

Abstract
Purpose
Abnormal DNA CpG island hypermethylation and transcriptionally repressive histone modifications are associated with the aberrant silencing of tumor suppressor genes. Lysine methylation is a dynamic, enzymatically-controlled process. Lysine-specific demethylase 1 (LSD1) has recently been identified as a histone lysine demethylase. LSD1 specifically catalyzes demethylation of mono- and dimethyl-lysine 4 of histone 3, key positive chromatin marks associated with transcriptional activation. We hypothesized that a novel class of oligoamine analogues would effectively inhibit LSD1 and thus cause the re-expression of aberrantly silenced genes.
Experimental Design
Human colorectal cancer cells were treated with the oligoamines and changes in mono- and dimethyl-lysine 4 of histone 3 (H3K4) and other chromatin marks were monitored. In addition, treated cells were evaluated for the re-expression of the aberrantly silenced secreted frizzled-related proteins (SFRPs) Wnt signaling pathway antagonist genes. Finally, the effects of the LSD1 inhibitors were evaluated in an in vivo xenograft model.
Results
Treatment of HCT116 human colon adenocarcinoma cells in vitro resulted in increased H3K4 methylation and re-expression of silenced SFRP genes. This re-expression is also accompanied by a decrease in H3K9me2 repressive mark. Importantly, co-treatment with low doses of oligoamines and a DNA methyltransferase (DNMT) inhibitor highly induces the re-expression of the aberrantly silenced SFRP2 gene and results in significant inhibition of the growth of established tumors in a human colon tumor model in vivo.
Conclusions
The use of LSD1-inhibiting oligoamine analogues in combination with DNMT inhibitors represents a highly promising and novel approach for epigenetic therapy of cancer.
Introduction
Epigenetics refers to heritable changes in gene expression patterns that are not regulated by changes in primary DNA sequence. In cancer, epigenetic silencing of gene expression, including of tumor suppressor genes, is a common occurrence (1) that is associated with abnormal DNA methylation patterns and changes in covalent histone modifications (2). The amino terminal tails of histones are subject to several post-translational modifications including acetylation, phosphorylation, and methylation that are closely tied to transcriptional regulation, DNA replication, and DNA repair (2). As demonstrated by the regulation of histone acetylation by histone acetyltransferases (HATs) and histone deacetylases (HDACs), the addition and removal of these post-translational modifications is a dynamic process. A similar dynamic regulation occurs for histone methylation with histone methyltransferases for addition of methyl groups, and recently discovered families of enzymes for specific histone demethylation. The first of these demethylating enzymes identified was the lysine-specific demethylase (LSD1/KDM1) (3), a FAD-dependent amine oxidase, which interacts directly with CoREST and HDAC1/2 proteins, forming a module found in several multiprotein co-repressor complexes and is known to act on intact chromatin as part of these complexes (3, 4). LSD1 demethylates H3K4me2/me1 through an oxidative reaction that leads to the reduction of the protein-bound FAD cofactor and the production of H2O2 and formaldehyde. More recently, a number of Jumonji (JmjC) domain-containing histone demethylases have been identified and shown to play important roles in concert with other histone modifying enzymes related to the control of transcriptional regulation, cellular differentiation, and animal development (5, 6).
As emerging data suggests that LSD1 has the capacity to broadly repress gene expression, and as dysregulation of epigenetic gene silencing is a common feature in all cancers, LSD1 is being actively pursued as an attractive drug target. In both normal cells, and colorectal cancer cells, increasing promoter region H3K4me2 is particularly tightly tied to increasing levels of active gene transcription (7, 8). Additionally, emerging data indicates that high expression of LSD1 may be a feature of specific human cancers (9). Several studies, including those from our laboratory, have reported the inhibitory effect of various small molecules on LSD1 (10-12). Structural analysis demonstrates that LSD1 is highly conserved across species and consists of an N-terminal SWIRM domain, a central protruding tower domain, and a C-terminal amine oxidase-like domain that contains an FAD-binding subdomain. The FAD-binding oxidase domain is highly homologous, with 20% similarity, to that of FAD-dependant monoamine oxidases (MAOs) and polyamine oxidases, such as spermine oxidase (SMO) and N1-acetylpolyamine oxidase (APAO) (6, 13-15). Furthermore, the catalytic domains of LSD1 and SMO possess over 60% similarity in amino acid sequences.
Although the natural polyamines are not substrates of LSD1 (3), the above structural and catalytic similarities of FAD-dependent oxidases, the strong association of polyamines with chromatin, and the structural similarity between the polyamines and the lysine tail of histones, led us to test the possibility that polyamine analogues might be potent inhibitors of LSD1 (10). Indeed, LSD1 inhibition by these unique compounds resulted in re-expression of multiple aberrantly silenced tumor suppressor genes in colorectal cancer cells, further validating LSD1 as a potential target for therapeutic intervention. Furthermore, the results suggested that other polyamine-like molecules might function as effective inhibitors of LSD1 and would thus be effective epigenetic targeting agents.
We previously found that specific members of a novel class of long chain polyamine analogues known as oligoamines (16) were not substrates of either APAO or SMO, but were potent inhibitors of these purified polyamine oxidases (17). Since these enzymes are highly homologous to the FAD-dependent LSD1, we hypothesized that specific members of the oligoamines would be effective inhibitors of LSD1. Additionally, because of their multivalent (+10) cationic structure we postulated that they would be targeted to chromatin (18), the site of action for LSD1, at least as efficiently as the biguanide or bisguanidine analogues we previously examined. We now report that that treatment of tumor cells with the oligoamines results in re-expression of specific aberrantly silenced genes in vitro. Furthermore, we demonstrate for the first time the use of LSD1 inhibitors in combination with a DNA methyltransferase inhibitor, a combination that is not only more efficacious in reactivating specific aberrantly silenced genes, but also leads to profound inhibition of the growth of established human colon cancer xenografts in a nude mouse model.
Materials and Methods
Compounds, Peptides, Histones, and Culture Conditions
Polyamine analogues were synthesized as previously reported (17). Stock solutions (10 mM in double distilled H2O) of each compound were diluted with medium to the desired concentrations for specific experiments. Synthetic H3K4me2 peptides were purchased from Millipore (Billerica, MA). The DNA methyltransferase (DNMT) inhibitors, 5-aza-2′-deoxycitidine (DAC) and 5-azacytidine (5-Aza), and bulk histones were purchased from Sigma (St. Louis, MO). HCT116 colorectal carcinoma cells were maintained in McCoy's 5A medium and RKO cells were maintained in MEM medium, each supplemented with 9% FBS (Atlanta Biologicals, Lawrenceville, GA) and 1% penicillin/streptomycin (Mediatech, Manassas, VA), and grown at 37°C in 5% CO2 atmosphere.
