

Abstract
The role of nutrition and balanced metabolism in normal growth, development, and health maintenance is well known. Patients affected with either acute or chronic diseases often show disorders of nutrient balance. In some cases, a devastating state of malnutrition known as cachexia arises, brought about by a synergistic combination of a dramatic decrease in appetite and an increase in metabolism of fat and lean body mass. Other common features that are not required for the diagnosis include decreases in voluntary movement, insulin resistance, and anhedonia. This combination is found in a number of disorders including cancer, cystic fibrosis, AIDS, rheumatoid arthritis, renal failure, and Alzheimer's disease. The severity of cachexia in these illnesses is often the primary determining factor in both quality of life, and in eventual mortality. Indeed, body mass retention in AIDS patients has a stronger association with survival than any other current measure of the disease. This has led to intense investigation of cachexia and the proposal of numerous hypotheses regarding its etiology. Most authors suggest that cytokines released during inflammation and malignancy act on the central nervous system to alter the release and function of a number of neurotransmitters, thereby altering both appetite and metabolic rate. This review will discuss the salient features of cachexia in human diseases, and review the mechanisms whereby inflammation alters the function of key brain regions to produce stereotypical illness behavior.
Introduction
The maintenance of energy homeostasis is one of the most crucial physiological tasks that animals perform to ensure long-term survival. Under normal conditions, energy intake and energy expenditure are precisely controlled, providing remarkable stability of body weight over an animals' lifetime. Because infectious illness is an acute threat to life, animals have evolved systems that sacrifice energy homeostasis in order to provide the greatest chance of survival. Indeed, nearly all of the behaviors and metabolic changes typical of sickness can be linked directly to a highly organized strategy of the organism to fight infection [1, 2]. One of the primary features of the host response to acute infection is fever. Increased body temperature has been strongly linked to survival from infection in numerous species, and appears to be one of the most critical non-specific defense systems animals employ to fight pathogens [3]. The increase in body temperature that is achieved during fever favors proliferation of immune cells and is unfavorable to the growth of many pathogens. Although it is generally an adaptive strategy for survival, the generation of fever comes at a very high metabolic cost. Most studies agree that the increase in metabolism per 1°C of fever is close to 13 percent [4]. Therefore, it is reasonable to view illness behavior in the context of an adaptive, but metabolically expensive, febrile response. In neurophysiological terms, fever represents an increase in hypothalamic thermoregulatory set point. In this instance, animals feel cold at previously normal body temperatures, and will seek out warmer environments to compensate. Furthermore, thermogenesis is increased (e.g. by shivering and activation of sympathetic outflow), and heat loss is reduced both behaviorally (e.g. by curling up, huddling, seeking warm environments) and physiologically (e.g. by shifting blood flow away from the skin and improving insulation through piloerection).
While these behaviors are relatively straightforward to justify from a teleological perspective, it is somewhat more difficult to understand how the anorexia typical of acute disease provides a survival benefit. In fact, one could argue that the increased metabolic demands imposed by the maintenance of fever and immune system activation should lead to relative hyperphagia rather than anorexia. On the other hand, an animal that is not hungry will not spend time foraging, thereby preserving energy reserves and reducing heat loss. In addition, prey animals are particularly vulnerable to predation when they are ill secondary to impaired locomotion, potentially providing another survival advantage of anorexia. It has also been suggested that decreased food intake during fever helps to maintain relatively low circulating levels of iron and zinc, both of which are known to be critical trace elements for bacterial growth (for review, see [5]). Overall, it appears that relative anorexia may also be an adaptive response to acute infection, an idea that is reinforced by the observation that maintaining basal food intake by force feeding infected mice doubles their mortality, though it is unclear whether this is a general phenomenon [6]. Coincident with the decrease in food intake, an overall hypermetabolic, catabolic state is maintained, thereby providing a ready fuel supply for the immune system, particularly in the form of muscle derived amino acids.
