

Abstract
Previous studies identified Inka1 as a gene regulated by AP-2α in the neural crest required for craniofacial morphogenesis in fish and frog. Here, we extend the analysis of Inka1 function and regulation to the mouse by generating a LacZ knock-in allele. Inka1-LacZ allele expression occurs in the cephalic mesenchyme, heart, and paraxial mesoderm prior to E8.5. Subsequently, expression is observed in the migratory neural crest cells and their derivatives. Consistent with expression of Inka1 in tissues of the developing head during neurulation, a low percentage of Inka1−/− mice show exencephaly while the remainder are viable and fertile. Further studies indicate that AP-2α is not required for Inka1 expression in the mouse, and suggest that there is no significant genetic interaction between these two factors during embryogenesis. Together, these data demonstrate that while the expression domain of Inka1 is conserved among vertebrates, its function and regulation are not.
Keywords: neural crest, AP-2α, Inka1, neural tube closure, PAK4
INTRODUCTION
Inka1 (previously known as Inca for Induced in Neural Crest by AP-2α) is a novel gene originally identified in Xenopus as a downstream target of the transcription factor AP-2α (Luo et al., 2005). Analysis of Inka1 expression in fish and frog demonstrated that Inka1 is primarily expressed in neural crest cells (NCCs) and their derivatives, overlapping with the presence of AP-2α in these tissues. Moreover, the loss or reduction of functional AP-2α in fish and frog results in a reduction in Inka1 expression (Luo et al., 2005; Luo et al., 2007). Genetic models for AP-2α function in mammals demonstrate that this gene is required for multiple developmental processes, including neural tube and ventral body wall closure, as well as limb, heart, eye, and craniofacial development (Schorle et al., 1996; Zhang et al., 1996; Nottoli et al., 1998; Brewer et al., 2004; Brewer and Williams, 2004). Consistent with a potential role for Inka1 as a downstream target of AP-2α, morpholino knockdown of Inka1 in fish and frog revealed that this gene has an important function in craniofacial development. Morphants exhibit a severe reduction in NC - derived cranial cartilage, reflecting a requirement for Inka1 in NC associated morphogenesis in these aquatic vertebrates (Luo et al., 2007).
Inka1 is relatively conserved among vertebrates, particularly in a 38 amino acid residue central domain called the “Inka1-box” and an adjacent 14-3-3 binding domain (Luo et al., 2007). The Inka1-box is also present in a distantly related gene, Inka2, but the 14-3-3 domain is not conserved between these orthologs. Molecular analysis has not revealed a function for the Inka1 14-3-3 domain, but the Inka1-box has been shown to interact with the p21-kinase protein PAK4 (Luo et al., 2007). Association of Inka1 with PAK4 modulates cytoskeletal dynamics and loss of these functional interactions is thought to be the mechanism responsible for the NCC defects observed in Inka1 morphants (Luo et al., 2007). A possible association between Inka1 mutation and human developmental defects has also been noted recently. In humans, INKA1 is located at 3p21.31, and a novel 3.1-Mb microdeletion within this region was shown to cause cortical blindness, cleft lip, CNS abnormalities, and developmental delay in a 3 year old patient (Haldeman-Englert et al., 2009). Together, these data suggest a role for INKA1 in mammalian development and led us to investigate the role of Inka1 during mouse embryogenesis. To this end, we have generated a novel Inka1-LacZ allele to examine the expression and function of Inka1 in the developing mouse. Our studies indicate that the absence of Inka1 can influence neural tube closure, but also reveal that the function and regulation of Inka1 in the mouse differs from that reported in fish and frog.
RESULTS
Generation and characterization of the Inka1-LacZ allele
Inka1 is located on mouse chromosome 9 and consists of two exons (Fig. 1). The majority of the 281 amino acid protein, including the conserved Inka1-box and the 14-3-3 binding domain, is encoded by exon 2 (Fig. 1A). Therefore, we targeted this region in our Inka1-LacZ knock-in construct to increase the probability of generating a non-functional allele. A portion of exon 2 encoding approximately 185 amino acids, encompassing both the Inka1-box and 14-3-3 binding domain, were deleted and replaced with an IRES-LacZ reporter construct (Fig. 1A, B), allowing us to simultaneously disrupt Inka1 function and follow expression of the locus via β-galactosidase (β-gal) activity. Following successful gene targeting in ES cells (Fig. 1B,C), we generated mice heterozygous for the Inka1-LacZ allele.
Figure 1. Generation of an Inka1-LacZ knock-in allele.

