;17:5560–5572. doi: 10.1523/JNEUROSCI.17-14-05560.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ. The nucleator. J Microsc. 1988;151:3–21. doi: 10.1111/j.1365-2818.1988.tb04609.x. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Hains BC, Black JA, Waxman SG. Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J Comp Neurol. 2003;462:328–341. doi: 10.1002/cne.10733. [DOI] [PubMed] [Google Scholar]
- Hollis ER, 2nd, Jamshidi P, Löw K, Blesch A, Tuszynski MH. Induction of corticospinal regeneration by lentiviral trkB-induced Erk activation. Proc Natl Acad Sci U S A. 2009;106:7215–7220. doi: 10.1073/pnas.0810624106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey DR, Corrie WS, Rietz R. Properties of the pyramidal tract neuron system within the precentral wrist and hand area of primate motor cortex. J Physiol (Paris) 1978;74:215–226. [PubMed] [Google Scholar]
- Jin Y, Fischer I, Tessler A, Houle JD. Transplants of fibroblasts genetically modified to express BDNF promote axonal regeneration from supraspinal neurons following chronic spinal cord injury. Exp Neurol. 2002;177:265–275. doi: 10.1006/exnr.2002.7980. [DOI] [PubMed] [Google Scholar]
- Jindrich DL, Courtine G, McKay H, Moseanko R, Bernot TJ, Roy R, Zhong H, Liu JJ, Rosenzweig ES, Havton LA, Tuszynski MH, Edgerton VR. Effects of cervical hemisection on locomotion and prehension in Rhesus monkeys. Soc Neurosci Abstr. 2005;31:784–9. [Google Scholar]
- Kadoya K, Tsukada S, Lu P, Coppola G, Geschwind D, Filbin MT, Blesch A, Tuszynski MH. Combined intrinsic and extrinsic neuronal mechanisms facilitate bridging axonal regeneration one year after spinal cord injury. Neuron. 2009;64:165–172. doi: 10.1016/j.neuron.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JM, Mouton PR, Jucker M, Ingram DK. What counts in brain aging? Design-based stereological analysis of cell number. J Gerontol A Biol Sci Med Sci. 1999;54:B407–B417. doi: 10.1093/gerona/54.10.b407. [DOI] [PubMed] [Google Scholar]
- Lu P, Blesch A, Tuszynski MH. Neurotrophism without neurotropism: BDNF promotes survival but not growth of lesioned corticospinal neurons. J Comp Neurol. 2001;436:456–470. doi: 10.1002/cne.1080. [DOI] [PubMed] [Google Scholar]
- Lu P, Yang H, Jones LL, Filbin MT, Tuszynski MH. Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J Neurosci. 2004;24:6402–6409. doi: 10.1523/JNEUROSCI.1492-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Jones LL, Tuszynski MH. BDNF-expressing marrow stromal cells support extensive axonal growth at sites of spinal cord injury. Exp Neurol. 2005;191:344–360. doi: 10.1016/j.expneurol.2004.09.018. [DOI] [PubMed] [Google Scholar]
- McQuarrie IG, Grafstein B. Axon outgrowth enhanced by a previous nerve injury. Arch Neurol. 1973;29:53–55. doi: 10.1001/archneur.1973.00490250071008. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. On Trk for retrograde signaling. Neuron. 2001;32:767–770. doi: 10.1016/s0896-6273(01)00529-3. [DOI] [PubMed] [Google Scholar]
- Murray EA, Coulter JD. Organization of corticospinal neurons in the monkey. J Comp Neurol. 1981;195:339–365. doi: 10.1002/cne.901950212. [DOI] [PubMed] [Google Scholar]
- Murray M, Kim D, Liu Y, Tobias C, Tessler A, Fischer I. Transplantation of genetically modified cells contributes to repair and recovery from spinal injury. Brain Res Brain Res Rev. 2002;40:292–300. doi: 10.1016/s0165-0173(02)00211-4. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Nudo RJ, Sutherland DP, Masterton RB. Variation and evolution of mammalian corticospinal somata with special reference to primates. J Comp Neurol. 1995;358:181–205. doi: 10.1002/cne.903580203. [DOI] [PubMed] [Google Scholar]