MTT Growth Inhibition Assay, Annexin V Staining, and PARP Cleavage
Cells were seeded at 5000 cells/well of a 96-well plate and allowed to attach overnight. Medium was aspirated and replaced with fresh media containing the appropriate concentrations of LSD1 inhibitor. Following incubation for 24 or 48 hr, 200 μl of a 1 mg/ml MTT solution (Sigma Chemical Co.), diluted in serum-free culture media, were added to each well. The plates were incubated at 37°C in 5% CO2 atmosphere for 4 h. At the end of the 4 hr incubation, the MTT solution was removed, and 200 μl of 1:1 (v/v) solution of DMSO:ethanol were added to each well to dissolve the formazan crystals. The absorbance in individual wells was determined at A540 nm..
Evidence for apoptotic cell death was determined by both PARP cleavage and annexin V staining as we previously published (19, 20).
Expression, Purification and Demethylase Activity of Recombinant Proteins
Human cDNAs coding either the full-length LSD1 protein or that with a deletion of the N-terminal 184 (Δ184 LSD1) amino acids were subcloned into the pET15b bacterial expression vector (Novagen, Madison, WI) in frame with an N-terminal 6× His-tag and transformed into the BL21(DE3) strain of Escherichia coli. Following selection, expression and purification of recombinant LSD1 protein were performed as previously described (10). Bound protein was eluted by imidazole and the eluate was dialyzed in PBS at 4°C. Enzymatic activity of LSD1 was examined using both a luminol-dependent chemiluminescence method to measure the production of H2O2, and a quantitative Western Blot method that measures the amount of H3K4me2 after incubation of purified protein with bulk histones as previously described (15).
Western Blotting
Nuclear fractions of treated cells were prepared for Western blot analysis using NE-PER Nuclear and Cytoplasmic Extraction reagents (Pierce, Rockford, IL). Equal amounts (30 μg/lane) of nuclear protein were fractionated on SDS-PAGE gels and transferred onto PVDF membranes. Primary antibodies against H3K4me2, H3K4me3, and H3K4me1 were from Millipore. The H3 polyclonal antibody used for normalization was purchased from Abcam (Cambridge, MA). Dye-conjugated secondary antibodies were used to quantify Western blot results with the Odyssey Infrared Detection system and software (LI-COR Biosciences, Lincoln, NE).
RNA Isolation, RT-PCR, qPCR, and Bisulfite Sequencing
RNA for RT-PCR was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). First-strand cDNA was synthesized using M-MLV reverse transcriptase with an oligo(dT) primer (Invitrogen). PCR was performed using the following custom primers: SFRP1 sense, 5′GGC CCA TCT ACC CGT GTC G; SFRP1 antisense, 5′GAT GGC CTC AGA TTT CAA CTC GT (annealing at 60°C); SFRP2 sense, 5′AAG CCT GCA AAA ATA AAA ATG ATG; SFRP2 antisense, 5′TGT AAA TGG TCT TGC TCT TGG TCT (annealing at 53°C); SFRP4 sense, TCT ATG ACC GTG GCG TGT GC; SFRP4 antisense, ACC GAT CGG GGC TTA GGC GTT TAC (annealing at 56°C); GATA5 sense, CCT GCG GCC TCT ACC ACA A; GATA5 antisense, GGC GCG GCG GGA CGA GGA C (annealing at 58°C). A total of 35 cycles of amplification was performed and GAPDH was amplified as an internal control. Amplified products were analyzed on 2% agarose gels with GelStar staining (Cambrex, Walkersville, MD).
qPCR of SFRP1 and SFRP2 was performed as published previously (7). The same forward and reverse primers that were used for RT-PCR were used for qPCR in a MyiQ single color real-time PCR machine (Bio-Rad, Hercules, CA) with GAPDH as an internal control. Amplification conditions for SFRP1 consisted of a 15-min denaturation step followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s. Identical conditions for SFRP2 were used except the annealing temperature was 53°C. To quantify relative expression, the comparative cycle threshold (Ct) method was used, normalizing the Ct values for the gene of interest to the Ct values of GAPDH relative to untreated control.
Bisulfite sequencing of the SFRP2 gene promoter was performed as we have previously published (10) using the following primers: sense, GGT AAT TTA GTA GAA ATT TCG GAT TG, antisense, ACT AAT CAC TAC TTT CTA AAT CTA ATT.
Chromatin immunoprecipitation (ChIP)
Control and analogue treated cells were exposed to 1% formaldehyde to cross-link proteins, and two million cells were used for each ChIP assay and performed as previously described (10). Antibody against H3 was purchased from Abcam. Primary antibodies against LSD1, H3K4me2, H3K4me1, H3K4me3, H3K9me2, acetyl-H3K9, or acetyl-H4K16 were from Millipore. PCR primer sets used for amplification of precipitated fragments were as follows: SFRP1 sense, 5′GGC CCA TCT ACC CGT GTC G; antisense, GAT GGC CTC AGA TTT CAA CTC GT; SFRP2(Fragment A): sense, 5′CTC CCT CCC AGC CTG CCC ATC TT; antisense, 5′ ACT GCC CAC CAT TTC CCC GTT TTG; SFRP2(Fragment B): sense, 5′CCT GTG TGA CTG GTG AGA CTC; antisense, 5′ CGG CGA ACT TCG TTT TCC CTC; SFRP2(Fragment C): sense, 5′GCT GCT GAA CGG TGG CTG GAG ATT; antisense, 5′ CAG GCA TGA GTG GGA GAG GGT GTG; SFRP2(Fragment D): sense, 5′TGG CAA CCC AGC AGA AAC TTC; antisense, 5′ACG CGC TTG CTG GAA GGG AAT TC; SFRP2(Fragment E): sense, 5′AGG GGC ATT CTC AAC GCAAAA CC; antisense, 5′GGG ACA CCG GGA GGA CAG C; SFRP2(Fragment F): sense, 5′GCT TCT CCG CGC CCC AGC CGC C; antisense, 5′AGC CCA GGC AGC AGT GCG AGG. Sheared genomic DNA was used as a positive control (Input). Quantitative ChIP confirmed changes in histone marks at the promoters of examined genes by using qPCR. qPCR was performed with the same primers used for standard ChIP on the MyiQ single color real-time PCR machine (Bio-Rad). Conditions used were 40 cycles of denaturation at 95°C for 30 s, annealing at 61°C for 30 s, and extension at 72°C for 30 s. DNA immunoprecipitated by H3 antibody was used for normalization.