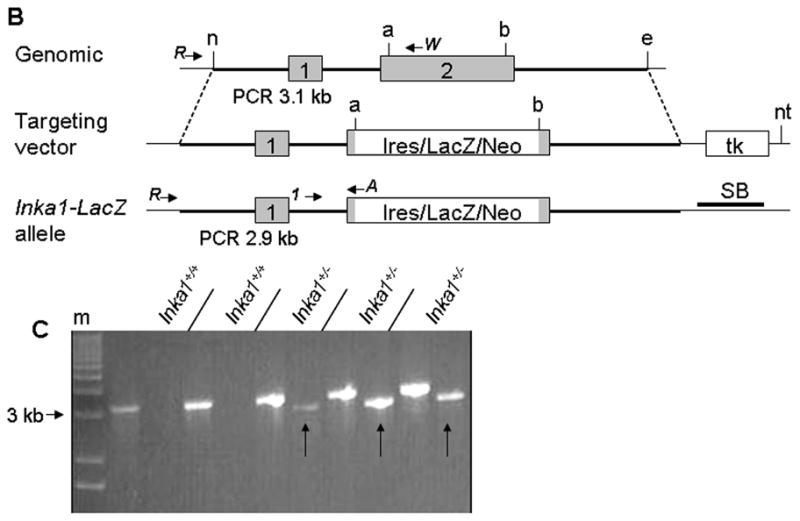
A) Protein alignment of Inka1 mouse and human sequences. Grey shaded region represents deletion in exon 2 of the targeting construct. Exons 1 and 2 are distinguished by the double back slash in the first line. Blue and yellow regions represent the 14-3-3 binding domain and “Inka1-box,” respectively. Alignment made using SIM (http://ca.expasy.org/tools/sim-prot.html). Human accession number BC012170.1, mouse accession number AK020090.1. B) Diagrammatic representation of the normal Inka1 locus (top), the targeting vector (middle), and the genomic locus following successful targeting. PCR primers used for screening and genotyping are illustrated as arrows. a – AvrII, b – BspHI, e – EcoRI, n – NheI, nt – NotI, tk – thymidine kinase, SB – Southern blot probe. Inka1-LacZ construct not drawn to scale. C) Representative PCR results from ES cell screening, m – marker, arrows indicate positive clones.
We next compared the expression of the Inka1-LacZ allele to that of endogenous mouse Inka1 (Fig. 2 & 3). Endogenous Inka1 transcripts were detected at embryonic day 8.5 (E8.5) in head mesenchyme (Fig. 2A, B), and shortly thereafter marked the migratory NCCs (Fig. 2C) and their derivatives including the dorsal root ganglia (DRG) and branchial arches (Fig. 2D, E). Starting at E9.5, expression could also be observed outside presumptive NCCs, in the developing limb buds (Fig. 2D, E). At later embryonic time points Inka1 expression became more diffuse, and by adulthood it was widespread, with particularly high levels in the heart (data not shown and (Luo et al., 2007)).
Figure 2. Inka1 expression during mouse embryogenesis.

A–E) Endogenous Inka1 expression detected by whole-mount in situ hybridization. F–M) Expression of the Inka1-LacZ allele detected via β-gal activity. All embryos shown in lateral views except for (B), ventral, and (F) ventral (left) and dorsal (right). BA1 & 2 – branchial arches one & two, CM – cephalic mesenchyme, DRG – dorsal root ganglia, FL – forelimb, H – heart, M – dorsal presomitc mesoderm, O – optic vesicle. Numbers in panel I refer to sections shown in Figure 3(A–D).
Figure 3. Tissue specific expression of the Inka1-LacZ allele.

A–H) Transverse sections of E8.0 (A–D) and E9.5 (E–H) embryos after whole-mount X-gal staining to detect expression of Inka1-LacZ. Plane of sections indicated by lines numbered in supplemental figure 1 (A–D) and Figure 2I(E–H). I–L) Sagittal sections of an E15.5 embryo stained with X-gal to show Inka1-LacZ expression and counterstained with nuclear fast red. a – aortic sac, acv – anterior cardinal vein, am – amnion, ba1– branchial arch 1, ce - conjunctival epithelium, cm – cephalic mesenchyme, d – dorsal aorta, e – esophagus, ec – endocardium, f – foregut, lc – left atrial chamber of heart, n – neural lumen, ne – neuroepithelium, mg – mammary gland, ope – optic evagination, opv – optic vesicle, otv – otic vesicle, p – paraxial mesoderm, pg – pigment layer of retina, rc – right atrial chamber of heart, sp – spinal cord, tc – throat cartilages, vb – vibrissae, yb – yolk sac blood island.
Examination of β-gal activity by whole-mount analysis in heterozygous Inka1-LacZ knock-in mice indicated that the targeted allele was expressed in the same spatiotemporal pattern as the endogenous gene (Fig. 2F–M). Consistent with the greater sensitivity of β-gal detection, we also noted additional areas of expression at E8.5 in the heart, and in the dorsal presomitic mesoderm, that were not as apparent using Inka1 RNA detection (compare Fig. 2B & 2F). At E8.75 β-gal activity was detected in the migratory NC streams populating branchial arches 1 and 2, and also around the optic vesicle, in the presumptive perioptic NC (Fig. 2G; compare with Fig. 2C). As development continued, Inka1-LacZ allele expression was also seen in the more posterior branchial arches, the DRG, and the limb buds (Fig. 2H – K). Note that due to technical issues with dye penetration, only superficial structures are stained using whole mount analysis from E12.5 onwards (Fig. 2L, M), although expression is still apparent in the face, limbs, and tail at E15.5 (Fig. 2M).
