

Abstract
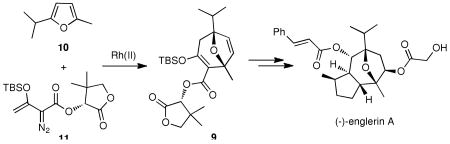
An enantioselective formal synthesis of (−)-englerin A (1) is reported. Key to the strategy is a Rh-catalyzed [4+3] cycloaddition reaction between furan 10 and diazoester 11 that, following an intramolecular aldol condensation, produces the tri-cyclic scaffold of englerin. This strategy also provides a rapid, efficient and stereoselective access to the biologically significant core motif of the guaiane sesquiterpenes.
Renal cell carcinoma (RCC also known as hypernephroma) is a type of kidney cancer that originates in the small tubes in the kidneys that filter the blood and remove waste products. This cancer ranks among the ten leading cancer types in the United States and only in 2009, it was responsible for about 58000 new cases and 13000 deaths.1 Importantly, RCC is resistant to radiation and chemotherapy leaving partial or radical nephrectomy as the only means of treatment. Therefore the search for new compounds that can safely and effectively block or reverse RCC remains a high priority.
Efforts to identify new small molecule leads against RCC led to the identification of englerin A (1) and B (2) (Figure 1) from the Tanzanian plant Phyllanthus engleri. 2 From the standpoint of biogenesis, the englerins are guaiane-type sesquiterpenes that contain an uncommon oxygenated motif (Figure 1). Structurally related members of this family include orientalols E (3) and F (4)3 and pubinernoid B (5)4 (Figure 1). Most likely, this motif originates from a more common bicyclic-sesquiterpenes core via a sequence of oxygenations and acid-induced oxo-cyclization reactions highlighted in Scheme 1.3
Figure 1.

Structures of englerin A, B and natural analogues
Scheme 1.

Proposed biosynthetic approach
Remarkably, englerin A (1) showed highly potent and selective cytotoxicities against various renal cancer cell lines at low nanomolar level.2 Preliminary biological investigations also showed a significant drop of potency and selectivity of englerin B (2), suggesting that the substitution at the C-9 position by the glycolate ester may be important for the activity and selectivity of englerins.2
Due to its promising anticancer activity and its scarcity from natural sources, englerin A has become an intriguing target in synthetic and medicinal chemistry. The absolute configuration of 1 was confirmed by Christmann et al. in their first total synthesis of ent-1 in 2009.5 More recently, three additional syntheses were reported, by Ma,6 Echavarren7 and Nicolaou8 the first two of which were enantioselective. In addition, a stereoselective approach to the guaianolide core of these sesquiterpenes has been disclosed.9
Inspired by the unusual tricyclic motif, we sought to design a general approach leading not only to the structures of 1 and 2 but also to various guaiane analogues, such as orientalols E (3) and F (4) and pubinernoid B (5). We envisioned that 1 could be derived from diol 6 via reversion of the C-6 stereocenter, hydrogenation and two esterifications. The C-9 hydroxyl group of the diol 6 could be easily obtained by the regio- and stereo-selective hydroboration on the disubstituted double bond of 7. Moreover, the cyclopentene moiety of 7 could be constructed from an intramolecular aldol condensation of diketone 8 which can be formed from oxa-tricyclic compound 9. In turn, the key oxa-tricyclic compound 9, could be achieved via the Davies Rh-catalyzed ring formation,10 from readily available di-substituted furan 10 and chiral diazo ester 11 (Scheme 2).
Scheme 2.

Retrosynthetic Analysis
Rhodium-triggered cyclization reactions have been widely applied in synthetic chemistry.11 In 1996, Davies et al. reported an elegant enantioselective Rh(II)-catalyzed [4+3] cycloaddition reaction between furans and diazo esters.10,12 With this in mind we prepared furan 10 from 2-methylfuran in 3 steps following the reported procedure;13 and diazo ester 11, which derived from (R)-pantolactone in 3 steps.10 Notably, both of these starting materials are readily available in more than 50 gram-scale. Slow addition of the chiral diazo ester 11 into furan 10 in refluxing hexane catalyzed with 2 mol% rhodium (II) octanoate, gave rise to key oxa-tricyclic motif 9 in an excellent yield (90%, Scheme 3) with moderate diastereoselectivity (dr = 3:1).14 This diastereomeric mixture can be easily separated through silica gel column chromatography. It is likely that a more bulky chiral auxiliary could provide better diastereoselectivity, although attempts at effecting this reaction under lower temperature or using less catalyst loading led to significantly poorer yield without any enhancement of diastereoselectivity.
Scheme 3.

