

Abstract
In this study, a putative N-acetylmuramyl-L-alanine amidase gene (bb0666) was identified in the genome of the Lyme disease spirochete Borrelia burgdorferi. This protein shares c. 30% identity with its counterparts from other bacteria. Reverse transcriptase-PCR analysis showed that bb0666 along with two other genes (bb0665 and bb0667) are cotranscribed with the motility and chemotaxis genes. This newly identified operon is termed as pami. Sequence and primer extension analyses showed that pami was regulated by a σ70-like promoter, which is designated as Pami. Transcriptional analysis using a gene encoding green fluorescence protein as a reporter demonstrated that Pami functions in both Escherichia coli and B. burgdorferi. Genetic studies showed that the Δbb0666 mutant grows in long chains of unseparated cells, whose phenotype is similar to its counterparts in E. coli. Taken together, these results demonstrate that bb0666 is a homolog of MurNac-LAAs that contributes to the cell division of B. burgdorferi.
Keywords: lyme disease, Borrelia burgdorferi, N-acetylmuramyl-L-alanine amidase
Introduction
Spirochetes are a medically significant but poorly understood group of bacteria. These organisms cause several human diseases such as syphilis, Lyme disease and leptospirosis (Charon & Goldstein, 2002). Spirochetes constitute a very diverse but monophyletic group of bacteria that have common morphological and structural attributes (Paster & Dewhirst, 2000). Compared with other bacterial species, the morphology of spirochete cells is quite unique (Li et al., 2000; Charon & Goldstein, 2002). They are either wave-like or helical, and the average size of spirochete cells is c. 10–20 μm in length and 0.1–0.3 μm in width, which is much slimmer than other bacterial species such as cocci- or bacilli-shaped bacteria. In addition, spirochetes share a unique cell structure: a protoplasmic cell cylinder surrounded by an outer membrane sheath. In the periplasm, between the peptidoglycan layer and the outer membrane sheath are periplasmic flagella (PF) that are attached at both ends of the cell (Charon & Goldstein, 2002; Limberger, 2004). Because of a paucity of genetic tools and their fastidious growth requirements, the basic biology of spirochetes is poorly understood (e.g. the molecular mechanisms of cell division and morphogenesis in spirochetes still remain unknown).
Cell division is an essential biological process for the growth and survival of bacterial cells (Margolin, 2006; Barak & Wilkinson, 2007; Lock & Harry, 2008). The molecular mechanisms involved in cell division have been most extensively studied in rod-shaped bacteria (Escherichia coli and Bacillus subtilis) (Barak & Wilkinson, 2007; Lutkenhaus, 2007). In these model organisms, cell division initiates from the formation of a Z-ring that occurs at the center of the cell. Following the formation of a Z-ring, the membrane-bound cell division proteins are recruited, resulting in the invagination of the cell wall and membrane to form a division septum. Once septum formation is complete, the peptidoglycan layer in the division septum is cleaved by specific peptidoglycan hydrolases, allowing two newborn cells to separate (Holtje & Heidrich, 2001; Vollmer et al., 2008). Bacterial peptidoglycan hydrolases form a vast and highly diverse group of enzymes that are capable of cleaving bonds in polymeric peptidoglycan. Among these enzymes, N-acetylmuramyl-L-alanine amidases (MurNac-LAAs) specifically hydrolyze the amide bond between N-acetylmuramic acid and the N-terminal L-alanine residue of the stem peptide (Holtje & Heidrich, 2001; Vollmer et al., 2008).
In E. coli, there are five amidases (AmiA, B, C, D and AmpD). Based on their sequence differences, these amidases can be divided into two groups (Bernhardt & de Boer, 2003; Vollmer et al., 2008). The first group is composed of AmiA, B and C enzymes, and the second contains AmiD and AmpD. The functions, enzymatic activity and cellular location of the first group are well studied (Heidrich et al., 2001; Bernhardt & de Boer, 2003). Genetic studies have shown that the deletion mutants in amiA and amiC grow in long chains of unseparated cells, indicating that these two amidases play an important role in cleaving the septum to release daughter cells after cell division (Heidrich et al., 2001; Bernhardt & de Boer, 2003). In contrast to E. coli, there is only one putative MurNac-LAA gene annotated in the sequenced spirochete genomes (bb0625 in Borrelia burgdorferi, TDE1714 in Treponema denticola, TP0247 in Treponema pallidum and LA3433 in Leptospira interrogans) (Fraser et al., 1997, 1998; Ren et al., 2003; Seshadri et al., 2004). The function of these genes in spirochetes remains unknown. Borrelia burgdorferi is the causative agent of Lyme disease, the most commonly reported tick-borne disease in the United States (Steere, 2001; Kurtenbach et al., 2006). In addition to being an important clinical entity, B. burgdorferi is one of the best understood spirochetes and one for which genetic tools have rapidly evolved in the past few years (Rosa et al., 2005), making it currently an ideal genetic model to investigate the role of MurNac-LAAs. In the present study, we report that an unannotated MurNac-LAA homolog (bb0666) contributes to the cell division of B. burgdorferi. This finding provided us with a starting point to further elucidate the roles of MurNac-LAAs in the cell division of B. burgdorferi and other spirochete species.
Materials and methods
Bacterial strains and growth conditions
A high-passage avirulent B. burgdorferi strain (B31A) was used as a parental strain to perform the transcriptional and genetic studies of bb0666 (Motaleb et al., 2000; Li et al., 2002; Sal et al., 2008). B31A and its derived strains were grown in Barbour–Stoenner–Kelly (BSK)-II liquid medium or on plates with or without kanamycin (300 μg mL−1) or streptomycin (80 μg mL−1) at 34 °C in 3% carbon dioxide as described before (Li et al., 2002; Sal et al., 2008). Escherichia coli strain JM109 (Promega) was used for plasmid construction, amplification and expression of the gfp gene. Escherichia coli strain M15 (Qiagen) was used for the preparation of the recombinant BB0666 protein.