We also examined β-gal activity in sectioned material from mice containing the Inka1-LacZ allele at E8.0, E9.5, and E15.5 (Fig. 3). Transverse sections of E8.0 embryos demonstrated expression of Inka1-LacZ in the cephalic mesenchyme (Fig. 3A, B; also see supplemental Fig. 1A, B). At this stage, expression was also observed in the paraxial mesoderm, dorsal aorta, and the yolk sac blood islands (Fig. 3C, D; also see supplemental Fig. 1A, B). Expression was not observed in the neural ectoderm along the entire rostral-caudal axis, nor was it prominent in neural crest precursors at the margins of the neural tube. However, Inka1 expression was observed in migratory NCCs, and NCC derived craniofacial mesenchyme from E8.75 onwards (Fig. 2C–K, Fig. 3E–H). Expression was also apparent in the endocardium of the heart at E9.5 (Fig. 3H). At E15.5 expression was prominent in the meninges of the developing brain, notably at the mesencephalic flexure and choroid plexus, but was not observed in the neural tissue of the CNS itself (Figure 3I & Supplementary Fig. 1C, D). With respect to the PNS, staining was seen in the DRG, consistent with their NC origin (Supplemental Fig. 1D). We also saw expression in mesenchymal tissues, including perichondrial membranes, NCC-derived corneal stroma of the eye, and in the skin surrounding epithelial structures such as the vibrissae and mammary gland (Fig. 3J–L). Taken together, these results demonstrate that the Inka1-LacZ allele functions as a faithful reporter of endogenous mouse Inka1 mRNA expression, and further illustrates that expression of Inka1 in migratory NCCs and their derivatives is conserved in fish, amphibians, and mammals.
Loss of Inka1 results in exencephaly in a subset of mouse embryos
Consistent with its normal expression in NCCs in fish and frog, morpholino knockdown of inka1 in these species results in severe craniofacial defects (Luo et al., 2007). Consequently, we next determined if the function if Inka1 in facial development was conserved in mouse. Mice heterozygous for the Inka1-LacZ allele were intercrossed to generate offspring lacking wild-type Inka1. We initially examined and genotyped pups at weaning age (Table 1), and unexpectedly, identified a number of Inka1−/− pups. These mice were indistinguishable in size and external appearance to their wild-type and heterozygous littermates (Fig. 4A–C). We next used RT-PCR on weanling tail RNA to determine if our gene targeting strategy had in fact caused the loss of normal Inka1 expression in homozygous mutants. Three separate reactions were employed to examine splicing between exon 1 and exon 2 in both wild-type and Inka1-LacZ alleles, and to ascertain if we had generated the appropriate deletion in exon 2 in the targeted allele. As shown in Fig. 4D–I, splicing occurred normally between exon 1 + 2 in all genotypes, and the expected deletion within exon 2 was present in Inka1-LacZ homozygous mice. These data confirm that mice lacking normal Inka1 transcripts can survive through weaning. Indeed, we have subsequently determined that such mice are still viable and healthy at 6 months of age, and that both males and females are fertile, though they generally have fewer pups than wild-type and Inka1-LacZ heterozygous mice (Table 1 and data not shown).
Table 1.
Frequency of Inka1 null progeny at weaning.
| Parental cross | Total at weaning | Genotype # observed (#expected) | X2 | P | ||
|---|---|---|---|---|---|---|
| Inka1+/+ | Inka1+/− | Inka1−/− | ||||
| Inka1+/− X Black Swiss | 67 | 32 (33.5) | 34 (33.5) | N/A | 0.061 | 0.806 |
| Inka1+/− X Inka1+/− | 69 | 24 (17.25) | 34 (34.5) | 11 (17.25) | 3.019 | 0.082 |
Chi-square test was applied to results of mating scheme shown in lines one and two to examine ‘goodness-of-fit’ to normal Mendelian ratios.
Figure 4. Inka1 function in mouse development.

A–C) Photographs of wild-type (A), Inka1 heterozygote (B), and Inka1 null (C) weanlings. D–F) Illustrations of RT-PCR reactions to examine splicing between exons 1 and 2 (D, PCR products shown in G), the deletion in exon 2 (E, PCR products shown in H), and splicing between exon 1 and exon 2 in the Inka1-LacZ allele (F, PCR products shown in I). Primers B, C, F, G, & H described in methods. J–K) Skeletal stains of PO wild-type (J) and Inka1−/− (K) heads. L) E15.5 Inka1-null mouse with exencephaly stained for LacZ expression. md – lower jaw, n – nasal bone, m – DNA marker, t – tympanic ring.
As Inka1 is strongly expressed in migratory NCCs, we next investigated NC migration in Inka1−/− embryos by means of expression of AP-2α, and found no change between heterozygotes and nulls (Supplemental Fig. 2A, B). We also examined the formation of NC derivatives such as the trigeminal and cranial ganglia and found these to be normal as well (Supplemental Fig. 2C, D). To examine the skeleton of Inka1−/− mice, we analyzed bone and cartilage in P0 pups. We found that the head skeleton in Inka1−/− mice was comparable to wild-type littermates (Fig. 4J, K; see also Supplemental Fig. 3A, B). All other skeletal structures, such as the vertebrae those associated with the limbs were also normal (Supplemental Fig. 3 & data not shown).