Although acute infection has provided a steady evolutionary pressure throughout most of human evolution, relatively recent medical developments have produced large numbers of individuals who are living with more chronic inflammatory insults. Although undoubtedly an oversimplification, the metabolic effects of chronic diseases can be viewed as an extended version of the acute response to infection that, while potentially beneficial in the acute setting, becomes pathological when prolonged. Cachexia (from the Greek kakos (bad) and hexis (condition)), or disease-associated wasting, is a common occurrence in cancer, renal failure, and infectious disease. Hippocrates wrote about the relationship between dropsy (chronic heart failure) and cachexia more than 2400 years ago: “The flesh is consumed and becomes water…the abdomen fills with water, the feet and legs swell, the shoulders, clavicles, chest and thighs melt away…This illness is fatal” [7]. This devastating state of malnutrition is brought about by a synergistic combination of a dramatic decrease in appetite and an increase in metabolism of fat and lean body mass. The severity of cachexia in many illnesses is the primary determining factor in both quality of life, and in eventual mortality [8, 9]. Other illness-induced morbidities including lethargy also directly compromise the ability of patients to recover from potentially life-saving or extending interventions, including surgery and cytotoxic chemotherapy, and can diminish the motivational drive to aggressively battle the condition. Although cachexia in chronic disease was described more than two thousand years ago, the central mechanisms underlying this disorder of energy homeostasis were until recently quite poorly understood. However, with the recent renaissance in understanding of appetite and body weight control, investigators have quickly begun to unravel many of the fundamental aspects of the physiology of cachexia (as described further below).
Unfortunately, despite our increased understanding of this process, there is currently no safe and effective treatment for this condition. Extensive trials with anti-inflammatory agents have shown modest benefit, and significant gastrointestinal and renal side effects are common [10]. Perhaps the most promising agents in early trials were the progestational agents (e.g., megestrol acetate), but more recent trials have shown that the modest weight gain produced by these agents is primarily due to gains in body fat and water retention, with little beneficial effects on lean body tissue [10, 11]. Glucocorticoids produce weight gain and improvement in quality of life scores, but they do not improve strength or lean body mass and are associated with numerous unwanted side effects typical of iatrogenic Cushing's syndrome [12]. Growth hormone has also received attention as a potential anti-cachexia agent, but its usefulness is limited by cost, difficulty of delivery, worsened insulin resistance, potential to promote the growth of tumors in individuals with cancer, and growth hormone resistance in most forms of cachexia [11, 13]. Our work and that of others has demonstrated that melanocortin antagonists and drugs that diminish endogenous melanocortin activity (e.g. ghrelin) show great promise in preclinical and early clinical trials [14-19]. However, there remains a critical need for the development of new agents with efficacy in treating various features of cachexia brought about by a variety of diseases.
Defining Cachexia
Sick individuals experience a variety of symptoms including weakness, malaise, anorexia, weight loss, and inability to concentrate. Perhaps because these symptoms are so common, they have in the past been underestimated as being simply an uncomfortable side effect of the illness process. However, as outlined above, these symptoms are not simply the result of debilitation, but rather represent an evolved adaptive strategy with important survival implications. On the other hand, when one views these adaptations in the context of chronic illness, the resulting metabolic state is not only debilitating, but strongly predicts both morbidity and mortality. Given its clinical importance it is not surprising that a great deal of effort has gone into developing therapeutics for cachexia. This effort quickly made it readily apparent that there was no real unifying definition of cachexia, nor were there any accepted guidelines for demonstrating efficacy of new therapeutic modalities. In December of 2007, a group of scientists, clinicians, and representatives of the Food and Drug Administration of the USA met to establish an operational definition of cachexia, as well as to begin discussions of possible therapeutic endpoints for clinical trials. The definition that emerged from this consensus conference was: “Cachexia is a complex metabolic syndrome associated with underlying illness and characterized by loss of muscle with or without loss of fat mass. The prominent clinical feature of cachexia is weight loss in adults (corrected for fluid retention) or growth failure in children (excluding endocrine disorders). Anorexia, inflammation, insulin resistance and increased muscle protein breakdown are frequently associated with cachexia. Cachexia is distinct from starvation, age-related loss of muscle mass, primary depression, malabsorption and hyperthyroidism and is associated with increased morbidity” [20]. Like any consensus statement, numerous details and nuances were debated, and the relative complexity of this definition reflects the real complexity of this metabolic derangement. Nonetheless, this definition provided a starting point for discussion of meaningful clinical trial endpoints.