- Pakkenberg B, Gundersen HJ. Neocortical number in humans: effect of sex age. J Comp Neurol. 1997;384:312–320. [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610–616. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- Richardson PM, Issa VM, Aguayo AJ. Regeneration of long spinal axons in the rat. J Neurocytol. 1984;13:165–182. doi: 10.1007/BF01148324. [DOI] [PubMed] [Google Scholar]
- Romero MI, Rangappa N, Li L, Lightfoot E, Garry MG, Smith GM. Extensive sprouting of sensory afferents and hyperalgesia induced by conditional expression of nerve growth factor in the adult spinal cord. J Neurosci. 2000;20:4435–4445. doi: 10.1523/JNEUROSCI.20-12-04435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Brock JH, Culbertson MD, Lu P, Moseanko R, Edgerton VR, Havton LA, Tuszynski MH. Extensive spinal decussation and bilateral termination of cervical corticospinal projections in rhesus monkeys. J Comp Neurol. 2009;513:151–163. doi: 10.1002/cne.21940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell L, Schneider R, Kolbeck R, Barde YA, Schwab ME. Neurotrophin-3 enhances sprouting of corticospinal tract during development and after adult spinal cord lesion. Nature. 1994;367:170–173. doi: 10.1038/367170a0. [DOI] [PubMed] [Google Scholar]
- Simic G, Kostovic I, Winblad B, Bogdanovic N. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer's disease. J Comp Neurol. 1997;379:482–494. doi: 10.1002/(sici)1096-9861(19970324)379:4<482::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Smith DE, Roberts J, Gage FH, Tuszynski MH. Age-associated neuronal atrophy occurs in the primate brain and is reversible by growth factor gene therapy. Proc Natl Acad Sci U S A. 1999;96:10893–10898. doi: 10.1073/pnas.96.19.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DE, Rapp PR, McKay HM, Roberts JA, Tuszynski MH. Memory impairment in aged primates is associated with focal death of cortical neurons and atrophy of subcortical neurons. J Neurosci. 2004;24:4373–4381. doi: 10.1523/JNEUROSCI.4289-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor L, Jones L, Tuszynski MH, Blesch A. Neurotrophin-3 gradients established by lentiviral gene delivery promote short-distance axonal bridging beyond cellular grafts in the injured spinal cord. J Neurosci. 2006;26:9713–9721. doi: 10.1523/JNEUROSCI.0734-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, Gabriel K, Gage FH, Suhr S, Meyer S, Rosetti A. Nerve growth factor delivery by gene transfer induces differential outgrowth of sensory, motor, and noradrenergic neurites after adult spinal cord injury. Exp Neurol. 1996;137:157–173. doi: 10.1006/exnr.1996.0016. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Grill R, Jones LL, McKay HM, Blesch A. Spontaneous and augmented growth of axons in the primate spinal cord: effects of local injury and nerve growth factor-secreting cell grafts. J Comp Neurol. 2002;449:88–101. doi: 10.1002/cne.10266. [DOI] [PubMed] [Google Scholar]
- Wannier T, Schmidlin E, Bloch J, Rouiller EM. A unilateral section of the corticospinal tract at cervical level in primate does not lead to measurable cell loss in motor cortex. J Neurotrauma. 2005;22:703–717. doi: 10.1089/neu.2005.22.703. [DOI] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH. Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc Natl Acad Sci U S A. 2001;98:3513–3518. doi: 10.1073/pnas.051626798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Ostergaard K, Andreassen OA, Finsen B. Estimation of the number of somatostatin neurons in the striatum: an in situ hybridization study using the optical fractionator method. J Comp Neurol. 1996;370:11–22. doi: 10.1002/(SICI)1096-9861(19960617)370:1<11::AID-CNE2>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]