Although Inka1−/− mice were present at weaning, we noted that our observed numbers of Inka1 null mice from monohybrid crosses were notably less than the expected 3:1 Mendelian ratio (Table 1). We therefore examined various embryological stages for developmental defects that would result in lethality. We found that 5–6% of Inka1−/− mice displayed mid-brain exencephaly (Fig. 4L). Inka1−/− mice with neural tube defects died shortly after birth and this pathology most likely accounted for the discrepancy in the expected Mendelian ratio. Thus, Inka1 may belong to a group of genes that predispose to cranial neural tube closure defects when they are mutated.
Inka1 expression is independent of AP-2α status in the mouse
Inka1 was initially identified in Xenopus as a downstream target of AP-2α (Luo et al., 2005; Luo et al., 2007). It was subsequently found to be down-regulated in low, a zebrafish AP-2α loss-of-function mutant (Knight et al., 2003; Luo et al., 2007). We therefore examined Inka1 expression in Tcfap2a-null mice. Surprisingly, we found that expression of endogenous Inka1 and the Inka1-LacZ allele in Tcfap2a null mice was comparable to that observed in wild-type (Fig. 5 and data not shown). These data stand in contrast to observations made in fish and frog, and suggest that Inka1 expression may not be regulated by AP-2α during mouse development. Additionally, the mouse breeding experiments required for the analysis of Inka1-LacZ expression in the Tcfap2a-null background required that we generate mice with various allelic combinations of Inka1 and Tcfap2a. We observed that mice heterozygous for both Inka1 and Tcfap2a were viable and fertile, and that loss of one or both wild-type alleles of Inka1 did not greatly exacerbate the Tcfap2a-null phenotype during embryogenesis (Fig. 5 and data not shown). Similarly, we found that the loss of one allele of Tcfap2a did not significantly alter the phenotype of Inka1-null mice in embryogenesis (data not shown). These findings strongly suggest that there is no significant genetic interaction between these two genes for mouse embryogenic development, despite their overlapping expression in NCCs and limb mesenchyme.
Figure 5. Analysis of Inka1 expression in Tcfap2a−/− mice.
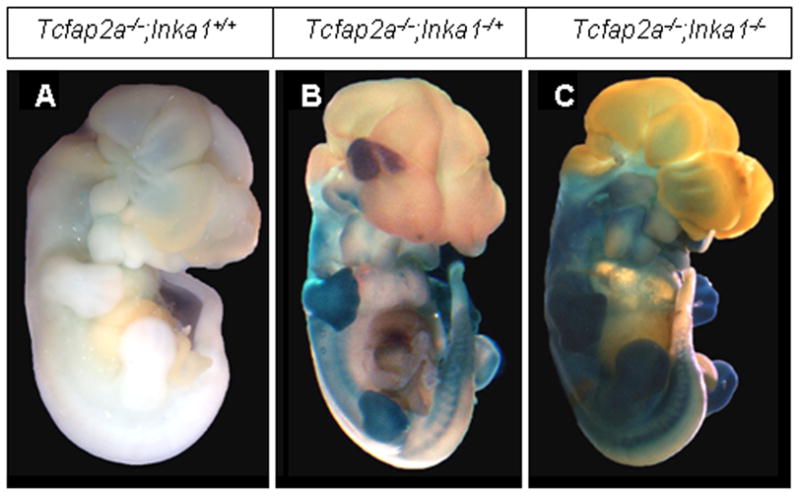
A–C) Lateral views of E12.5 Tcfap2a null mice that are wild-type (A), heterozygous (B) or homozygous (C) for the Inka1-LacZ allele. Embryos are stained with X-gal to show expression of Inka1.
DISCUSSION
In this study we have generated a novel Inka1-LacZ allele to examine the expression and function of Inka1 in the developing mouse. We show that the expression pattern of mouse Inka1 is conserved with that of fish and frog, and is initially concentrated in the head mesenchyme, paraxial mesoderm, heart, and the migrating NCCs and their derivatives. Though the Inka1-LacZ allele is not a pan-NCC marker, it is a good marker for early cephalic NCC migration, and may be useful for following the development of this cell population during mouse embryogenesis. As mouse development proceeds, Inka1 expression is also observed in the limb buds and perichondrial tissue. The expression domains of Inka1 in the NC and limbs are also shared with Tcfap2a, and it has been shown that the AP-2α transcription factor is a major regulator of Inka1 expression in fish and frog. However, our studies reveal that mouse Inka1 expression does not change significantly in the absence of Tcfap2a, indicating that this aspect of Inka1 regulation is not conserved in the mouse. Since several members of the AP-2 gene family, particularly AP-2β and AP-2γ, are expressed in a similar spatiotemporal pattern to that of AP-2α, it is possible that there is functional redundancy between these family members with respect to mouse Inka1 expression. However, preliminary analysis of mice lacking both Tcfap2a and Tcfap2b indicates that Inka1 expression is still maintained in these mutants, too (B. R., unpublished). Data obtained in fish also indicate that expression of inka1 is not solely dependent upon the AP-2α transcription factor, as some inka1 transcript is still present in low fish, particularly at later stages (Luo et al., 2007). A thorough understanding of these species-specific differences with respect to AP-2 regulation will require a detailed analysis of how Inka1 mRNA levels are regulated during development in fish, frog, and mouse.