PCR, reverse transcriptase (RT)-PCR and primer extension analyses
These analyses were performed as described previously (Ge et al., 1997a, b). All the primers used for PCR, RT-PCR and primer extension analyses are listed in Table 1. For PCR, either Taq (Invitrogen) or Vent (New England Biolabs) DNA polymerases were used. For RNA isolation, 100 mL of mid-exponential-phase B. burgdorferi cells were harvested and washed twice with cold phosphate-buffered saline (pH 8.0). Total RNA was isolated with Tris Reagent (Sigma-Aldrich) according to the manufacturer's instructions, followed by a treatment with RNAse-free DNAse I (Promega). Further purification of RNA samples was performed using a Qiagen RNA isolation kit. The AMV Reverse Transcriptase Primer Extension System (Promega) was used to generate cDNA for RT-PCR or for primer extension analysis.
Table 1.
Oligonucelotide primers used in this study
| Primer | Description | Sequences |
|---|---|---|
| P1 | bb0666 (F), inactivation | 5′-TACAGCTTGGAGGTTTTGACG-3′ |
| P2 | bb0666 (R), inactivation | 5′-TGCAAATTCCGACCGTTC-3′ |
| P3 | kan (F), inactivation | 5′-AAGCTTTAATACCCGAGCTTCAAG-3′ |
| P4 | kan (R), inactivation | 5′-CAATTGTCAAGTCAGCGTAATGCT-3′ |
| P5 | bb0666 (F), expression | 5′-GGATCCATTTTGTTTTCATATTTAAGC-3′ |
| P6 | bb0666 (R), expression | 5′-CTGCAGCATTAAAGCTTTAAGTATTAG-3′ |
| P7 | bb0664 (F), RT-PCR | 5′-ATAAAGTCATTTTCTGAG-3′ |
| P8 | bb0665 (R), RT-PCR | 5′-AAATTTTTGTTCCTCTGC-3′ |
| P9 | bb0665 (F), RT-PCR | 5′-TTTTTGTTGGAAATGGCG-3′ |
| P10 | bb0666 (R), RT-PCR | 5′-CTTCAAATATCCCTTATC-3′ |
| P11 | bb0666 (F), RT-PCR | 5′-AACAGCAGTATGCCTGCT-3′ |
| P12 | bb0667 (R), RT-PCR | 5′-AACAAAAAGCTTACTGCC-3′ |
| P13 | bb0667 (F), RT-PCR | 5′-TAAGGTCAATCTTTTTGC-3′ |
| P14 | bb0668 (R), RT-PCR | 5′-TAGTTGAACTTGGATCTC-3′ |
| P15 | bb0665 (R), primer extension | 5′-AAAGAAGATTTTTGTG-3′ |
| P16 | bb0666 (F), complementation | 5′-CATATGCCATTGAGCTTTGGGAAAATG-3′ |
| p17 | bb0666 (R), complementation | 5′-CTGCAGAGCACCCTAATCATATGC TC-3′ |
| P18 | flgBp (F), complementation | 5′-GGATCCTAATACCCGAGCTTCAAG-3′ |
| P19 | flgBp (R), complementation | 5′-CATATGACCTCCCTCATTTAAAATTGC-3′ |
| P20 | gfp (F), expressing GFP | 5′-GGATCCAAGAAGGAGATATACATATG-3′ |
| P21 | gfp (R), expressing GFP | 5′-CTGCAGTTTGTATAGTTCATCCATGCC-3′ |
| P22 | flaAp (F), flaA promoter | 5′-CTGCAGTGCGCTTTAACTATCCTG-3′ |
| P23 | flaAp (R), flaA promoter | 5′-GGATCCCATGTAAACCAACTCCTT-3′ |
| P24 | pami (F), pami promoter | 5′-CTGCAGAAAGTCATTTTCTGAGTC-3′ |
| P25 | pami (R), pami promoter | 5′-GGATCCAAATTACTTTTAAAATCC-3′ |
| P26 | flgBp (F), flgB promoter | 5′-CTGCAGTAATACCCGAGCTTCAAG-3′ |
| P27 | flgBp (R), flgB promoter | 5′-GGATCCACCTCCCTCATTTAAAATTGC-3′ |
| P28 | flaBp (F), flaB promoter | 5′-CTGCAGTGTCTGTCGCCTCTTGTGG-3′ |
| P29 | flaBp (R), flaB promoter | 5′-GGATCCATATCATCCCTCCATGAT-3′ |
| P30 | aadA (F), pKFSS1 | 5′-TATCAGAGGTAGTTGGCGTC-3′ |
| P31 | aadA (R), pKFSS1 | 5′-TGTCTAGCTTCAAGTATGACG-3′ |
F, forward; R, reverse. The underlined sequences are engineered restriction enzymes for cloning.
Primer extension analysis was performed as before with slight modification (Ge et al., 1997a, b). Briefly, 20 pmol of the primer P15 (Table 1) was labeled with 32P-ATP at 37 °C for 30 min and purified with a Qiagen Nucleotide Removal Kit. For reverse transcription, 1 μL of labeled primer was mixed with 20 μg RNA and incubated at 58 °C for 20 min. AMV reverse transcriptase and other reagents were then added, and the extension reaction was carried out at 42 °C for 45 min. The resultant cDNA products were precipitated with ethanol and dissolved in 5 μL standard DNA loading buffer. The obtained samples, along with an 35S-labeled DNA ladder, which was generated from sequencing the B. burgdorferi flaA gene, were separated on a 6% polyacrylamide–8M urea denaturing gel. The gels were dried and analyzed on a PhosphorImager system (Storm 860, Molecular Dynamics). The identified promoter consensus was designated Pami.