Animal Studies
BALB c nu/nu athymic nude mice (Harlan Bioproducts for Science Inc., Indianapolis, IN) were implanted with the human colorectal cancer HCT-116 cells. By 20 days post-implantation average tumor size was > 350 mm3 and mice were randomized into treatment (n = 5) and control (n = 5) groups. The single agent treatment groups were: vehicle (saline control at qdx5), PG-11144 (10 mg/kg, 2× wk), 2d (10 mg/kg qdx5), and 5-Aza (2 mg/kg qdx5). The combination treatment groups were: PG-11144 (10 mg/kg, 2× wk)/5-Aza (2 mg/kg qdx5) and 2d (10 mg/kg qdx5)/5-Aza (2 mg/kg qdx5). Test agents were administered by intraperitoneal (i.p.) injection for 3 weeks. The drugs administered on a 2× wk schedule (PG-11144) were given on days 20, 23, 27, 30, 34, and 37 after implantation and drugs given on the qdx5 schedule (2d, 5-Aza) were injected on days 20-24, 27-31 and 34-38. The control group was treated with vehicle (sterile saline) using the same qdx5 treatment schedule. The animals were observed for adverse clinical signs after each dose and once daily thereafter throughout the course of the study. Morbidity and mortality was monitored daily following the initial dose and continued throughout the course of the study. The experiment was terminated on day 38 after tumor implantation and the tumors were harvested for Western blotting analysis of H3K4me2. It should be noted that 5-Aza was chosen over DAC for in vivo experiments, as 5-Aza has been the drug of choice for ongoing clinical trials in our institution (21); 5-Aza has been shown to have efficacy in the pre-leukemic disease, myelodysplasia and is approved by the FDA for use in patients with this disorder (22).
Results
Inhibition of recombinant LSD1 activity by oligoamines
We tested a series of conformationally restricted and saturated polyamine and oligoamine analogues, including pentamines, hexamines, octamines and decamines (Fig. 1A), for their effect on recombinant LSD1 activity. The conformationally restricted analogues tested incorporate molecular alterations that restrict the free rotation of specific bonds in otherwise flexible molecules, and are longer than the natural polyamines. Most of those tested here have four methylene (CH2) residues between each imine nitrogen (16). While the pentamine and hexamine analogues marginally affect LSD1 activity, octamine and decamine analogues inhibited this activity by >50% at 10 μM using 5μM H3K4me2 peptide (1-21 aa) as a substrate (Fig. 1B). The two most potent inhibitors, the decamine analogues PG-11144 and PG-11150, which are cis and trans isomers, were chosen for further study.
Figure 1.

Inhibition of LSD1 by polyamine analogues. (A) Structures of natural polyamines and members of the oligoamine family of polyamine analogues. (B) 0.038 μg/μl of purified Δ184 LSD1 protein were incubated with 5 μM H3K4me2 (1-21 aa) as substrate in the presence of 10 μM of the indicated analogue. Effect of oligoamines on enzymatic activity of LSD1 was examined using luminol-dependent chemiluminescence to measure the production of H2O2. The integral values were calibrated against standards containing known concentrations of H2O2, and the activities expressed as picomoles of H2O2 per milligram of protein per minute. Shown are means ± SD of independent experiments performed in triplicate.
We confirmed that PG-11144 and PG-11150 inhibit LSD1 demethylase activity by Western blot using bulk histone as a substrate (Fig. 2A). Both analogues inhibit recombinant LSD1 activity in a concentration-dependent manner with IC50s around 5μM using 5μM H3K4me2 peptide (1-21aa) as substrate (Fig. 2B). Using purified recombinant LSD1, PG-11144 exhibited competitive inhibition kinetics at concentrations <10 μM (Fig. 2C), suggesting that PG-11144 may compete with H3K4me2 at the LSD1 active site. Similar kinetics were observed for PG-11150 (data not shown).
Figure 2.

Inhibition of recombinant human LSD1 by oligoamines. (A) 0.05 μg/ul of purified bulk histones were incubated with or without 0.01 μg/μl purified full-length LSD1 for 3 hr at 37°C. Effects of LSD1 enzymatic activity were analyzed by Western blotting using antibodies that specifically recognize the dimethyl group of H3K4. Relative levels were determined by quantitative Western analysis using the Odyssey infrared detection system. Total H3 was used as loading control. The results represent the mean of three determinations ± SD. (B) 0.038 μg/μl of purified Δ184 LSD1 protein were incubated with 5 μM H3K4me2 (1-21 aa) as substrate in the presence of increasing concentrations of PG-11144 and PG-11150. Effect of oligoamines on enzymatic activity of LSD1 was examined using luminol-dependent chemiluminescence to measure the production of H2O2. The vertical bars indicate mean ± SD. (C) The effects of increasing concentrations of PG-11144 on LSD1 activity in the presence of increasing substrate concentrations. Double reciprocal plots indicate inhibition of LSD1 by PG-11144 to be competitive. Shown are means ± SD of independent experiments performed in triplicate.
Inhibition of growth by oligoamines
The sensitivity of the HCT-116 and RKO cell lines to the oligoamines, PG-11144 and PG-11150, was assessed with the MTT cell proliferation assay. Both cell lines exhibited time- and concentration-dependent growth inhibition by oligoamines (Supplementary Fig. S1). The IC50 values for both cell lines are about 2.5-5 μM for a 48h treatment.
To determine the cause of the observed decrease in proliferation, PG-11144-treated cells (5 μM) were assessed for apoptotic cell death by annexin V staining and PARP cleavage. Annexin V staining analysis indicates that 24 h treatment with PG-11144 induces an increase in apoptotic cell death (19.3% annexin V positive cells in treated versus 4% in controls, 10,000 events each), but DAC treatment (100 nM), either alone (5% annexin V positive) or in combination with PG-11144 (20.4% annexin V positive) did not significantly increase apoptotic cell death. The PARP cleavage data (Fig. S2) are entirely consistent with the annexin V results indicating the observed apoptosis is a result of PG-11144 treatment. It should be noted that the 24 h treatment time was chosen for analsyis as this treatment time was used for the chromatin and gene expression analyses presented below.