General Mechanisms in Cachexia: Inflammation
A shared characteristic of chronic diseases associated with the development of cachexia is increased production of pro-inflammatory cytokines. Cells of the innate immune system, such as macrophages and dendritic cells, act as sentinels that detect microbial infection and tissue damage by recognizing common molecular patterns. In response, these cells release pro-inflammatory cytokines, which act in a paracrine and endocrine fashion. In this way, a local signal of damage or danger is amplified and distributed to distal sites of action, resulting in common immunological and behavioral responses to sickness. For example, elevations in interleukin-1 beta (IL-1β), leukemia inhibitory factor (LIF), interleukin-6 (IL-6), and tumor necrosis factor alpha (TNF-α) are found in rats, mice, and sheep following peripheral administration of lipopolysaccharide (LPS), a potently inflammatory bacterial endotoxin known to induce the full spectrum of sickness behavior [6, 21-26]. It is precisely these cytokines that appear to mediate disease-associated wasting syndromes. Indeed, elevated circulating levels of TNF-α and IL-6 in patients with cardiac cachexia are the strongest predictor for pathological weight loss [27, 28]. In cancer cachexia, IL-1β, IL-6, LIF and TNF-α have been shown to be produced by human tumor cells in tissue culture and in patients [29, 30]. This rise in circulating cytokines has been implicated in the physiologic and behavioral responses to inflammation in rodents, including anorexia [31], HPA axis activation [32], fever [33], increased non-REM sleep [34], and anhedonia [35]. Indeed, intracerebroventricular (i.c.v.) administration of several pro-inflammatory cytokines in rodents is effective in recapitulating the cardinal features of cachexia including: anorexia, weight loss, increased energy expenditure and catabolism of fat and lean body mass (LBM) [36, 37]. These findings suggest that there are systems within the brain that are sufficient to promote the cachectic state in response to cytokines.
It is important to note that doses and routes of administration of endotoxin and cytokines used in these and subsequently cited studies vary considerably between experiments. Because different doses or injection sites of immune-modulating agents may recruit distinct inflammatory mechanisms, caution in interpreting these data together must be exercised. In lieu of standardizing dosing regimens, however, evaluation of existing studies can identify important common mediators of sickness behavior. Given that these different inflammatory methods induce similar cytokine induction patterns in the brain, common mechanisms presumably underlie the conserved effects. Remarkably, the induction of cytokine expression in the brain appears to be conserved through mammalian evolution, affording us the opportunity to integrate observations from several species into a common understanding of sickness behavior. Taken together, these studies strongly suggest an important role for cytokine signaling, both in the central nervous system (CNS) and the periphery, in mediating the sickness response.
Peripheral Inflammation
Cachexia research has traditionally focused on the local mechanisms of inflammation-induced muscle wasting. In the periphery, cytokines act directly on muscle cells to disrupt the normal balance that exists between anabolic and catabolic pathways [38]. Activation of nuclear factor-kappa B (NF-κB) in muscle is increased by IL-1β and TNF-α [39]. Increased NF-κB activation in muscle promotes muscle degeneration by accelerating protein breakdown through ubiquitin-dependent proteolysis [40] and preventing muscle repair by decreasing the expression of the transcription factor MyoD [41]. Further, muscle explants, when treated with inflammatory cytokines, exhibit dysregulated protein turnover leading to catabolism [42]. These studies define an unambiguous peripheral role for cytokine signaling in mediating inflammation-induced lean mass catabolism. Though muscle wasting eventually leads to weakness and lethargy, sick patients and animals exhibit behavioral depression long before significant catabolism can occur. Collectively, these data support the hypothesis that the coordinated behavioral and metabolic responses that occur in the clinical syndrome of cachexia likely involve the synergistic effects of combinations of cytokines acting in both the brain and periphery. However, a full discussion of the effects of cytokines in the periphery is beyond the scope of this review.
Central Inflammation
In the absence of infection or tissue damage, cytokine expression in the brain remains very low, though basal cytokine levels may play a role in sleep and energy balance [43-46]. Central expression of pro-inflammatory cytokines is strongly induced by peripheral inflammation. Peripheral injections of LPS have been shown to increase the expression of IL-1α, IL-1β, IL-6, TNF-α and LIF in the rodent brain [47, 48]. Central administration of several of these cytokines, including IL-1β, TNF-α, and LIF, stimulate their own production as well as induce the synthesis of other cytokines in the brain [49-51]. This provides a mechanism for local amplification and maintenance of inflammatory signaling in the brain in response to peripheral inflammation. The importance of this central inflammation was recently supported by research using MyD88 (a common intracellular mediator of inflammatory signaling upstream of NF-κB) knockout mice and bone marrow transplants to demonstrate that the endocrine action of circulating cytokines is insufficient to elicit anorexia in response to peripheral LPS without functional inflammatory pathways in the brain or endothelium [52]. Although one must always consider developmental adaptation when interpreting knockout data, this finding suggests that intact inflammatory signaling to the CNS is necessary to promote the full cachectic phenotype.