Fluorescent reporter constructs
To test whether Pami was functional, a mutant gfp gene was used as a transcriptional reporter (Eggers et al., 2002). In addition, previously identified flaA, flaB and flgB promoters (PflaA, PflaB and PflgB, respectively) were used as controls to evaluate the strength of Pami (Ge & Charon, 1997a; Ge et al., 1997a, b). To fuse Pami to gfp, the fragment containing Pami was PCR amplified using primers P24/P25 with engineered PstI and BamHI restriction cut sites at the 5′ and 3′ ends, respectively. The obtained PCR product was then cloned into the pGEM-T Easy vector (Promega). Meanwhile, the full-length gfp gene was PCR amplified with engineered BamHI and PstI restriction at 5′ and 3′ ends, respectively. The gfp gene was fused to Pami at the BamHI cut site by restriction digestion and religation. The fused Pami-gfp fragment was digested with PstI and further cloned into the shuttle vector pKFSS1 (Frank et al., 2003), creating the vector Pami-gfp/pKFSS1. The same method was used to fuse other promoters to gfp, and created vectors PflaA-gfp/pKFSS1, PflaB-gfp/pKFSS1 and PflgB-gfp/pKFSS1. These vectors were, respectively, transformed into either E. coli strain JM109 or B. burgdorferi strain B31A to monitor the expression level of green fluorescence protein (GFP). The expression of GFP was visualized with fluorescent microscopy (Axiostar Plus, Ziess), and the levels of GFP in transformed strains were evaluated by either Western blot using a monoclonal GFP antibody (Invitrogen) or a fluorometer model LPS-220B at excitation wavelength 494 nm (Photon Technology International Inc.).
Preparation of recombinant BB0666 and antiserum against BB0666
The full-length bb0666 gene was PCR amplified using primers P5/P6 with engineered BamHI and PstI cut sites at the 5′ and 3′ ends, respectively. The obtained PCR product was then cloned into the BamHI–PstI-restricted pQE30 vector (Qiagen), which expresses a six-histidine tag at the N-terminus of the recombinant protein. The overexpression of bb0666 was induced with 1 mM isopropyl-β-D-thiogalactoside. His-tagged BB0666 was purified with nickel agarose columns and concentrated in 10-kDa molecular weight cut-off Amicon ultracentrifugal concentrators (Millipore). To raise the antiserum against BB0666, rabbits were immunized with 400μg of purified recombinant protein over 1 month. Polyclonal antiserum was further purified with a Protein A IgG Purification Kit (Pierce).
Construction of the bb0666 deletion mutant and complementation of the mutant
Similar methods described before were used to inactivate the bb0666 gene (Motaleb et al., 2000; Li et al., 2002; Sal et al., 2008). As described in Fig. 1, the DNA fragment containing bb0666 was PCR amplified and the obtained PCR product was cloned into the pGEM-T Easy vector, yielding pbb0666-Easy. Meanwhile, a kanamycin-resistance marker (kan) described before (Bono et al., 2000) was PCR amplified to engineer HindIII (5′) and MunI (3′) restriction sites. A 96-bp HindIII–MunI fragment within bb0666 was deleted and replaced with kan, creating the vector pbb0666∷kan. This plasmid was linearized and transformed into B31A strain by electroporation as described before (Samuels, 1995). Transformants were selected on soft BSK-II agar containing 300 μg mL−1 kanamycin. The targeted mutagenesis was confirmed by PCR and the loss of the cognate gene product was detected by Western blot as described before (Motaleb et al., 2000; Li et al., 2002; Sal et al., 2008). The obtained mutant was designated as Δbb0666.
Fig. 1.

Constructing plasmids for the inactivation of bb0666 and the complementation of ΔBBB066. (a) The plasmid pbb0666∷kan was used to inactivate bb0666. (b) The plasmid FlgB666/pKFSS1 was used to complement Δbb0666. Arrows indicate the relative positions of the primers used for constructing these plasmids. The sequences of these primers are listed in Table 1. The symbol `Δ'shows that a 96-bp HindIII/MunI fragment within bb0666 was deleted and replaced with the kan cassette.
To complement Δbb0666, the entire length of bb0666 was PCR amplified, and the resultant product was fused to PflgB at an NdeI site and the resultant PflgB-bb0666 fragment was further cloned into the shuttle vector pKFSS1 at BamHI and PstI sites, creating the complemented vector, FlgB666/pKFSS1 (Fig. 1b). To complement Δbb0666, the vector FlgB666/pKFSS1 was transformed into the mutant cells by electroporation. The selection and characterization of the complemented mutant was performed as described previously. The obtained mutant was referred to as Δbb0666+.
Gel electrophoresis and Western blot analysis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with an enhanced chemiluminescent detection method (ECL-Amersham Pharmacia) were carried out as reported previously (Motaleb et al., 2004; Sal et al., 2008). The concentration of cellular proteins in lysates was determined by a Bio-Rad protein assay kit. Ten micrograms of whole cell lysates were run by SDS-PAGE and were subjected to Western blot using specific antibodies. For quantitative Western blot, the B. burgdorferi flagella filament protein, FlaB, and E. coli flagella motor switch protein, FliG, were used as internal controls. Monoclonal antibody against FlaB (H9724) and polyclonal antibody against FliG were kindly provided by A.G. Barbour (University of California, Irvine) and R. Macnab (Yale University), respectively. The monoclonal antibody against GFP was purchased from Invitrogen. The amounts of immunoreactive proteins in the gels were determined using FluorChem spot densitometry as before (Motaleb et al., 2004; Sal et al., 2008).
Light and electron microscopy
The mutant and complemented strains were visually examined under dark-field microscope for altered cell morphology. The images were captured and processed with the software AXIOVISION AC 4.1 (Zeiss). The membrane-disrupted cells were prepared and visualized under an electron microscope as described before (Motaleb et al., 2000; Li et al., 2002; Sal et al., 2008).