Oligoamines increase global H3K4 methylation in colorectal tumor cells
To determine whether the in vitro inhibition of LSD1 activity by oligoamines translates into cellular response, the effects on global H3K4 methylation were examined after exposure of human colon cancer HCT116 and RKO cells to increasing concentrations of each compound. This exposure did not reduce protein levels of nuclear LSD1, but produced significant global increases of both H3K4me1 and H3K4me2, both of which are substrates of LSD1 (Fig. 3A). H3K4me3 levels were not affected in treated cells (not shown). These results indicate that the oligoamines effectively inhibit LSD1 enzymatic activity in situ. Furthermore, these results suggest that the oligoamines are selective for the FAD-dependent LSD1 and do not inhibit the JmjC histone demethylases, as no changes in H3K4me3 levels were observed.
Figure 3.
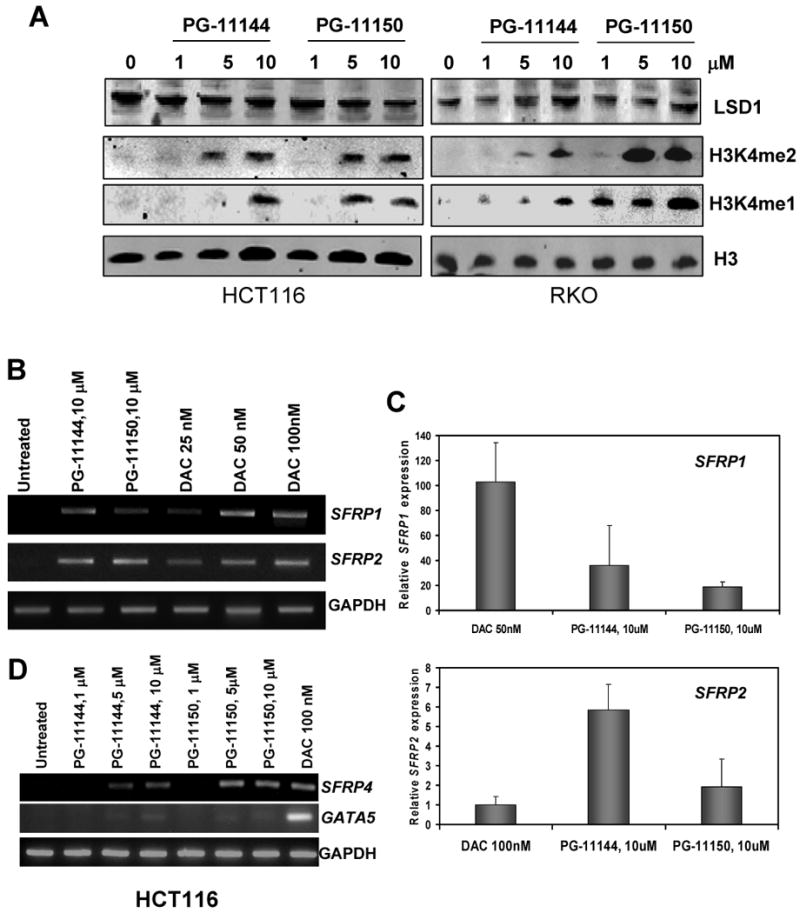
Inhibition of LSD1 by oligoamines increases global H3K4me2, resulting in re-expression of aberrantly silenced genes in treated human colon cancer cells. (A) HCT116 cells and RKO cells were exposed to increasing concentrations of PG-11144 or PG-11150 for 24 h or 48 hr, and 30 μg of nuclear protein/lane were analyzed for expression of LSD1, H3K4me1, H3K4me2, and H3 as a loading control. (B) HCT116 cells were treated for 24 h with the indicated concentrations of oligoamines and DAC. Total RNA was extracted for RT-PCR analysis of SFRP1 and SFRP2 expression. GAPDH is included as an internal control. (C) The levels of re-expression of SFRP1 and SFRP2 induced by oligoamines in HCT116 cells were compared to levels resulting from DAC treatment using SYBR Green/qPCR with GAPDH as an internal control. (D) HCT116 cells were treated for 24 h with the indicated concentrations of oligoamines or DAC. Total RNA was extracted for RT-PCR analysis of SFRP4 and GATA5 expression. GAPDH is included as an internal control.
Oligoamines reactivate the expression of multiple aberrantly silenced genes
In colorectal cancer cells promoter region H3K4me2 is usually associated with open chromatin and active transcription (7, 8). The occupancy of H3K4me2 is typically found to be at a low level in the promoters of some frequently DNA hypermethylated and epigenetically silenced genes important in tumorigenesis (1, 8). Multiple such silenced genes are present in HCT116 cells as well as in primary human colon carcinomas. These genes include members of the Wnt signaling pathway antagonists, the secreted frizzled-related protein family (SFRPs), and the GATA family of transcription factors (1, 23-25). Therefore, we examined whether such genes could be re-expressed following the exposure of colorectal cancer cells to oligoamine treatment. In HCT116 cells, treatment with 10 μM PG-11144 or PG-11150 for 24h led to substantial re-expression of aberrantly silenced SFRP1 and SFRP2 genes (Fig. 3B). The gene re-expression achieved with oligoamine treatment was compared with that achieved with the DNMT inhibitor 5-aza-2′-deoxycitidine (DAC), as determined by quantitative real-time PCR. Treatment of cells with oligoamines resulted in robust re-expression of SFRP2 (≈150-550% that achieved by 100 nM DAC treatment) and modest re-expression of SFRP1 (≈20-40% that achieved by 50 nM DAC treatment) (Fig. 3C). Oligoamine treatment also led to the detectable re-expression of the SFRP4 gene and modest re-expression of the GATA5 gene in HCT116 cells (Fig. 3D). These results demonstrate that oligoamines are effective at producing significant re-expression of multiple epigenetically silenced genes in human colorectal cancer cells.
Oligoamine treatment increases activating H3K4 methylation and decreases repressive H3K9 methylation marks at the promoters of re-expressed genes
To investigate whether oligoamine-induced gene re-expression was accompanied by changes in regulatory chromatin marks at specific gene promoters, quantitative chromatin immunoprecipitation (ChIP) analysis was performed on HCT116 cells exposed to 10 μM PG-11144 for 24h. Results revealed that treatment-induced gene re-expression was accompanied by increased enrichment of H3K4me1 and H3K4me2 at the promoters of both SFRP1 (upstream proximal region to transcription start site) and SFRP2 genes (spanning transcription start site, Fig. 4A). Consistent with the results observed above for global methylation, no change in promoter H3K4me3 levels was detected, implying that oligoamines do not affect the activity of the JmjC domain-containing histone demethylases that act on H3K4me3. The increased H3K4 methylation was accompanied by decreases in the repressive mark H3K9me2 in both genes and a modest increase in the active mark acetyl-H3K9 in SFRP2. The level of LSD1 occupancy remains unchanged after PG-11144 treatment, suggesting that the oligoamine actually inhibits LSD1 activity without decreasing its presence in the promoter of specific genes. Although increases in H3K4 methylation were consistently observed after exposure to the oligoamines, changes in the activating histone acetylation marks, H3K9 and H4K16 were more variable. Treatment with PG-11144 led to a decrease of both acetyl-H3K9 and acetyl-H4K16 at the promoter of SFRP1, whereas the level of acetyl-H3K9 was slightly increased by PG-11144 and acetyl-H4K16 remained unchanged at the promoter of SFRP2.