Hypothalamic Involvement in Cachexia
Several lines of evidence strongly implicate the hypothalamus as a critical site of action for inflammatory cytokines. Early lesioning studies confirmed an essential role for several hypothalamic nuclei in the regulation of energy homeostasis [53, 54]. Inflammation within neuronal tissue, as represented by markers for NF-κB activation, is localized exclusively to the hypothalamus and brainstem following systemic immunologic challenge [55]. These neurons and glia within the hypothalamus respond to inflammatory stimuli by initiating local production of cytokines in nuclei known to be involved in the regulation of ingestive behavior [56-59]. These findings point toward the hypothalamus as a unique site of inflammatory amplification within the CNS. Furthermore, feeding centers in the hypothalamus, including the paraventricular (PVN) and arcuate nuclei (ARC), express receptors for these cytokines [60-63]. The immediate early gene, c-fos, a marker of neuronal activation, is induced in the ARC and PVN in response to peripheral inflammatory stimuli, suggesting that cytokines may exert their cachexigenic effects by directly influencing the activity of hypothalamic centers controlling energy balance [64-66]. The relationship of acute inflammation-induced c-fos to the chronic cachexia syndrome is not clear, however these studies identify candidate inflammation-responsive loci in the CNS. It is important to note that feeding nuclei in the brainstem, such as the nucleus tractus solitarus (NTS), also show strong c-fos induction and NF-κB pathway activation in response to peripheral inflammatory challenges. Though these brainstem nuclei clearly play a significant role in the behavioral response to inflammation, this review will focus on hypothalamic mechanisms in cachexia.
The Role of Central Melanocortins in Cachexia
The ARC is one hypothalamic region that is essential in regulating energy homeostasis. This nucleus is linked with other hypothalamic and brainstem metabolic regulation centers by a complex web of reciprocal innervation [67]. The ARC contains two populations of neuropeptide-expressing neurons with opposing actions on energy balance. One population expresses the anorexigenic peptide, alpha-melanocyte-stimulating hormone (α-MSH), a cleavage product of the pro-opiomelanocortin (POMC) precursor, which is also expressed by brainstem neurons of the nucleus tractus solitarus (NTS) [68]. ARC POMC neurons are opposed by adjacent neurons expressing the orexigenic neuropeptides agouti-related protein (AgRP) and neuropeptide-Y (NPY) [69]. α-MSH derives its anorectic effects by binding to the type-3 melanocortin receptors (MC3-R) located on POMC neurons and within other areas of the hypothalamus, and the type-4 melanocortin receptors (MC4-R) distributed widely throughout the brain [68, 70-73]. AgRP neurons project to a majority of MC4-R expressing neurons through out the brain and act as an endogenous inverse agonist at the receptor [74]. Together, these populations of POMC- and AgRP-expressing neurons in the ARC and NTS make up the central melanocortin system. This neural network plays a critical role in regulating feeding behavior, linear growth, metabolic rate, and insulin sensitivity (recently reviewed in [75]). While the role of the MC3-R in energy balance remains somewhat obscure, a great deal is now known about the MC4-R. MC4-R is expressed in hypothalamic and brainstem feeding centers as well as on parvocellular neurons in the medial PVN, which project to brainstem nuclei controlling the activity of the sympathetic nervous system. Experimental administration of MC4-R agonists, such as α-MSH or the small molecule agonist, melanotan-II (MTII) results in a dramatic decrease in food intake coupled with an increase in energy expenditure, primarily as resting thermogenesis [76]. Conversely, administration of melanocortin antagonists leads to positive energy balance by increasing food consumption and reducing energy expenditure and physical activity [68, 77-79]. The relative activity of each of these neuronal populations is modulated by circulating signals of energy status as well as afferent neuronal input from the brainstem and other hypothalamic areas. In this way the central melanocortin system can orchestrate energy balance in accord with the metabolic demands of the organism.