Results
BB0666 is a homolog of MurNac-LAAs
The genes encoding MurNac-LAAs are widely distributed among bacteria, and many bacterial species contain multiple MurNac-LAAs (Heidrich et al., 2002; Kajimura et al., 2005; Vollmer et al., 2008). For instance, E. coli has five MurNac-LAAs (Heidrich et al., 2001; Bernhardt & de Boer, 2003). In contrast, only one MurNac-LAA homolog (BB0625) was annotated in the genome of B. burgdorferi (Fraser et al., 1997). To detect whether there are other potential MurNac-LAA homologs, the E. coli MurNac-LAAs were used as queries to search the genome of B. burgdorferi. From these searches, BB0666 was found to have significant similarity to AmiA, B and C (e-values < 10−7). In contrast, a previously annotated putative MurNac-LAA (BB0625) showed less similarity to the E. coli MurNac-LAAs (e-values > 0.05), indicating that BB0666 is likely a homolog of MurNac-LAA, but BB0625 is not.
BB0666 consists of 342 amino acids and its predicted molecular weight is c. 40 kDa. To further confirm whether this protein belongs to the family of MurNac-LAAs, BB0666 was used to query the database of Pfam (http://pfam.sanger.ac.uk), and the results show that BB0666 belongs to the family of amidase_3 (PF01520) (e-values < 10−40). The C-terminus (118–335 amino acids) of BB0666 contains a well-conserved catalytic domain that is present in MurNac-LAAs from a variety of different bacteria (Fig. 2) (Shida et al., 2001; Mishima et al., 2005). Sequence alignment analyses further revealed that some residues within the catalytic domains are well conserved among different bacterial species. Genetic and biochemical studies of CwlC, a MurNac-LAA of B. subtilis, indicate that some of those conserved residues are essential for the enzymatic activity of CwlC. For example, the substitution of residues E24, D55, H79 and E141 completely abolished the activity of CwlC (Shida et al., 2001). Consistently, those four residues are well conserved in BB0666 and other MurNac-LAAs (Fig. 2). Taken together, these results have suggested that BB0666 is most likely a MurNac-LAA that is involved in the cleavage of peptidoglycan during the cell division of B. burgdorferi.
Fig. 2.

Multiple-sequence alignment of the catalytic domains of amidases. Residues with 100% identity are shaded in black. GenBank accession numbers for the aligned sequences are as follows: Bacillus subtilis CwlC (BAA03500), Borrelia burgdorferi BB0666 (NP_212800), Treponema pallidum AmiA (NP_218687), Chlamydia pneumoniae AmiA (NP_224617), Escherichia coli AmiB (NP_290799), Salmonella typhimurium AmiB (NP_463219), Neisseria meningitidis AmiC (AF194079) and Helicobacter pylori AmiA (NP_207565). The residues labeled with * and+have been studied by amino acid substitutions in CwlC. *Essential residues for the catalytic activity of CwlC (Shida et al., 2001). The alignments were performed using the program ALIGNX (Invitrogen).
The bb0666 gene is cotranscribed with the genes of the flaA operon
The two genes adjacent to bb0666 (bb0665 and bb0667; Fig. 3a) encode two conserved hypothetical proteins (Fraser et al., 1997), and these three genes are adjacent to a previously identified flaA operon (Ge & Charon, 1997a, b). There is only 71 bp of intergenic space between bb0667 and the flaA gene (bb0668) (Fraser et al., 1997). In addition, the ORFs encoding these genes reside on the same DNA strand as the genes of the flaA operon (Fig. 3a) (Fraser et al., 1997), implying that these three genes may be cotranscribed with the flaA operon. To test this hypothesis, a previously described RT-PCR analysis was performed (Ge et al., 1997a, b). As shown in Fig. 3a, four pairs of primers from adjacent genes spanning bb0664 to flaA (bb0668) region were used for the RT-PCR analysis. As expected, no product was detected between bb0664 and bb0665 genes because they are divergently transcribed. In contrast, all other primers yielded PCR products of predicted sizes (Fig. 3b). These results indicate that bb0665, bb0666 and bb0667 are cotranscribed with the genes of the flaA operon, thus constituting an eight-gene operon that is c. 9 kb long (Fraser et al., 1997). Because bb0666 is a homolog of E. coli amiA, B and C (ami), this newly identified operon is designated as pami.
Fig. 3.
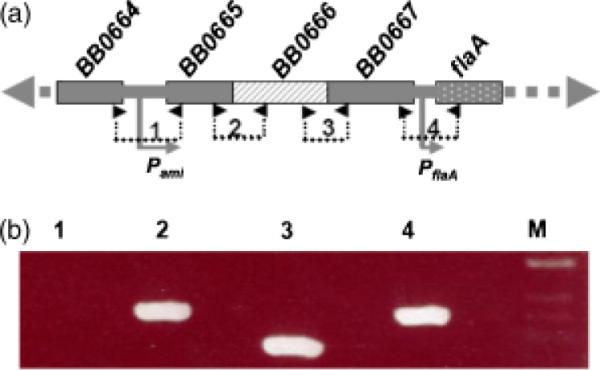
Characterization of the pami operon. (a) The diagram showing the genes upstream of the flaA operon. The numbers and small arrows show the primers used for RT-PCR analysis. The relative positions of the promoters Pami and PflaA are labeled. (b) RT-PCR analysis. The numbered RT-PCR products were correspondingly produced by the primers labeled in (a). The sequences of these primers are described in Table 1. M, 1-kb DNA ladder.
The pami operon is initiated by a σ70-like promoter
There is a 726-bp intergenic space upstream of bb0665 (Fraser et al., 1997). To understand the genetic regulation of pami, this region was searched for different promoter consensus sequences that typically exist in B. burgdorferi (e.g. σ70- and σ54-like promoters). A highly conserved σ70-like promoter was found upstream of bb0665: a −10 region showing 100% identity to the consensus sequence (TATAAT), with a less-conserved −35 region (TTGTAT) (Fig. 4b) (Ge et al., 1997a, b). To further confirm that transcription of pami is initiated from this putative promoter, a primer extension analysis was performed. As expected, the transcriptional initiation site is 7 bp downstream of the −10 region and 21 bp from the start codon of bb0665 (Fig. 4). Sequence comparison showed that this promoter is very similar to other σ70-like promoters identified in B. burgdorferi (e.g. flgB and flgK promoters: their −10 regions are 100% identical; Fig. 4b) (Ge et al., 1997a, b). Taken together, these results show that the identified promoter is a σ70-like promoter, which is referred to as Pami.