Figure 4.

Inhibition of LSD1 by oligoamines increases activating H3K4me2 and H3K4me1 marks and decreases repressive H3K9me2 marks at the promoters of re-expressed genes. (A) HCT116 cells were treated with 10 μM of PG-11144 for 24 hr. Quantitative chromatin immunoprecipitation analysis was used to determine the occupancy of the indicated promoters by multiple activating and repressive marks. (B) Composite graph showing enrichment of H3K4me2 in the proximal promoter region of SFRP2, spanning ∼-1000 bp to +300 bp relative to the transcriptional start site (TSS). The quantified results are the means of three independent experiments with S.D. as indicated.
To more precisely determine the effect of oligoamines on the promoter histone marks, we extended the ChIP analyses of the SFRP2 gene to cover the proximal promoter region from ∼ -1000 bp to +300 bp relative to the transcriptional start site (TSS). SFRP2 was selected since it is the gene that shows the most significant induction by the oligoamines in HCT116 cells. Oligoamine treatment led to significant increase of H3K4me2 enrichment at promoter regions B, C and E (TSS site), and modest increase of H3K4me2 enrichment at region (F) downstream of the TSS site (Fig. 4B). This analysis more precisely defines the effects of oligoamines on the histone marks in the critical proximal promoter regions that are likely responsible for the oligoamine-induced re-expression of SFRP2.
Combination treatment with oligoamines and a DNMT inhibitor results in greater re-expression of specific aberrantly silenced genes than when either agent is used alone
Promoter CpG island DNA hypermethylation typically collaborates with specific histone marks in the transcriptional silencing of specific genes in cancer cells. Previous studies have demonstrated synergy between DNA methylation inhibition and histone deacetylase inhibition in the re-expression of silenced genes in colorectal cancer cells (26). To determine whether the combination of LSD1-inhibiting oligoamines with DNMT inhibitors can lead to enhanced expression of epigenetically silenced genes in cancer cells, we investigated the expression status of the SFRP1 and SFRP2 genes in HCT116 cells following a 24h treatment with low doses of oligoamines and the DNMT inhibitor, DAC, alone and in combination. The combination resulted in a striking synergistic increase in expression of SFRP2 (Fig. 5), but not SFRP1 (data not shown). This selectivity suggests that similar to other agents targeting epigenetic regulation, the genes that are re-expressed may be tumor type- and agent-specific (27). However, a broader-based analysis of genome wide effects of the combination treatments will be necessary to understand the underlying mechanism resulting in selective re-expression of genes.
Figure 5.

Synergy of oligoamines and DNA methyltransferase inhibitor in the re-expression of an aberrantly silenced gene. HCT116 cells were treated for 24 hr with the indicated doses of DAC and oligoamines alone or simultaneously. RNA was extracted and cDNA was synthesized and subjected to quantitative real time PCR for the SFRP2 gene using the DNA-intercalating SYBR green reagent. GAPDH expression was used as an internal standard. The quantified results are the mean of three independent experiments performed in triplicate in the MyiQ single color real-time PCR machine (Bio-Rad). S.D. is indicated by the error bars.
To determine if the synergistic response of SFRP2 expression to the combination treatment with PG-11144 and DAC was a result of a greater reduction of promoter CpG methylation, bisulfite sequencing was performed on cells treated with either PG-11144 or DAC alone or in combination. The results indicate that PG-11144 treatment has no effect on CpG methylation and that the combination treatment did not result in greater demethylation of the SFRP2 promoter than treatment with DAC alone (Fig. S3).
PG-11144 combined with a DNMT inhibitor increases H3K4 methylation and profoundly inhibits growth of established tumors in vivo
The preceding in vitro results demonstrating apparent synergy between the LSD1-inhibiting polyamine analogues and a DNMT inhibitor for gene re-expression raised the important question of whether such results might also translate to therapeutic efficacy. As previously stated, the concept of combination epigenetic therapies is an emerging theme for ongoing clinical trials. Therefore we tested the in vivo therapeutic effect of polyamine analogues as LSD1 inhibitors alone, or in combination with the DNMT inhibitor 5-Aza, using human colorectal cancer cell HCT116 xenografts in athymic nude mice. As a comparison, the in vivo effect of PG-11144 was compared to the effects of a previously identified potent LSD1 inhibitor, the biguanide polyamine analogue 2d (10). Increasing doses of either PG-11144 or 2d alone, or in combination with the DNMT inhibitor, were administered i.p. as described in Materials and Methods. Western blot analyses demonstrate that both PG-11144 and 2d increase H3K4me2 in treated HCT116 tumors (Fig. 6A). Treatment with either PG-11144 or 5-Aza alone each generally displayed significant anti-tumor effects against growth of HCT116 xenografts, while treatment with 2d alone did not do so. However, for both polyamine analogues a marked increase in the inhibition of tumor growth was noted when combined with the DNMT inhibitor (Fig. 6B). Importantly, for future therapeutic translation of these data, there was no significant overall toxicity seen in the treatment groups as indicated by animal weight. These results indicate that the polyamine analogues effectively inhibit LSD1 and exhibit significantly increased growth inhibition when used in combination with a DNMT inhibitor.
Figure 6.