Initial functionalization of this oxa-tricyclic compound 9 was successfully accomplished by removing the auxiliary. This was achieved by treating 9 with DIBAL-H to afford the corresponding unstable β-hydroxyl enol ether, followed by the Lewis acid induced rearrangement15 to afford enone 12 in 59% yield. For the next step, attempts of α-hydroxylation with Davis oxaziridines16 of enone 12 were not successful. Gratifyingly, Rubottom oxidation17 of enone 12 led to the desired α-hydroxyl enone 13 in very good yield (87% brsm). Not surprisingly, the hydroxylation occurred on the less hindered face of this molecule, leading exclusively to the opposite stereochemistry at C-6. Nonetheless, the C-6 stereocenter can be easily reversed in a latter stage. Notably, m-CPBA selectively oxidized the electronic richer TMS enol-ether without touching any of other double bonds of our substrate. The absolute stereochemistry of α-hydroxyl enone 13 was confirmed by a single crystal Cu-mediated X-ray analysis,18 thus establishing the desired absolute stereochemistry of oxa-tricyclic compound 9.19
The next stage of the synthesis involved elaboration of the enone system to the 5-membered ring, which eventually forms the tricyclic englerin core. After a simple TBS protection on the C-6 hydroxyl group, the enone Michael system was subjected to a 1,4-addition with propanal catalyzed by thiazolium salt 14 under basic conditions (Scheme 4).20 The diketone 8 was cleanly obtained in good yield (75% over two steps) as a single diastereomer. Having the precursor 8 on hand, the ensuing aldol condensation proved to be unexpectedly difficult. Extensive studies on this intramolecular aldol condensation were then applied, with different substituents on the C-6 hydroxyl (H-, MOM-, TES-, etc.), various bases (tBuOK, KOH, NaOMe, LDA, etc.) and several reaction conditions. Eventually, we were pleased to discover that treatment of diketone 8 with NaHMDS followed by heating in NaOMe/MeOH could provide the desired key tri-cyclic intermediate 7 in acceptable yield (43% brsm).21 Reduction of enone 7 furnished the corresponding allylic alcohol as a single diastereomer, which was then directly protected with benzyl group to give 15, paving the way for the following hydroboration.
Scheme 4.

The next task was the installation of a hydroxyl group at the C-9 position. This was accomplished in a regio- and stereo-selective manner by treating 15 with borane•THF complex followed by basic H2O2 oxidative cleavage to give the desired C-9 β-alcohol in 60% yield (Scheme 5). After removal of the minor (∼10%) C-8 regio-isomer via column chromatography, the C-9 β-alcohol was converted to the corresponding di-TBS-ether. After removal of the benzyl group, initial efforts to hydrogenate the tetra-substituted double bond met with failure presumably due to the steric hindrance of this alkene.22 This prompted us to deoxygenate the C-3 stereocenter under standard Barton-McCombie conditions (40% yield).23 However, a more efficacious route from 16 to 18 was achieved by the dehydration with Burgess reagent24 followed by a simple hydrogenation (90% over 2 steps). While the cleavage of the di-TBS-ether gave only messy results under regular deprotection conditions (TBAF/THF, rt or 80 °C, overnight), a microwave accelerated deprotection proved efficient and produced 6 in excellent yield (TBAF/THF, 80 °C, 45 min, 93%).
Scheme 5.

The final stages of this synthesis would require the esterification of the C-9 hydroxyl group, reversion of the stereocenter at C-6, hydrogenation and esterification. In fact, during the course of our work, an alternative route to (+)-6 and its successful elaboration to (−)-englerin A was reported by Ma et al.6 Our synthetic work to (+)-6 provided another novel and facile access to the englerin core. This approach with 15 steps from readily available compound 10 and 11 in 5% overall yield, constitutes a formal synthesis of (−)-englerin A. More importantly, this Rh-catalyzed [4+3] cycloaddition, followed by an intramolecular aldol condensation, provides a unique and facile method for construction of a common guaiane-type tricyclic-sesquiterpene skeleton. This strategy is broadly applicable to an enantioselective synthesis of englerin and related natural products from the key intermediate 7 (Scheme 6). The applications of this strategy to the synthesis of natural and designed tricyclic sesquiterpenes could facilitate the SAR and chemical biology studies of this unexplored class of natural products.
Scheme 6.