Fig. 4.

The pami operon is initiated by a σ70-like promoter, Pami. (a) Primer extension assay. The DNA ladder was generated with a DNA-sequencing reaction using Borrelia burgdorferi flaA gene as a template. The number is the size of the cDNA product generated from the primer extension assay. (b) Comparison of the Pami sequence with other σ70-like promoters identified in Escherichia coli and B. burgdorferi (Ge et al., 1997a, b). The asterisk indicates transcriptional initiation sites.
Transcriptional analysis of Pami in E. coli and B. burgdorferi
To examine whether Pami functions in E. coli and B. burgdorferi, the gfp gene was used as a transcriptional reporter as described previously (Carroll et al., 2003; Eggers et al., 2006). We found that both E. coli and B. burgdorferi cells harboring the vector Pami-gfp/pKFSS1 expressed GFP protein (Fig. 5) and were fluorescent (data not shown), indicating that this promoter is functional in both E. coli and B. burgdorferi. As shown in Fig. 4, several σ70-like promoters have been identified in B. burgdorferi, and some of these promoters (e.g. PflaB and PflgB) have been well characterized and used to initiate the expression of antibiotic resistance and gfp genes, as well as to complement mutated genes (Ge et al., 1997b; Bono et al., 2000; Eggers et al., 2002; Carroll et al., 2003). To further understand the function of Pami, the strength of this promoter was compared with other σ70-like promoters such as PflaB, PflgB and PflaA. The promoter PflaA is located immediately upstream of the flaA gene (bb0668). However, a previous study using the chloramphenicol-resistance marker (cat) as a report showed that PflaA failed to express CAT in E. coli (Ge & Charon, 1997a; Ge et al., 1997a, b). These three promoters were used as controls to evaluate the strength of the newly identified Pami promoter.
Fig. 5.

Transcriptional analysis of PflaA, Pami, PflgB and PflaB promoters using gfp as a reporter. The levels of GFP in (a) Escherichia coli and (b) Borrelia burgdorferi were detected by Western blot analysis. Approximately 10 μg of whole cell lysates were run by SDS-PAGE. The flagella motor switch complex protein, FliG, and the flagella filament protein, FlaB, were used as internal controls in E. coli and B. burgdorferi, respectively. The monoclonal antibodies against GFP and FlaB, and polyclonal antiserum against E. coli FliG, were used. (c) Quantitative Western blot analysis of GFP levels in E. coli and B. burgdorferi. Protein density was determined by FluorChem spot densitometry (Bio-Rad). Data were expressed as mean of density unit±SD of the mean from three independent experiments.
Promoter activities were evaluated by measuring the level of GFP protein expressed in the transformed cells or the fluorescent intensities of the transformed cells. As shown in Fig. 5, PflaA failed to express the gfp gene in both E. coli and B. burgdorferi, which is consistent with the previous analysis, suggesting that PflaA may be a pseudo- or conditional promoter that only functions at a certain stage (e.g. within a tick or mammalian host) during the life cycle of B. burgdorferi (Ge et al., 1997). The remaining three promoters are functional in both E. coli and B. burgdorferi (Fig. 5). Based on the levels of GFP expressed in the transformed cells, the activity of PflaB is the strongest, Pami is the weakest and PflgB is intermediate. The ratio between these three promoters (PflaB : PflgB : Pami) is c. 3 : 2 : 1 in both E. coli and B. burgdorferi (Fig. 5). A similar pattern was observed by comparing the fluorescent intensities of transformed cells (data not shown).
Inactivation of bb0666 represses the cell division of B. burgdorferi
In E. coli, the deletion mutants in amiA and amiC grow in long chains of unseparated cells (Tomioka et al., 1983; Heidrich et al., 2001; Bernhardt & de Boer, 2003). To determine whether bb0666 has a function similar to its counterparts in E. coli, the gene was inactivated as described previously (Motaleb et al., 2000; Li et al., 2002). PCR analysis demonstrated that Δbb0666 contained the kan insert as expected (data not shown), and Western blot analysis showed that the cognate gene product was absent in the mutant (Fig. 6). These results show that bb0666 is expressed in the wild type, but is abrogated in the mutant. Microscopic analysis showed that the mutant grew in chains of multiple cells (Fig. 7), and such a phenotype was most pronounced in the stationary phase (Table 2). For example, in the early log phase, <6% of the mutant cells grew in chains (≥3 cells per chain), and this percentage was increased to c. 80% in the stationary phase. In contrast, <2% of the wild-type cells grow in chains (≥3 cells per chain). Electron microscopic analysis showed that the division septum was still formed between individual daughter cells (data not shown). This phenotype suggests that although the division septum can be formed in the mutant, it cannot be separated due to the inactivation of bb0666. A similar phenotype was observed in the E. coli amiA and amiC mutants (ami-) (Tomioka et al., 1983; Heidrich et al., 2001; Bernhardt & de Boer, 2003).
Fig. 6.

Characterization of Δbb0666 and the complemented strain Δbb0666+ by Western blot analysis. Approximately 10 μg of whole cell lysates were run by SDS-PAGE, and the polyclonal antiserum against Borrelia burgdorferi BB0666 was used for Western blots. WT, wild type.
Fig. 7.

Phenotypic analysis of Δbb0666. The middle stationary phase cultures of the wild type, Δbb0666 and the complemented strain Δbb0666+ were observed by dark-field microscopy. At least 20 fields were examined for each strain.
Table 2.