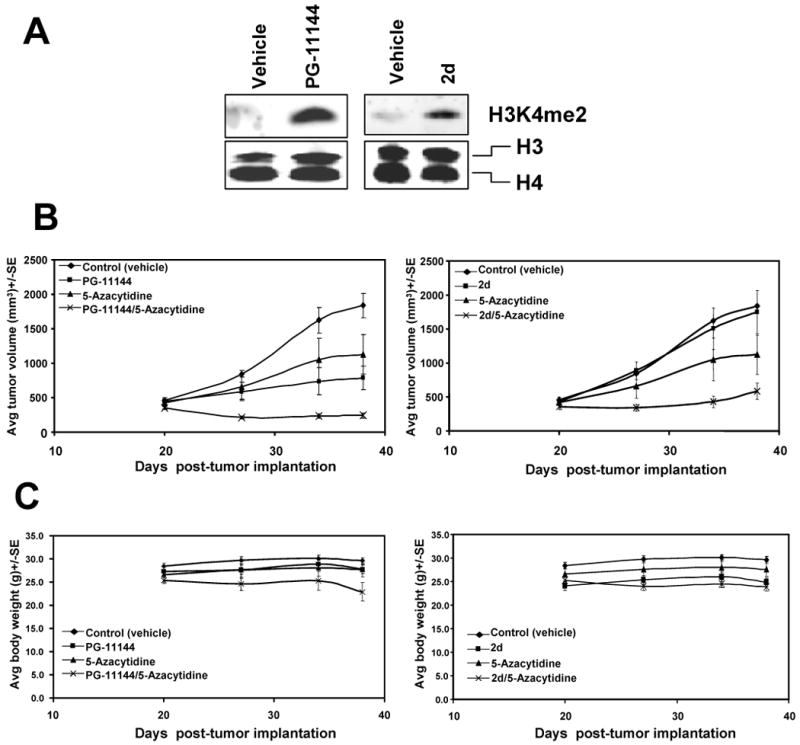
Effects of polyamine analogues on LSD1 and tumor growth in nude mice bearing HCT116 xenografts. Mice were randomized into treatment (n = 5) and control (n = 5) groups by 20 days post-implantation of HCT116 xenograft. The treatment groups given as single agents were: vehicle (saline control at qdx5), PG-11144 (10 mg/kg, 2× wk), 2d (10 mg/kg qdx5), and 5-Aza (2 mg/kg qdx5). The combination treatment groups were: PG-11144 (10 mg/kg, 2× wk)/5-Aza (2 mg/kg qdx5), and 2d (10 mg/kg qdx5)/5-Aza (2 mg/kg qdx5). (A) Tumor sample of HCT116 xenografts treated with vehicle (saline control at qdx5), PG-11144 (10 mg/kg, 2× wk), or 2d (10 mg/kg qdx5) for 37 days. 30 μg of nuclear protein/lane were analyzed for expression of H3K4me2, and H3/H4 as a loading control. (B) Tumor volumes of mice measured twice weekly. The vertical bars indicate mean tumor size (in mm3) ± SE. (C) Weights of mice were measured weekly. The vertical bars indicate mean mouse weight (g) ± SE.
Discussion
In this study we merged the rapidly increasing interest in epigenetic abnormalities in cancer, and the possibilities for reversing these changes as a cancer therapy target, with the latest discoveries of enzymes that reverse key histone lysine modifications. The discovery of the amine oxidase homolog LSD1 as the first of these enzymes, created an exciting new epigenetic target for which there has been an increasing effort to identify and design effective inhibitors (9-12). Our initial strategy for identifying effective inhibitors of LSD1 (3, 10, 14, 15, 28) was based on the structural similarity between LSD1 and the FAD-dependent SMO. This strategy proved to be effective and resulted in the successful identification of biguanide and bisguanidine polyamine analogues as LSD1 inhibitors (10). In the present study we explored the oligoamines, another class of polyamine analogues that inhibit polyamine oxidases and that have high affinity for DNA based on the increased number of positively charged nitrogens as compared to the biguanide and bisguanidine analogues (10, 15, 16). The oligoamines have demonstrated efficacy in inhibiting growth and inducing apoptosis in multiple types of cancer cells in vitro and in vivo (29, 30). In human prostate carcinoma cells, oligoamines are efficacious in the 10-100 nM dose range and are markedly more cytotoxic against tumor cells in culture than any previously described polyamine analogue (16). In human breast cancer cell lines, oligoamines can effectively inhibit growth in culture and in nude mouse xenografts without inducing significant weight loss (17).
We now show, in context with the above, that the oligoamine analogues are competitive inhibitors of recombinant LSD1, suggesting that the oligoamines may directly compete with the substrate at the active site. Although the precise mechanism of LSD1 inhibition by oligoamines remains unclear, our findings suggest that an understanding of the structural and functional similarities between amine oxidases can assist in identifying mechanism-based molecules that can specifically target LSD1. It should also be noted that it appears that the inhibitory activity of the oligoamines increases somewhat proportionally to increased charge and chain length. More rigorous structure/activity relationship studies will be required to determine whether or not this observation holds true over a wide range of analogues.
Abnormal epigenetic silencing of tumor suppressor and growth regulatory genes due to promoter CpG hypermethylation is usually associated with the initiation and progression of multiple human cancers (1). CpG island hypermethylation frequently acts in concert with abnormal histone mark activities in silencing these genes. The active histone mark H3K4me2 is associated with open chromatin and increases track uniformly with increasing levels of active gene transcription (7, 8, 23, 31), Importantly, occupancy of H3K4me2 is globally found at low levels in the promoters of DNA hypermethylated and epigenetically silenced genes important in tumorigenesis (8, 32). Critically, such depressed levels of this histone mark have been found to provide a suitable milieu for recruitment of DNMT's to nucleosomes (33-35). Studies of the important role that these epigenetic changes play in tumorigenesis, and of how DNA methylation and the H3K4me2 mark may be tightly related, thus fuel active efforts to develop anticancer drugs that can target these epigenetic mechanisms and restore expression of important growth regulatory genes. The data presented here demonstrate that treatment of colorectal cancer cells with oligoamines indeed increases both global and gene promoter levels of H3K4me2 and H3K4me1. This increase in histone methylation is accompanied by a substantial re-expression of multiple important genes that are aberrantly silenced in tumorigenesis including the Wnt signaling pathway antagonist family members, secreted frizzled-related proteins, SFRP1, SFRP2 and SFRP4, and the GATA family transcription factor, GATA5. Importantly, ChIP analysis not only confirms that LSD1 is present at the promoter of each gene examined, but that treatment of cells with oligoamines has no obvious effect on this promoter occupancy. This latter finding suggests that the effects seen for the oligoamines result from inhibition of LSD1 enzymatic activity rather than displacement of the protein from its target locations. Another key finding is that levels of another histone modification associated with active genes, H3K4me3, remain unchanged with oligoamine treatment, suggesting that these compounds are selective for LSD1 inhibition and do not affect the JmjC domain demethylases that target H3K4me3 (36-38). Interestingly, the increased H3K4me2 that results from oligoamine treatment was accompanied by a decrease in the repressive mark H3K9me2 in both genes examined. These results are similar to the down-regulation of H3K9me2 that is observed with gene re-activation in colon cancer cells treated with DNMT inhibitors or biguanide and bisguanidine polyamine analogues, as well as in colon cancer cells in which two key DNMT's have been genetically disrupted (8, 10, 23, 39). This suggests that reduced levels of H3K9me2 might be a common mechanism contributing to re-expressing genes silenced in colorectal cancer cells treated with various epigenetic reagents. However, it should be noted that in contrast to results with the biguanide and bisguanidine LSD1 inhibitors, no consistent increase in H3K9 acetylation was observed after oligoamine treatment (10) demonstrating that oligoamine-induced expression of previously silenced genes occurs in the absence of substantial increases in this activating mark. These differences suggest the classes of LSD1 inhibitors may have varied off-target effects that contribute to reactivation of aberrantly silenced genes, including their ability to alter chromatin structure based on their affinity for DNA (16, 40, 41).