Supplementary Material
Acknowledgments
We gratefully acknowledge the National Institutes of Health (NIH) for financial support of this work through Grant Number R01 GM081484-01. We thank the National Science Foundation for instrumentation grants CHE9709183 and CHE0741968. We also thank Dr. Anthony Mrse (UCSD NMR Facility); Dr. Yongxuan Su (UCSD MS Facility) and Dr. Arnold L. Rheingold and Dr. Curtis E. Moore (UCSD X-Ray facility). We also appreciate the technical assistance from Weng K. Chang and Steven D. E. Sullivan (UCSD, Theodorakis group).
Footnotes
Supporting Information Available: Experimental procedures and characteristic data of new compounds, as well as X-ray data of compound 13. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer Statistics 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ratnayake R, Covell D, Ransom TT, Gustafson KR, Beutler JA. Org Lett. 2009;11:57–60. doi: 10.1021/ol802339w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beutler JA, Ratnayake R, Covell D, Johnson TR. WO 2009/088854. 2009 [Google Scholar]
- 3.Peng GP, Tian G, Huang XF, Lou FC. Phytochemistry. 2003;63:877–881. doi: 10.1016/s0031-9422(03)00222-x. [DOI] [PubMed] [Google Scholar]
- 4.Huang SX, Yang J, Xiao WL, Zhu YL, Li RT, Li LM, Pu JX, Li X, Li SH, Sun HD. Helv Chim Acta. 2006;89:1169–1175. [Google Scholar]
- 5.Willot M, Radtke L, Könning D, Fröhlich R, Gessner VH, Strohmann C, Christmann M. Angew Chem Int Ed. 2009;48:9105–9108. doi: 10.1002/anie.200905032. [DOI] [PubMed] [Google Scholar]
- 6.Zhou QF, Chen XF, Ma DW. Angew Chem Int Ed. 2010;49:3513–3516. doi: 10.1002/anie.201000888. [DOI] [PubMed] [Google Scholar]
- 7.Molawi K, Delpont N, Echavarren AM. Angew Chem Int Ed. 2010;49:3517–3519. doi: 10.1002/anie.201000890. [DOI] [PubMed] [Google Scholar]
- 8.Nicolaou KC, Kang Q, Ng SY, Chen DYK. J Am Chem Soc. 2010;132:8219–8222. doi: 10.1021/ja102927n. [DOI] [PubMed] [Google Scholar]
- 9.(a) Jiménez-Núñez E, Claverie CK, Nieto-Oberhuber C, Echavarren AM. Angew Chem Int Ed. 2006;45:5452–5455. doi: 10.1002/anie.200601575. [DOI] [PubMed] [Google Scholar]; (b) Jiménez-Núñez E, Echavarren AM. Chem Commun. 2009;45:7327–7329. doi: 10.1039/b920119j. [DOI] [PubMed] [Google Scholar]
- 10.Davies HML, Ahmed G, Churchill MR. J Am Chem Soc. 1996;118:10774–10782. [Google Scholar]
- 11.For an excellent review, see: Jeong N, Robinson JE, Wender PA, Gamber GG, Williams TJ, Davies HML, Walji AM. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Chapter 11-14. Wiley-VCH Verlag GmbH & Co. KGaA; 2005. pp. 215–340.
- 12.For a recent application of this strategy on natural product synthesis, see also: Jackson KL, Henderson JA, Motoyoshi H, Phillips AJ. Angew Chem Int Ed. 2009;48:2346–2350. doi: 10.1002/anie.200806111.
- 13.Weyerstahl P, Brendel J. Liebigs Ann Chem. 1988;1015-1016 [Google Scholar]
- 14.Calculated via 1H-NMR spectroscopy.
- 15.Poirier JM, Hennequin L. Tetrahedron. 1989;45:4191–4202. [Google Scholar]
- 16.Davis FA, Vishwakarma LC, Billmers JM, Finn J. J Org Chem. 1984;49:3241–3243. [Google Scholar]
- 17.Rubottom GM, Vazquez MA, Pelegrina DR. Tetrahedron Lett. 1974;15:4319–4322. [Google Scholar]
- 18.CCDC782508 contains the supplementary crystallographic data for compound 13. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/products/csd/request/
- 19.Hooft WWR, Straver LH, Spek AL. J Appl Cryst. 2008;41:96–103. doi: 10.1107/S0021889807059870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stetter H. Angew Chem Int Ed. 1976;15:639–647. [Google Scholar]
- 21.To our surprise, compared to as regular intramolecular reactions, this intramolecular aldol reaction requires quite high concentration (0.4 ∼ 0.7 M).
- 22.The C4-C5 double bond stay untouched even under high pressure (up to 130 bar) with Pd/C, Crabtree's catalyst or PtO2.
- 23.Barton DHR, McCombie SW. J Chem Soc, Perkin Trans 1. 1975;16:1574–1585. [Google Scholar]
- 24.Atkins GM, Burgess EM. J Am Chem Soc. 1968;90:4744–4745. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.