Percentile of unseparated cells in the wild type and the Δbb0666 mutant
| WT |
Δbb0666 |
|||||
|---|---|---|---|---|---|---|
| 2* | 3–6 | >6 | 2 | 3–6 | >6 | |
| Early log-phase (<105) | 10–20% | <1% | NA | 30–40% | <5% | <1% |
| Middle log-phase (<107) | 10–20% | <1% | NA | 40–50% | 20–30% | <10% |
| Stationary phase (>108) | 20–30% | <2% | NA | 5–10% | 50–60% | <20% |
Number of cells per chain. The remaining percentages are attributed to single cell per chain.
WT, wild type.
Because there are several genes downstream of bb0666, the insertion of kan in the mutant may repress the expression of these genes, and the observed phenotype may be the result of a polar effect on other genes (Fraser et al., 1997; Ge & Charon, 1997b). To exclude this possibility, the mutant was complemented in trans- using the vector pBB0666/pKFSS1 (Fig. 1). The complementation successfully restored the expression of bb0666 (Fig. 6), and the process of separating daughter cells was rescued in the complemented strain Δbb0666+ (Fig. 7). Taken together, these results have shown that bb0666 contributes to the cell division of B. burgdorferi.
Discussion
Peptidoglycan hydrolyases, including MurNac-LAAs, are potentially lethal enzymes capable of disrupting bacterial cell walls. Some of these enzymes are autolysins, which are involved in programmed bacterial cell death (Holtje & Heidrich, 2001; Rice & Bayles, 2003; Vollmer et al., 2008). Thus, these enzymes must be highly regulated to prevent adventitious cell lysis. This regulation occurs at multiple levels from the transcriptional to the posttranslational level (Vollmer et al., 2008). In B. subtilis, the expression of lytC and lytD, which encode two amidases, is coregulated with flagellation and chemotaxis (Blackman et al., 1998; Serizawa et al., 2004). The expression of these two genes is regulated by σD, a homologof σ28 factors that are responsible for transcription of the genes involved in flagella assembly, motility and chemotaxis in E. coli and other bacteria (Hughes et al., 1993; Hughes & Mathee, 1998). In this report, we demonstrate that the bb0666 gene is cotranscribed with the flaA operon (Fig. 3), which includes a flagella filament gene (flaA) and four chemotaxis genes (cheA2, cheW3, cheX and cheY3).
In many flagellated bacteria, the expression of flagellin and chemotaxis genes is regulated by σ28 and anti-σ28 factors (Aldridge & Hughes, 2002; Aldridge et al., 2006). However, the regulation of flagellation and chemotaxis is quite unique in B. burgdorferi, because σ28 and anti-σ28 factors are absent in the genome of B. burgdorferi (Fraser et al., 1997). All motility and chemotaxis genes so far identified are regulated by σ70 (a homolog of σA, the major housekeeping σ factor of B. subtilis) (Li et al., 2000; Charon & Goldstein, 2002). This study shows that the newly identified pami operon is also regulated by σ70 (Fig. 3). This is consistent with our previous studies (Charon & Goldstein, 2002), but differs from the regulation of lytC and lytD in B. subtilis, which is regulated by σD (Blackman et al., 1998; Serizawa et al., 2004). These results suggest that the coregulation between cell division and flagella formation also exists in B. burgdorferi. The advantage of such coregulation may allow bacterial cells to simultaneously cleave the chain between daughters, break up into single cells and swim efficiently after the flagella formation is completed. Consistent with this assumption, the lytC and lytD mutants of B. subtilis are unable to effectively execute swarming motility and chemotaxis due to the so-called pushmi-pullyu effect in unseparated cells (Blackman et al., 1998).
In E. coli, AmiA, B and C cleave the amide bond between MurNAc and the N-terminal L-alanine residue of the stem peptide, and they are involved in the splitting of the murein septum during cell division (Heidrich et al., 2001; Bernhardt & de Boer, 2003). Inactivation of amiA or amiC partially represses cell division (<30% of mutant cells grow in chains). Inactivation of amiB does not influence cell division. However, up to 40% of the double-mutant (with each possible combination) cells grow in chains, and up to 90% of triple-mutant cells grow in chains (Heidrich et al., 2001, 2002). Because cell division is an essential biological process for the growth and survival of bacterial cells, such redundancy might be able to secure cell division. Although the cell division is significantly repressed in Δbb0666 (Table 2), the mutant is still able to divide at a very slow rate, indicating that there may be another peptidoglycan hydrolyase in B. burgdorferi.
Previous studies have shown that lytic transglycosylases (e.g. Slt70 of E. coli, and LytC and LytD of B. subtilis) are exoglucosidases that are able to degrade murein strands and contribute to bacterial cell division (Heidrich et al., 2001, 2002; Powell et al., 2006). BLAST analysis revealed that there is a lytic transglycosylase homolog (bb0625) in the genome of B. burgdorferi. This gene was previously annotated as a putative MurNac-LAA (Fraser et al., 1997). However, BLAST analysis showed that BB0625 shares little similarity with the E. coli MurNac-LAAs (e-values > 0.05). Instead, it contains multiple lysin domains (data not shown). The lysin domain is typically found in lytic transglycosylases (e.g. Slt70 of E. coli, and LytC and LytD of B. subtilis) (van Asselt et al., 1999; Powell et al., 2006). Taken together, these results suggest that BB0625 is most likely a homolog of lytic transglycosylases but not MurNac-LAAs. Thus, the remaining cell division observed in Δbb0666 could be due to the presence of this enzyme. Further genetic studies will help us to clarify the role of bb0625 in B. burgdorferi (e.g. constructing double mutations in bb0625 and bb0666).