It is notable that the local pattern of H3K4me2 observed in the promoter of the SFRP2 gene after treatment with PG-11144 mirrors what has been reported for other silenced genes whose expression has been induced by DAC, suggesting this local pattern is a common occurrence when previously silenced genes are re-expressed (8). Additionally, this pattern of a biphasic H3K4me2 peak proximal to the transcriptional start site is commonly seen in actively transcribed genes and is likely the result of a combination of nucleosomal positioning and the positioning of transcriptional repressing or activating complexes (42-44)
The combination of DNMT and HDAC inhibitors has demonstrated synergistic effect in re-expressing epigenetically silenced genes in cultured cancer cells and resulted in clinical response in patients with leukemias (21, 26). Therefore, we hypothesized that the combination of the LSD1-inhibiting oligoamines would be more effective when combined with DNA methyltransferase inhibitors in re-expression of silenced genes and in tumor growth inhibition. Here we report the first attempt to combine LSD1 inhibitors with agents that target DNA CpG island methylation. The results of these experiments demonstrate that the in vitro combination of low dose oligoamine and DNMT inhibitor also results in robust re-expression of the SFRP2 gene. In vivo, treatment with either PG-11144 or the previously identified LSD1 inhibitor, 2d (10), produced increases in tumor H3K4me2 levels. Treatment with PG-11144 alone also significantly inhibited the growth of HCT116 xenograft tumors in nude mice. Importantly, the most efficacious inhibition of tumor growth was observed when either of the LSD1-inhibiting analogues was used in combination with the DNMT inhibitor 5-Aza. These results indicate that, similar to what has been observed with the HDAC inhibitors, LSD1 inhibitors may be most effective when used in combination with agents that target other epigenetic regulatory mechanisms and provide the basis for clinical trial combining agents targeting these two epigenetic regulatory pathways.
In summary, we demonstrate that a novel class of polyamine analogues, the oligoamines, inhibits LSD1 activity and results in the reactivation of epigenetically silenced genes important in tumorigenesis. The results of these studies are consistent with the hypothesis that LSD1 represents a rational and important epigenetic target for drug development. Additionally, these studies demonstrate the promise of using LSD1 inhibitors in combination with other chromatin-modifying agents as a novel approach to cancer treatment with considerable clinical potential.
Supplementary Material
Supplementary Figure S1. Effects of oligoamines on growth of human colon cancer cells. HCT116 and RKO cells were seeded at 5,000 cells per well of a 96-well plate and allowed to attach overnight. Cells were treated with increasing concentrations of oligoamines for 24 or 48 h. MTT assays were performed as described in “Materials and Methods”. Shown are means ± SD of independent experiments performed in quadruplicate.
Supplementary Figure S2. Induction of PARP cleavage by treatment with PG-11144 alone or in combination with DAC. HCT116 cells were treated for 24 h with DAC (100 nM) and PG-11144 (5μM) alone or in combination. Total protein was extracted and analyzed by Western blotting using an antibody that recognizes full length and cleaved PARP. The positions of the 116 kDa full length and 85 kDa cleaved PARP are indicated on the left. Actin was used as loading control.
Supplementary Figure S3. Effects of oligoamines and DAC treatment on promoter CpG island methylation. The methylation status of 29 CpG sites in the promoter region of SFRP2 was assessed via bisulfite sequencing. Each circle represents one CpG site that is methylated (filled circle) or unmethylated (open circle) and each horizontal line shows the methylation status of CpG sites for a single cloned allele. The number of unmethylated sites is listed to the right of each allele. Treatment with DAC (100 nM, 24 hr) alone or in combination with PG-11144 (5 μM, 24 hr) resulted in an increase in the frequency of unmethylated CpG sites from 3.0% to 52.5% and 20.4% respectively.
Acknowledgments
This work is supported by NIH grants CA 51085, CA 98454, the Susan G. Komen for the Cure Foundation KG 088923, and the Samuel Waxman Cancer Research Foundation.
Footnotes
Translational relevance: Aberrant epigenetic silencing of important tumor suppressor genes has been demonstrated in essentially all human cancers. Efforts are ongoing to target this aberrant epigenetic regulation for therapeutic benefit and clinical trials combining agents that inhibit DNA CpG methylation with agents that inhibit histone deacetylase have yielded promising results. The discovery of lysine-specific demethylase 1 (LSD1) and the dynamic nature of histone methylation suggested the potential for inhibiting this transcriptional repressor as a means of re-expressing aberrantly silenced genes. The results of the initial in vitro studies from our laboratory and others were consistent with the hypothesis that LSD1 was a rational target for inducing the re-expression of aberrantly silenced genes. Here we present critical in vitro and in vivo data firmly establishing LSD1 as a target for therapy, and that demonstrate inhibitors of this enzyme have considerable promise, both when used alone and in combination with other agents that target epigenetic regulation.
Disclosure of Potential Conflicts of Interest: Dr. Marton is the Chief Scientific Officer of Progen Pharmaceuticals. Drs. Casero, Baylin, and Woster serve as scientific advisors to Progen Pharmaceuticals. Drs. Casero, Woster, Marton, and Baylin are inventors on patents/patent applications related to this article.