Our analysis of Inka1 function in mouse embryogenesis also reveals significant differences from its role in fish and frog development. Inka1 gene knockdown in frog and fish results in major alterations of the craniofacial skeleton. Additionally, over-expression of Inka1 in these species also leads to severe developmental defects (Luo et al., 2007). In contrast, no defects were apparent in the external morphology of the Inka1 null mouse face, nor in the underlying craniofacial skeleton. Instead, the major gross morphological defect seen in a significant fraction of the Inka1 null mice was exencephaly. This phenotype was not observed in fish and frog inka1 morphants. The absence of neural tube closure defects in fish compared to mouse is not surprising given the different mechanisms of neurulation between these species. However, while reduced expression of inka1 in frog did not cause exencephaly, neural tube closure defects were reported in Xenopus embryos over-expressing inka1 (Luo et al., 2007). Several mechanisms can be invoked to explain the influence of Inka1 expression on mouse neural tube closure. One possibility is that the loss of Inka1 expression in NCCs results in a failure of appropriate neural fold morphogenesis. Indeed, the early expression of AP-2α in the NCCs induced at the boundary between the neural and non-neural ectoderm appears to be the mechanism by which Tcfap2a modulates neural tube closure (Schorle et al., 1996; Zhang et al., 1996; Brewer et al., 2004). Our finding that Inka1 expression is not prominent in NCCs until their migratory phase represents one argument against this hypothesis. Additionally, we have not observed a strong genetic interaction between Inka1 and Tcfap2a with respect to neurulation. These data imply that Inka1 and AP-2α act independently in the early phase of mouse neural tube closure.
Two other possible tissues in which Inka1 may act to modulate neural tube closure are the neuroectoderm and the cranial mesenchyme (Harris and Juriloff, 2007; Zohn and Sarkar, 2008). Since Inka1 is not expressed in the neural tube, we do not favor the neuroectoderm as the site of Inka1 action. Instead, the most likely site of Inka1 function in neurulation is the cranial mesenchyme, which displays strong expression of Inka1 during the appropriate developmental window. In this regard, analyses of mice with mutations in Twist, Hectd1, and Cart1 have shown that the cephalic mesenchyme has a critical role in neural tube closure (Chen and Behringer, 1995; Zhao et al., 1996; Juriloff and Harris, 2000; Soo et al., 2002; Zohn et al., 2007). Recent studies suggest a potential mechanism by which Inka1 may influence cell behavior, and ultimately neural tube closure – via protein:protein interaction with the p21-activated kinase 4 (PAK4). The PAKs are involved in the regulation of several cellular processes, including cell death, cell motility and morphology, and transcription through the MAP kinase cascade (Gnesutta et al., 2001; Bokoch, 2003; Arias-Romero and Chernoff, 2008; Eswaran et al., 2008). Furthermore, PAK4 is necessary for mouse embryogenesis, and has a role in neural tube closure (Qu et al., 2003; Tian et al., 2009). Inka1 interacts with the C-terminal domain of PAK4, and in frog, inka1 and PAK5 (the ortholog of mouse PAK4) work synergistically to modulate cytoskeletal dynamics associated with processes including gastrulation and wound healing (Luo et al., 2007). Given these findings, it may be interesting to examine wound healing dynamics in the Inka1 null mice and to determine if there is any interaction between Pak4 and Inka1 during mouse development.
Our results indicate that Inka1 belongs to a subset of genes whose mutation predisposes mice to neural tube closure defects (Fleming and Copp, 2000; Nadeau, 2001) and raises the possibility that it may be involved in modifying the phenotypes seen in specific human chromosomal deletion syndromes (Haldeman-Englert et al., 2009). Nevertheless, a significant number of Inka1 null mice are viable and fertile indicating that this gene is not essential for mouse embryogenesis, in contrast to the conclusions reached in fish and frog. While it is possible that the Inka1 allele we have generated is not a true null allele, we believe that this is unlikely as we have deleted a significant portion of the protein, including the most conserved sequences. Another possibility is that the function of Inka1 in the fish and frog system is shared between the two Inka genes present in mouse, Inka1 and Inka2, leading to functional redundancy. Therefore, in the future, it will be of interest to determine how the loss of Inka2 – either alone or in association with Inka1 – influences mouse embryogenesis.
EXPERIMENTAL PROCEDURES
Inka1-LacZ plasmid construction
To isolate a genomic clone of Inka1, BAC pools (ResGen, CITB Mouse BAC plates #96023) were screened with primers located in exon 2 (Inka1 fwd: 5′-ctc tag act ggg act ctg gct tct ccg agg-3′ & Inka1 rvs: 5′-gcc gtc cag tcc tct gct tcc aat cct cg-3′). After a single library pool was identified (plate #525) individual bacterial colonies were spotted onto a nylon membrane using a 384 pin MicroWell Plate Copier. After an overnight incubation at 37°C, colonies were lysed and UV-cross-linked (Sambrook and Russell, 2001). Confirmation of the presence of a BAC containing Inka1 in a single colony (F6) was achieved using a P−32 labeled probe corresponding to exon 2. We employed restriction digests of F6 BAC DNA followed by Southern blot to analyze the genomic structure of that region, and identified a 14kb EcoRI fragment that contained Inka1 homology. Utilizing a unique AscI site located just upstream of exon 1, the EcoRI fragment was cloned as two AscI-EcoRI pieces of approximately 8.6kb and 5.8kb, which were then confirmed by sequencing. With these genomic clones, and several other plasmids that contained selectable markers (Brewer et al., 2002), we constructed the Inka1-LacZ plasmid which was used for gene targeting (Fig. 1).