Biochemical analyses show that the E. coli AmiA and C are able to cleave the amide bond between MurNAc and the N-terminal L-alanine residue of the stem peptide (Heidrich et al., 2001; Bernhardt & de Boer, 2003). The Δbb0666 mutant has a phenotype similar to the ami- mutants of E. coli, implying that BB0666 probably belongs to the family of MurNac-LAAs (Heidrich et al., 2001; Bernhardt & de Boer, 2003). To further confirm whether BB0666 is a MurNac-LAA, we attempted to analyze its enzymatic activity. However, the recombinant BB0666 formed inclusion bodies that could not be renatured. Thus, such biochemical analysis cannot be performed. A similar phenotype was observed in the E. coli AmiB (Heidrich et al., 2001).
Acknowledgements
We thank S. Samuels for providing the shuttle vectors, J. Radolf for providing the mutant gpf gene and W. Zhang for measuring the fluorescence of GFP. This research was supported by the United States Public Health Service AI073354, AI078958, and the American Heart Association 0735236N to C.L., and AI29743 to N.W.C.
References
- Aldridge P, Hughes KT. Regulation of flagellar assembly. Curr Opin Microbiol. 2002;5:160–165. doi: 10.1016/s1369-5274(02)00302-8. [DOI] [PubMed] [Google Scholar]
- Aldridge PD, Karlinsey JE, Aldridge C, Birchall C, Thompson D, Yagasaki J, Hughes KT. The flagellar-specific transcription factor, sigma28, is the Type III secretion chaperone for the flagellar-specific anti-sigma28 factor FlgM. Gene Dev. 2006;20:2315–2326. doi: 10.1101/gad.380406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak I, Wilkinson AJ. Division site recognition in Escherichia coli and Bacillus subtilis. FEMS Microbiol Rev. 2007;31:311–326. doi: 10.1111/j.1574-6976.2007.00067.x. [DOI] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman SA, Smith TJ, Foster SJ. The role of autolysins during vegetative growth of Bacillus subtilis 168. Microbiology. 1998;144:73–82. doi: 10.1099/00221287-144-1-73. [DOI] [PubMed] [Google Scholar]
- Bono JL, Elias AF, Kupko JJ, III, Stevenson B, Tilly K, Rosa P. Efficient targeted mutagenesis in Borrelia burgdorferi. J Bacteriol. 2000;182:2445–2452. doi: 10.1128/jb.182.9.2445-2452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, Stewart PE, Rosa P, Elias AF, Garon CF. An enhanced GFP reporter system to monitor gene expression in Borrelia burgdorferi. Microbiology. 2003;149:1819–1828. doi: 10.1099/mic.0.26165-0. [DOI] [PubMed] [Google Scholar]
- Charon NW, Goldstein SF. Genetics of motility and chemotaxis of a fascinating group of bacteria: the spirochetes. Annu Rev Genet. 2002;36:47–73. doi: 10.1146/annurev.genet.36.041602.134359. [DOI] [PubMed] [Google Scholar]
- Eggers CH, Caimano MJ, Clawson ML, Miller WG, Samuels DS, Radolf JD. Identification of loci critical for replication and compatibility of a Borrelia burgdorferi cp32 plasmid and use of a cp32-based shuttle vector for the expression of fluorescent reporters in the Lyme disease spirochaete. Mol Microbiol. 2002;43:281–295. doi: 10.1046/j.1365-2958.2002.02758.x. [DOI] [PubMed] [Google Scholar]
- Eggers CH, Caimano MJ, Radolf JD. Sigma factor selectivity in Borrelia burgdorferi: RpoS recognition of the ospE/ospF/elp promoters is dependent on the sequence of the −10 region. Mol Microbiol. 2006;59:1859–1875. doi: 10.1111/j.1365-2958.2006.05066.x. [DOI] [PubMed] [Google Scholar]
- Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. aadA confers streptomycin resistance in Borrelia burgdorferi. J Bacteriol. 2003;185:6723–6727. doi: 10.1128/JB.185.22.6723-6727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Casjens S, Huang WM, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Norris SJ, Weinstock GM, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- Ge Y, Charon NW. An unexpected flaA homolog is present and expressed in Borrelia burgdorferi. J Bacteriol. 1997a;179:552–556. doi: 10.1128/jb.179.2.552-556.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Charon NW. Molecular characterization of a flagellar/chemotaxis operon in the spirochete Borrelia burgdorferi. FEMS Microbiol Lett. 1997b;153:425–431. doi: 10.1111/j.1574-6968.1997.tb12606.x. [DOI] [PubMed] [Google Scholar]
- Ge Y, Old IG, Girons IS, Charon NW. The flgK motility operon of Borrelia burgdorferi is initiated by a sigma70-like promoter. Microbiology. 1997a;143:1681–1690. doi: 10.1099/00221287-143-5-1681. [DOI] [PubMed] [Google Scholar]
- Ge Y, Old IG, Saint GI, Charon NW. Molecular characterization of a large Borrelia burgdorferi motility operon which is initiated by a consensus sigma70 promoter. J Bacteriol. 1997b;179:2289–2299. doi: 10.1128/jb.179.7.2289-2299.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, Holtje JV. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol. 2001;41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- Heidrich C, Ursinus A, Berger J, Schwarz H, Holtje JV. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J Bacteriol. 2002;184:6093–6099. doi: 10.1128/JB.184.22.6093-6099.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtje JV, Heidrich C. Enzymology of elongation and constriction of the murein sacculus of Escherichia coli. Biochimie. 2001;83:103–108. doi: 10.1016/s0300-9084(00)01226-8. [DOI] [PubMed] [Google Scholar]
- Hughes KT, Mathee K. The anti-sigma factors. Annu Rev Microbiol. 1998;52:231–286. doi: 10.1146/annurev.micro.52.1.231. [DOI] [PubMed] [Google Scholar]
- Hughes KT, Gillen KL, Semon MJ, Karlinsey JE. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science. 1993;262:1277–1280. doi: 10.1126/science.8235660. [DOI] [PubMed] [Google Scholar]