The remarkably similar physiologic signature of increased melanocortin signaling and cachexia, specifically anorexia, increased metabolic rate, and alterations in tissue mass, suggest a potential role for the MC4-R in mediating some aspects of sickness behavior. Indeed, our own work and that of others has demonstrated that blockade of signaling through MC4-R by genetic and pharmacologic means prevents many of the features of cachexia that would normally occur during acute inflammation and in several models of chronic disease [17-19, 80, 81]. In our initial studies, we demonstrated that mice with genetic or pharmacologic blockade of melanocortin signaling (MC4-RKO and AgRP treated mice, respectively) had attenuated responses to non-specific inflammation (LPS) as well as to tumor growth (Figure 1). Similar results were found in tumor bearing rats treated with AgRP and in LPS-treated sheep treated with AgRP [19, 82]. Alternatively, analogues of the orexigenic hormone ghrelin, which derives its appetite stimulating effects at least partly through AgRP neuron activation, improve food intake and body weight in rat models of cancer cachexia and chronic renal failure [14, 15]. This has directly led to the development of drugs that dampen or block central melanocortin signaling that are currently being tested as therapeutics for cachexia. Although these observations provided an exciting therapeutic target, they left the mechanistic details of cytokine feedback to central melanocortin signaling largely unexplored.
Figure 1.
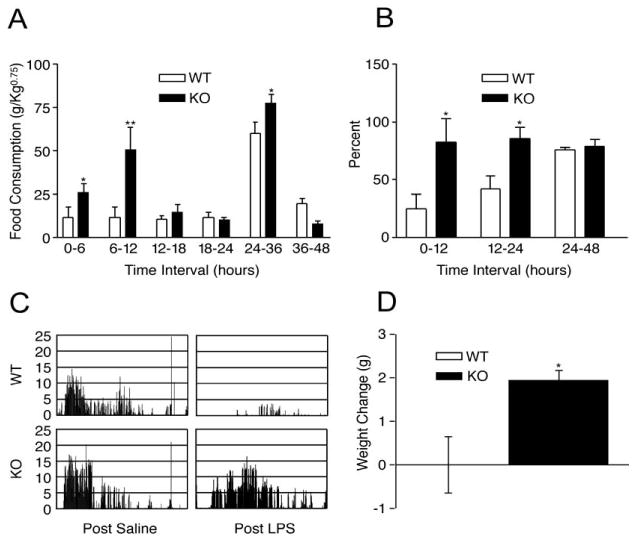
MC4-RKO mice are resistant to LPS-induced cachexia and illness behavior. LPS reduces food intake for approximately 36 h in WT mice, but not MC4-RKO mice, when expressed as a normalized value (A) or percent basal intake (B). LPS reduces wheel running activity below baseline in WT mice, but not MC4-RKO (C). Normal nocturnal increase in wheel running activity is maintained in MC4-RKO mice despite LPS-challenge. LPS induced growth failure in young WT mice, while young MC4-RKO mice are resistant (D). *, p < 0.05, ** p < 0.01 v. WT control. (Adapted with permission from Marks, D.L.; Ling, N.; and Cone, R.D. Role of the central melanocortin system in cachexia. Cancer Res, 2001. 61: 1432-8. Figure 2.)
Neuropeptide Y
Studies have also shown that NPY mRNA expression is diminished in several inflammatory states, providing an alternative mechanism for inflammation-induced negative energy balance (reviewed in [83]). Megestrol acetate is thought to derive its orexigenic effects by increasing NPY production, though this leads to a gain primarily in fat mass and little advantage in lean mass retention [10, 84]. Further, in contrast to AgRP administration, treatment of tumor bearing rats with i.c.v. NPY worsens their anorexia, suggesting that cachexia does not result from a selective reduction in NPY release [85]. Thus, the role of NPY in cachexia, if any, is unclear.