Targeting vector plasmid DNA was linearized at a unique NotI site and purified by phenol:chloroform extraction followed by ethanol precipitation. 30ug of DNA was electroporated into CJ-7 ES cells and these were then grown in media plus G418, a drug that selects for the presence of the neo gene, and ganciclovir, which selects for the absence of HSV TK. ES cell colonies that passed both modes of selection were then screened via PCR using external primers (Inka1-A: 5′-gat ctg ggc cca tgc atg tat acc cta gga cat cc-3′, Inka1-W: 5′-ggt aca tct cca tca ctc ccc tca ctg gtt-3′, Inka1-R: 5′-tcc ctt cgc tgg ttt cat agt cct-3′). PCR reactions were performed using Elongase (Invitrogen #10480-028) in the presence of 1.5mM [Mg2+], using an annealing temperature of 60 C and an extension temperature of 68 C for 3.5 minutes. Positive clones from the PCR reactions were confirmed by Southern blot analysis (data not shown) prior to karyotyping. One euploid clone was selected for injection into C57Bl/6 blastocysts and subsequently chimeric males were bred to Black Swiss females (Taconic) to obtain germ line transmission of mice heterozygous for the Inka1-LacZ allele.
Inka1-LacZ expression analysis
Timed matings were set and embryos were isolated at various development time points for expression analysis. The presence of a vaginal plug was scored as E0.5. DNA isolated from yolk sac or tail using REDextract (Sigma # XNATS) was used to determine genotype by PCR. For Inka1, primers Inka1-A and Inka1-W were used in combination with an internal primer, (Inka1-1: 5′-cgt aat gca tcc cgg gaa aga agt gctg-3′) (Fig. 1). PCR reactions were performed using an annealing temperature of 65 C and an extension temperature of 72 C for one minute; 35 cycles. PCR products of 880bp are indicative of the wild-type allele and PCR products of 650bp are indicative of the Inka1-LacZ allele. Inka1-LacZ expression was detected by X-gal staining as follows.
Embryos of the desired time-points were dissected in PBS, and fixed for 15–20 minutes at room temperature in a solution of 0.2% glutaraldehyde in 0.1M phosphate buffer pH 7.5, 5mM EGTA and 2mM MgCl2. The embryos were thenwashed briefly in PBT, and placed in staining cocktail for 2–3 hours at 37°C or at room temperature overnight (Nagy et al., 2003). All embryos were stained with gentle rocking in the dark. A selection of embryos which stained positive for LacZ were embedded in OCT for cryosectioning (procedure follows).
Cryosectioning
Embryos were fixed overnight in 0.2% paraformaldehyde and then placed in freshly made 30% sucrose/PBS overnight or until the embryos sank. After a 15 minute incubation in 1:1 30% sucrose/OCT, embryos were placed in 100% OCT and set on dry ice until frozen. Blocks were sectioned on a cryotome at 7μm, 10μm, and 12 μm, and representative sections counterstained with nuclear fast red.
RNA expression analysis
RNA was extracted from tails using the RNeasy kit (Qiagen, #74104). To make cDNA, 5ug of RNA, 200ng of random primers and 10mM dNTP (total volume of 28μl) were incubated at 65°C for 5 minutes, and then put on ice for 1 minute. The following mix was then added to each sample: 8μl of 5X first strand buffer, 2μl of 0.1M DTT, 2μl RNase out, and 2μl of Superscript III RT (Invitrogen). Samples were then incubated sequentially at 25°C for 5 minutes, 50°C for 90 minutes, and 70° C for 15 minutes, then chilled to 4°C before storing at −20°C. The following primers were used to detect Inka1 transcript: For splicing between exons 1 and 2: (Inka1-B 5′-gct tcc tga gcc agc tcc gct-3′, & Inka1-C 5′-cca agg gta aac cct tgt ctt ctgc-3′); For the deletion in exon 2: (Inka1-F 5′-gca tgc tct aga ctg ggac-3′, & Inka1-G 5′-cga gcc ctg gcc agc cta cg-3′); For splicing between exon 1 and exon 2 in the Inka1-LacZ allele: (Inka1-B (see above sequence) & Inka1-H 5′-gat ctg ggc cca tgc atg tat acc cta gga cat cc-3′). PCR reactions were performed using Elongase (Invitrogen #10480-028) in the presence of 1.5mM [Mg2+], using an annealing temperature of 60 C and an extension temperature of 68 C for 45 seconds.
Tcfap2a-null mice were generated and genotyped as previously described (Zhang et al., 1996). Endogenous Inka1 was detected in embryos using an in situ probe of 550bp located in exon 2, cloned from genomic DNA into TOPO-TA vector (Invitrogen). Endogenous AP-2α expression was detected as previously described (Zhang et al., 2003). In situ hybridization procedure is described in (Feng et al., 2009).