- Kajimura J, Fujiwara T, Yamada S, Suzawa Y, Nishida T, Oyamada Y, Hayashi I, Yamagishi J, Komatsuzawa H, Sugai M. Identification and molecular characterization of an N-acetylmuramyl-L-alanine amidase Sle1 involved in cell separation of Staphylococcus aureus. Mol Microbiol. 2005;58:1087–1101. doi: 10.1111/j.1365-2958.2005.04881.x. [DOI] [PubMed] [Google Scholar]
- Kurtenbach K, Hanincova K, Tsao JI, Margos G, Fish D, Ogden NH. Fundamental processes in the evolutionary ecology of Lyme borreliosis. Nat Rev Microbiol. 2006;4:660–669. doi: 10.1038/nrmicro1475. [DOI] [PubMed] [Google Scholar]
- Li C, Motaleb A, Sal M, Goldstein SF, Charon NW. Spirochete periplasmic flagella and motility. J Mol Microb Biotech. 2000;2:345–354. [PubMed] [Google Scholar]
- Li C, Bakker RG, Motaleb MA, Sartakova ML, Cabello FC, Charon NW. Asymmetrical flagellar rotation in Borrelia burgdorferi nonchemotactic mutants. P Natl Acad Sci USA. 2002;99:6169–6174. doi: 10.1073/pnas.092010499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limberger RJ. The periplasmic flagellum of spirochetes. J Mol Microb Biotech. 2004;7:30–40. doi: 10.1159/000077867. [DOI] [PubMed] [Google Scholar]
- Lock RL, Harry EJ. Cell-division inhibitors: new insights for future antibiotics. Nat Rev Drug Discov. 2008;7:324–338. doi: 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J. Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu Rev Biochem. 2007;76:539–562. doi: 10.1146/annurev.biochem.75.103004.142652. [DOI] [PubMed] [Google Scholar]
- Margolin W. Bacterial division: another way to box in the ring. Curr Biol. 2006;16:R881–R884. doi: 10.1016/j.cub.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Mishima M, Shida T, Yabuki K, Kato K, Sekiguchi J, Kojima C. Solution structure of the peptidoglycan binding domain of Bacillus subtilis cell wall lytic enzyme CwlC: characterization of the sporulation-related repeats by NMR. Biochemistry. 2005;44:10153–10163. doi: 10.1021/bi050624n. [DOI] [PubMed] [Google Scholar]
- Motaleb MA, Corum L, Bono JL, Elias AF, Rosa P, Samuels DS, Charon NW. Borrelia burgdorferi periplasmic flagella have both skeletal and motility functions. P Natl Acad Sci USA. 2000;97:10899–10904. doi: 10.1073/pnas.200221797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motaleb MA, Sal MS, Charon NW. The decrease in FlaA observed in a flaB mutant of Borrelia burgdorferi occurs posttranscriptionally. J Bacteriol. 2004;186:3703–3711. doi: 10.1128/JB.186.12.3703-3711.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paster BJ, Dewhirst FE. Phylogenetic foundation of spirochetes. J Mol Microb Biotech. 2000;2:341–344. [PubMed] [Google Scholar]
- Powell AJ, Liu ZJ, Nicholas RA, Davies C. Crystal structures of the lytic transglycosylase MltA from N. gonorrhoeae and E. coli: insights into interdomain movements and substrate binding. J Mol Biol. 2006;359:122–136. doi: 10.1016/j.jmb.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Ren SX, Fu G, Jiang XG, et al. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature. 2003;422:888–893. doi: 10.1038/nature01597. [DOI] [PubMed] [Google Scholar]
- Rice KC, Bayles KW. Death's toolbox: examining the molecular components of bacterial programmed cell death. Mol Microbiol. 2003;50:729–738. doi: 10.1046/j.1365-2958.2003.t01-1-03720.x. [DOI] [PubMed] [Google Scholar]
- Rosa PA, Tilly K, Stewart PE. The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol. 2005;3:129–143. doi: 10.1038/nrmicro1086. [DOI] [PubMed] [Google Scholar]
- Sal MS, Li C, Motalab MA, Shibata S, Aizawa S, Charon NW. Borrelia burgdorferi uniquely regulates its motility genes and has an intricate flagellar hook-basal body structure. J Bacteriol. 2008;190:1912–1921. doi: 10.1128/JB.01421-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS. Electrotransformation of the spirochete Borrelia burgdorferi. Methods Mol Biol. 1995;47:253–259. doi: 10.1385/0-89603-310-4:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serizawa M, Yamamoto H, Yamaguchi H, Fujita Y, Kobayashi K, Ogasawara N, Sekiguchi J. Systematic analysis of SigD-regulated genes in Bacillus subtilis by DNA microarray and Northern blotting analyses. Gene. 2004;329:125–136. doi: 10.1016/j.gene.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Seshadri R, Myers GS, Tettelin H, et al. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. P Natl Acad Sci USA. 2004;101:5646–5651. doi: 10.1073/pnas.0307639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shida T, Hattori H, Ise F, Sekiguchi J. Mutational analysis of catalytic sites of the cell wall lytic N-acetylmuramoyl-L-alanine amidases CwlC and CwlV. J Biol Chem. 2001;276:28140–28146. doi: 10.1074/jbc.M103903200. [DOI] [PubMed] [Google Scholar]
- Steere AC. Lyme disease. New Engl J Med. 2001;345:115–125. doi: 10.1056/NEJM200107123450207. [DOI] [PubMed] [Google Scholar]
- Tomioka S, Nikaido T, Miyakawa T, Matsuhashi M. Mutation of the N-acetylmuramyl-L-alanine amidase gene of Escherichia coli K-12. J Bacteriol. 1983;156:463–465. doi: 10.1128/jb.156.1.463-465.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Asselt EJ, Thunnissen AM, Dijkstra BW. High resolution crystal structures of the Escherichia coli lytic transglycosylase Slt70 and its complex with a peptidoglycan fragment. J Mol Biol. 1999;291:877–898. doi: 10.1006/jmbi.1999.3013. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Joris B, Charlier P, Foster S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev. 2008;32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]