References
- 1.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–16. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 2.Jenuwein T, Allis C. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 3.Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Lee M, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–5. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 5.Tsukada Y, Fang J, Erdjument-Bromage H, Warren M, Borchers C, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–6. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8:829–33. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 7.Liang G, Lin J, Wei V, et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A. 2004;101:7357–62. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGarvey KM, Van Neste L, Cope L, et al. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res. 2008;68:5753–9. doi: 10.1158/0008-5472.CAN-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulte JH, Lim S, Schramm A, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–71. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Greene E, Murray-Stewart T, Goodwin A, Baylin S, Woster P, Casero R., Jr Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A. 2007;104:8023–8. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee M, Wynder C, Schmidt D, McCafferty D, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–7. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Culhane J, Szewczuk L, Liu X, Da G, Marmorstein R, Cole P. A mechanism-based inactivator for histone demethylase LSD1. J Am Chem Soc. 2006;128:4536–7. doi: 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Devereux W, Woster P, Stewart T, Hacker A, Casero R., Jr Cloning and characterization of a human polyamine oxidase that is inducible by polyamine analogue exposure. Cancer Res. 2001;61:5370–3. [PubMed] [Google Scholar]
- 14.Wang Y, Hacker A, Murray-Stewart T, et al. Properties of recombinant human N1-acetylpolyamine oxidase (hPAO): potential role in determining drug sensitivity. Cancer Chemother Pharmacol. 2005;56:83–90. doi: 10.1007/s00280-004-0936-5. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Murray-Stewart T, Devereux W, Hacker A, Frydman B, Woster P, Casero R., Jr Properties of purified recombinant human polyamine oxidase, PAOh1/SMO. Biochem Biophys Res Commun. 2003;304:605–11. doi: 10.1016/s0006-291x(03)00636-3. [DOI] [PubMed] [Google Scholar]
- 16.Valasinas A, Reddy VK, Blokhin AV, et al. Long-chain polyamines (oligoamines) exhibit strong cytotoxicities against human prostate cancer cells. Bioorg Med Chem. 2003;11:4121–31. doi: 10.1016/s0968-0896(03)00453-x. [DOI] [PubMed] [Google Scholar]
- 17.Huang Y, Hager E, Phillips D, et al. A novel polyamine analog inhibits growth and induces apoptosis in human breast cancer cells. Clin Cancer Res. 2003;9:2769–77. [PMC free article] [PubMed] [Google Scholar]
- 18.Carruthers LM, Marton LJ, Peterson CL. Polyamine analogues: potent inducers of nucleosomal array oligomerization and inhibitors of yeast cell growth. Biochem J. 2007;405:541–5. doi: 10.1042/BJ20061347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ha HC, Woster PM, Casero RA., Jr Unsymmetrically substituted polyamine analogue induces caspase-independent programmed cell death in Bcl-2-overexpressing cells. Cancer Res. 1998;58:2711–4. [PubMed] [Google Scholar]
- 20.Angstreich GR, Matsui W, Huff CA, et al. Effects of imatinib and interferon on primitive chronic myeloid leukaemia progenitors. Br J Haematol. 2005;130:373–81. doi: 10.1111/j.1365-2141.2005.05606.x. [DOI] [PubMed] [Google Scholar]
- 21.Gore SD, Baylin S, Sugar E, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–9. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 22.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGarvey K, Fahrner J, Greene E, Martens J, Jenuwein T, Baylin S. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006;66:3541–9. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki H, Gabrielson E, Chen W, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31:141–9. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- 25.Akiyama Y, Watkins N, Suzuki H, et al. GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol Cell Biol. 2003;23:8429–39. doi: 10.1128/MCB.23.23.8429-8439.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cameron E, Bachman K, Myohanen S, Herman J, Baylin S. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–7. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 27.Schuebel KE, Chen W, Cope L, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3:1709–23. doi: 10.1371/journal.pgen.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005;579:2203–7. doi: 10.1016/j.febslet.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 29.Reddy V, Valasinas A, Sarkar A, Basu H, Marton L, Frydman B. Conformationally restricted analogues of 1N,12N-bisethylspermine: synthesis and growth inhibitory effects on human tumor cell lines. J Med Chem. 1998;41:4723–32. doi: 10.1021/jm980172v. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y, Pledgie A, Casero RA, Jr, Davidson NE. Molecular mechanisms of polyamine analogs in cancer cells. Anticancer Drugs. 2005;16:229–41. doi: 10.1097/00001813-200503000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–7. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 32.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ooi SK, Qiu C, Bernstein E, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terranova R, Agherbi H, Boned A, Meresse S, Djabali M. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc Natl Acad Sci U S A. 2006;103:6629–34. doi: 10.1073/pnas.0507425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–66. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 36.Shi X, Hong T, Walter KL, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–9. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scibetta AG, Santangelo S, Coleman J, et al. Functional Analysis of the Transcription Repressor PLU-1/JARID1B. Mol Cell Biol. 2007;27:7220–35. doi: 10.1128/MCB.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamane K, Tateishi K, Klose RJ, et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell. 2007;25:801–12. doi: 10.1016/j.molcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Rhee I, Bachman KE, Park BH, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–6. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 40.Basu HS, Pellarin M, Feuerstein BG, Shirahata A, Samejima K, Deen DF, Marton LJ. Interaction of a polyamine analogue, 1,19-bis-(ethylamino)-5,10,15- triazanonadecane (BE-4-4-4-4), with DNA and effect on growth, survival, and polyamine levels in seven human brain tumor cell lines. Cancer Res. 1993;53:3948–55. [PubMed] [Google Scholar]
- 41.Feuerstein BG, Williams LD, Basu HS, Marton LJ. Implications and concepts of polyamine-nucleic acid interactions. J Cell Biochem. 1991;46:37–47. doi: 10.1002/jcb.240460107. [DOI] [PubMed] [Google Scholar]
- 42.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schones DE, Cui K, Cuddapah S, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–98. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Effects of oligoamines on growth of human colon cancer cells. HCT116 and RKO cells were seeded at 5,000 cells per well of a 96-well plate and allowed to attach overnight. Cells were treated with increasing concentrations of oligoamines for 24 or 48 h. MTT assays were performed as described in “Materials and Methods”. Shown are means ± SD of independent experiments performed in quadruplicate.
Supplementary Figure S2. Induction of PARP cleavage by treatment with PG-11144 alone or in combination with DAC. HCT116 cells were treated for 24 h with DAC (100 nM) and PG-11144 (5μM) alone or in combination. Total protein was extracted and analyzed by Western blotting using an antibody that recognizes full length and cleaved PARP. The positions of the 116 kDa full length and 85 kDa cleaved PARP are indicated on the left. Actin was used as loading control.
Supplementary Figure S3. Effects of oligoamines and DAC treatment on promoter CpG island methylation. The methylation status of 29 CpG sites in the promoter region of SFRP2 was assessed via bisulfite sequencing. Each circle represents one CpG site that is methylated (filled circle) or unmethylated (open circle) and each horizontal line shows the methylation status of CpG sites for a single cloned allele. The number of unmethylated sites is listed to the right of each allele. Treatment with DAC (100 nM, 24 hr) alone or in combination with PG-11144 (5 μM, 24 hr) resulted in an increase in the frequency of unmethylated CpG sites from 3.0% to 52.5% and 20.4% respectively.