Skeletal stains
E18.5 embryos were dissected in tap water and fixed in 95% ethanol at room temperature for 24 hours. Samples were then skinned, eviscerated, and placed in alcian blue stain (15mg in 80% ethanol, 20% glacial acetic acid) for 2 days at room temperature. After a 10 minute rinse in ethanol, embryos were stained for bone with 0.1% alizarin red in ethanol for 2 days, and then washed one day at room temperature in each of the following solutions: 1% KOH; 20% glycerol in 1% KOH; 50% glycerol in 1% KOH; and 80% glycerol in 1% KOH. Embryos were photographed and stored in 100% glycerol.
Supplementary Material
A, B) Whole-mount X-gal stained E8.0 embryos shown in ventral (A) and dorsal (B) views. Numbered lines indicate planes of sections shown in Figure 3A–D. C–D) Sagittal sections of E15.5 embryos stained with X-gal and counterstained with nuclear fast red. cp – choroid plexus, drg – dorsal root ganglia, mf – mesencephalic flexure, nsp – nasopharanyx, orp – oropharanyx, pit – pituitary, ttr – tubo-tympanic recess, v –vertebrae.
AB) Lateral views of Tcfap2a RNA expression in E9.5 wild-type (A) and Inka1−/− embryos (B). C–D) Lateral views of E10.5 wild-type (C) and Inka1−/− embryos (D) stained for neurofilament to show peripheral nervous system. cg – cranial ganglia, drg – dorsal root ganglia, tg – trigeminal ganglia. Numbers 7–10 indicate cranial nerves.
A, D) Skeletal stains of wild-type (A, C) and Inka1 null (B, D) mice shown in norma verticalis (A, B) and dorsal (C, D) views. E, F) Alcian blue cartilage stains of E15.5 wild-type (E) and Inka1 null (F) forelimbs. a – anterior, hl – hindlimb, ip – interparietal, n, nasal bone, p – posterior, pr – parietal, r – ribs, rd – radius, u – ulna, v – vertebrae, z – zygomatic.
Acknowledgments
NIDCR
Grant number: DE12728
We would like to thank Dr. Vida Melvin for support and advice, Dr. Brian Parr and Dr. Timothy Nottoli for assistance with gene targeting, and Dr. Ting Luo for discussions. The 2H3 monoclonal antibody developed by Jessell T. M. & Dodd J. was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
Contributor Information
Bethany S. Reid, Department of Craniofacial Biology and Cell and Developmental Biology, University of Colorado Denver, 12801 East 17th Avenue, Aurora, CO 80045
Thomas D. Sargent, Laboratory of Molecular Genetics, NICHD, NIH, Bethesda, MD 20892
Trevor Williams, Department of Craniofacial Biology and Cell and Developmental Biology, University of Colorado Denver, 12801 East 17th Avenue, Aurora, CO 80045.
References
- Arias-Romero LE, Chernoff J. A tale of two Paks. Biol Cell. 2008;100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- Brewer S, Feng W, Huang J, Sullivan S, Williams T. Wnt1-Cre-mediated deletion of AP-2alpha causes multiple neural crest-related defects. Dev Biol. 2004;267:135–152. doi: 10.1016/j.ydbio.2003.10.039. [DOI] [PubMed] [Google Scholar]
- Brewer S, Jiang X, Donaldson S, Williams T, Sucov HM. Requirement for AP-2alpha in cardiac outflow tract morphogenesis. Mech Dev. 2002;110:139–149. doi: 10.1016/s0925-4773(01)00579-2. [DOI] [PubMed] [Google Scholar]
- Brewer S, Williams T. Loss of AP-2alpha impacts multiple aspects of ventral body wall development and closure. Dev Biol. 2004;267:399–417. doi: 10.1016/j.ydbio.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Eswaran J, Soundararajan M, Kumar R, Knapp S. UnPAKing the class differences among p21-activated kinases. Trends Biochem Sci. 2008;33:394–403. doi: 10.1016/j.tibs.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Feng W, Simoes-de-Souza F, Finger TE, Restrepo D, Williams T. Disorganized olfactory bulb lamination in mice deficient for transcription factor AP-2epsilon. Mol Cell Neurosci. 2009;42:161–171. doi: 10.1016/j.mcn.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A, Copp AJ. A genetic risk factor for mouse neural tube defects: defining the embryonic basis. Hum Mol Genet. 2000;9:575–581. doi: 10.1093/hmg/9.4.575. [DOI] [PubMed] [Google Scholar]
- Gnesutta N, Qu J, Minden A. The serine/threonine kinase PAK4 prevents caspase activation and protects cells from apoptosis. J Biol Chem. 2001;276:14414–14419. doi: 10.1074/jbc.M011046200. [DOI] [PubMed] [Google Scholar]
- Haldeman-Englert CR, Gai X, Perin JC, Ciano M, Halbach SS, Geiger EA, McDonald-McGinn DM, Hakonarson H, Zackai EH, Shaikh TH. A 3.1-Mb microdeletion of 3p21.31 associated with cortical blindness, cleft lip, CNS abnormalities, and developmental delay. Eur J Med Genet. 2009;52:265–268. doi: 10.1016/j.ejmg.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res A Clin Mol Teratol. 2007;79:187–210. doi: 10.1002/bdra.20333. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. Mouse models for neural tube closure defects. Hum Mol Genet. 2000;9:993–1000. doi: 10.1093/hmg/9.6.993. [DOI] [PubMed] [Google Scholar]