Neurohumoral Link
Though early ablation studies identified the hypothalamus as a critical regulator of energy balance [86, 87], it was not until parabiosis experiments between two spontaneously obese strains of mice, the obese (ob/ob) mouse and diabetic (db/db) mouse, that the existence of a neurohumoral link regulating food intake and adiposity was confirmed. These experiments found that ob/ob partners of db/db mice displayed fatal metabolic exhaustion, characterized by hypophagia, hypoglycemia, and weight loss, within four weeks of surgery [88]. This seminal experiment demonstrated that excess in a circulating satiety factor can subvert normal energy homeostasis and lead to a fatal wasting disorder. Subsequently the ob gene encoding this factor was cloned and the protein was named leptin from the Greek leptos, meaning thin [89]. When the leptin receptor (Lep-R) was cloned from the db locus, it was found that Lep-R was highly expressed in the hypothalamus, particularly in feeding and satiety centers [90]. This finding, when taken together with lesion studies, strongly suggested that the hypothalamus was the critical point for the integration of neural and humoral factors controlling energy balance. Subsequent studies have confirmed that the active isoform of Lep-R is expressed by POMC and AgRP neurons in the ARC, and that leptin specifically activates POMC neurons while inhibiting AgRP neurons [91-93]. These studies describe an elegant neurohumoral network that is capable of supporting normal energy homeostasis, while being susceptible to changes in circulating factors.
Though it is released by adipocytes in proportion to total body fat, leptin is a class I helical cytokine, like IL-6, LIF, and ciliary neurotrophic factor (CNTF). Therefore, leptin research offers potentially valuable insights into the mechanism of cytokine transfer into the CNS and its actions there. Leptin levels in the cerebrospinal fluid are strongly correlated to plasma leptin levels indicating that the molecule crosses the blood brain barrier (BBB) in proportion to its circulating concentration [94]. The ARC is located immediately adjacent to the median eminence, a circumventricular organ with an attenuated BBB. It has long been thought that this anatomic location allows leptin and other cytokines access to ARC neurons. Recent work confirms that the microvasculature in the mediobasal hypothalamus have a specialized fenestrated endothelium, which could explain the transfer of cytokines from the circulation to the CNS, where it could stimulate the local production of cytokines described above [95]. Brain endothelial cells also directly participate in the transfer of inflammatory signals into the CNS. The circulating satiety factors leptin and insulin show saturable transport into the CNS, suggesting that passive diffusion is not solely responsible for their actions in the brain (reviewed in [96]). Similarly, active transport of several inflammatory cytokines into the CNS, including IL-1β, IL-6, and TNF-α, and LIF has been described, and these cytokines facilitate the transfer of each other across the BBB [97-100].
Endothelial cells are further known to release cytokines and diffusible inflammatory signals, such as prostaglandins (PG) and nitric oxide (NO) into the CNS in response to circulating cytokines. These small molecules readily cross the BBB and are sufficient and under some conditions necessary to provoke several aspects of the physiological response to sickness [101], including the induction of cytokine expression, fever, and short-wave sleep (reviewed in [102]). Of particular interest is the synthesis of the prostanoid, PGE2 by endothelial cells and perivascular phagocytes in immune-sensitive brain nuclei in response to inflammation. PGE2 is necessary for the induction of fever by LPS [103], and may be necessary for the induction of anorexia by central or peripheral IL-1β [104]. PGE2 synthesis requires both arachadonic acid cleavage by the rate-limiting cyclooxygenase enzymes (COX-1 and COX-2) common to the production of all prostanoids, as well as microsomal prostaglandin E synthases (mPGES-1 and mPGES-2) [105]. Studies have shown that both COX-2 and mPGES-1 are coexpressed and induced by inflammatory stimuli [106, 107]. Recent work demonstrates that mPGES-1-KO mice exhibit attenuated c-fos induction by LPS in immunosensitive CNS nuclei [108] and are relatively resistant to anorexia in a murine cancer model [109]. Recent work by Serrats et al. has eloquently described the role for endothelial cells and perivascular macrophages in transducing inflammatory signals from the circulation to the CNS [110]. In inflammatory conditions, endothelial cell activation induces the production of PGE2 by perivascular macrophages, which is in turn necessary for the induction of the hypothalamic-pituitary-adrenal (HPA) response. However, this cascade does not appear to be responsible for the induction of fever or behavioral depression by circulating IL-1β or LPS. The HPA response can be blocked by inhibition of either COS-1 or COX-2, suggesting that both enzymes may be necessary to mediate HPA activation by LPS [110, 111]. Together these data support a vital role of prostaglandins, particularly PGE2, in relaying and amplifying inflammatory information in the CNS. Though key mediators, the central mechanisms of prostaglandin action remain poorly understood. Specifically, it is unclear whether these molecules directly influence neuronal activation or whether they only act to transmit the inflammatory messages. Interestingly, in cachectic patients COX inhibition has shown limited effectiveness at ameliorating anorexia or lethargy, indicating that either this mechanism is not conserved in humans or that COX inhibition alone is insufficient to block transmission of the inflammatory signals into the CNS. Certainly, more research investigating the mechanism of central prostaglandin signaling and its relevance to sickness behavior is warranted.