- Knight RD, Nair S, Nelson SS, Afshar A, Javidan Y, Geisler R, Rauch GJ, Schilling TF. lockjaw encodes a zebrafish tfap2a required for early neural crest development. Development. 2003;130:5755–5768. doi: 10.1242/dev.00575. [DOI] [PubMed] [Google Scholar]
- Luo T, Xu Y, Hoffman TL, Zhang T, Schilling T, Sargent TD. Inca: a novel p21-activated kinase-associated protein required for cranial neural crest development. Development. 2007;134:1279–1289. doi: 10.1242/dev.02813. [DOI] [PubMed] [Google Scholar]
- Luo T, Zhang Y, Khadka D, Rangarajan J, Cho KW, Sargent TD. Regulatory targets for transcription factor AP2 in Xenopus embryos. Dev Growth Differ. 2005;47:403–413. doi: 10.1111/j.1440-169X.2005.00809.x. [DOI] [PubMed] [Google Scholar]
- Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2:165–174. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo, A Laboratory Manual. Cold Spring Harbor, New York: John Inglis; 2003. [Google Scholar]
- Nottoli T, Hagopian-Donaldson S, Zhang J, Perkins A, Williams T. AP-2-null cells disrupt morphogenesis of the eye, face, and limbs in chimeric mice. Proc Natl Acad Sci U S A. 1998;95:13714–13719. doi: 10.1073/pnas.95.23.13714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Li X, Novitch BG, Zheng Y, Kohn M, Xie JM, Kozinn S, Bronson R, Beg AA, Minden A. PAK4 kinase is essential for embryonic viability and for proper neuronal development. Mol Cell Biol. 2003;23:7122–7133. doi: 10.1128/MCB.23.20.7122-7133.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2001. Molecular Cloning. [Google Scholar]
- Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235–238. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- Soo K, O’Rourke MP, Khoo PL, Steiner KA, Wong N, Behringer RR, Tam PP. Twist function is required for the morphogenesis of the cephalic neural tube and the differentiation of the cranial neural crest cells in the mouse embryo. Dev Biol. 2002;247:251–270. doi: 10.1006/dbio.2002.0699. [DOI] [PubMed] [Google Scholar]
- Tian Y, Lei L, Cammarano M, Nekrasova T, Minden A. Essential role for the Pak4 protein kinase in extraembryonic tissue development and vessel formation. Mech Dev. 2009;126:710–720. doi: 10.1016/j.mod.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Zhang J, Brewer S, Huang J, Williams T. Overexpression of transcription factor AP-2alpha suppresses mammary gland growth and morphogenesis. Dev Biol. 2003;256:127–145. doi: 10.1016/s0012-1606(02)00119-7. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hagopian-Donaldson S, Serbedzija G, Elsemore J, Plehn-Dujowich D, McMahon AP, Flavell RA, Williams T. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature. 1996;381:238–241. doi: 10.1038/381238a0. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Behringer RR, de Crombrugghe B. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat Genet. 1996;13:275–283. doi: 10.1038/ng0796-275. [DOI] [PubMed] [Google Scholar]
- Zohn IE, Anderson KV, Niswander L. The Hectd1 ubiquitin ligase is required for development of the head mesenchyme and neural tube closure. Dev Biol. 2007;306:208–221. doi: 10.1016/j.ydbio.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohn IE, Sarkar AA. Modeling neural tube defects in the mouse. Curr Top Dev Biol. 2008;84:1–35. doi: 10.1016/S0070-2153(08)00601-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, B) Whole-mount X-gal stained E8.0 embryos shown in ventral (A) and dorsal (B) views. Numbered lines indicate planes of sections shown in Figure 3A–D. C–D) Sagittal sections of E15.5 embryos stained with X-gal and counterstained with nuclear fast red. cp – choroid plexus, drg – dorsal root ganglia, mf – mesencephalic flexure, nsp – nasopharanyx, orp – oropharanyx, pit – pituitary, ttr – tubo-tympanic recess, v –vertebrae.
AB) Lateral views of Tcfap2a RNA expression in E9.5 wild-type (A) and Inka1−/− embryos (B). C–D) Lateral views of E10.5 wild-type (C) and Inka1−/− embryos (D) stained for neurofilament to show peripheral nervous system. cg – cranial ganglia, drg – dorsal root ganglia, tg – trigeminal ganglia. Numbers 7–10 indicate cranial nerves.
A, D) Skeletal stains of wild-type (A, C) and Inka1 null (B, D) mice shown in norma verticalis (A, B) and dorsal (C, D) views. E, F) Alcian blue cartilage stains of E15.5 wild-type (E) and Inka1 null (F) forelimbs. a – anterior, hl – hindlimb, ip – interparietal, n, nasal bone, p – posterior, pr – parietal, r – ribs, rd – radius, u – ulna, v – vertebrae, z – zygomatic.