Several studies have examined a role for the vagus nerve in transducing inflammatory signals from the periphery. Blockade of endothelial IL-1R signaling prevents the induction of fever or lethargy by IL-1β administered centrally or intravenously, but not intraperitoneally, implicating the vagus in transducing inflammatory signals from the viscera to the CNS [112]. Subdiaphragmatic vagotomy attenuates IL-1β expression and c-fos induction in the hypothalamus, brainstem and amygdala resulting from immune challenge, though treated animals still display significantly different c-fos expression and cytokine expression compared to controls [113, 114]. The role of the vagus in mediating the sickness response is controversial, as vagotomy has been shown to attenuate behavioral depression [114-116] and anorexia in some studies [117], while others report that vagotomy does not block the anorectic component of sickness behavior [118]. Given the different doses and routes of administration used in these studes, the vagus appears to be more important in mediating the behavioral response to small doses of immune modulating agents delivered i.p. The abdominal vagus has a unique relationship with adjacent immune cells, increasing its discharge rate in response to locally released cytokines. This leads to altered firing of excitatory catecholeminergic neurons originating in the NTS and projecting to several hypothalamic nuclei [119, 120]. These studies reinforce the importance of brainstem participation in the development of sickness behavior and hypothalamic inflammation. How subdiaphragmatic vagotomy alters CNS cytokine expression in response to inflammation remains unclear, but it may remove an essential co-factor for cytokine production by neural cells. Unfortunately, no anatomic studies have been performed to identify whether cytokine expression in specific brain areas are uniquely affected by vagotomy, so the influence of ascending autonomic tone on the hypothalamus is not well defined. Collectively, these data illustrate the complex exchange from systemic inflammation to cytokine induction in the brain and further support a critical role for hypothalamic inflammation in the development of sickness behavior.
Interaction of Cytokines with the Central Melanocortin System
While the primary cytokine signals producing cachexia during illness have been studied in detail, we have limited understanding of the specific hypothalamic cell groups involved in processing these signals. The success of melanocortin antagonism in ameliorating the catabolic features induced by several disease models, suggests that POMC and AgRP neurons are plausible targets for CNS cytokine action. This idea is supported by data showing that ARC POMC mRNA was increased following peripheral LPS and TNF-α while AgRP mRNA expression was simultaneously decreased by TNF-α [121, 122]. Surprisingly, as recently as 2005, very little additional research had investigated whether POMC and AgRP neurons were directly influenced by pro-inflammatory cytokines. To elucidate the mechanism whereby inflammation may lead to excessive activation of central melanocortin signaling, we undertook a series of studies designed to demonstrate activation of POMC neurons by pro-inflammatory cytokines.
IL-1β
Perhaps the most obvious target to investigate is the “prototypic” pro-inflammatory cytokine, IL-1β. In a healthy brain, trace expression of IL-1β is detected in glia, neurons, cerebrovascular cells and circulating immune cells [reviewed in [123]]. Expression of IL-1β in the brain, mainly in the hypothalamus, is markedly increased in response to disease and inflammation both in the periphery and within the CNS itself [124-127]. Inflammation-induced brain IL-1β is primarily produced by microglial cells and perivascular and meningeal marcrophages [128]. Two isoforms of IL-1, IL-1α and IL-1β, act as an agonist at the IL-1 receptor (IL-1RI), but only IL-1β is cleaved and secreted while IL-1α remains mainly intracellular [129, 130]. Another member of the IL-1 family, the IL-1 receptor antagonist (IL-1ra), binds the IL-1RI, but does not initiate signaling [131].
Binding of IL-1β to IL-1RI increases cellular production and nuclear translocation of NF-κB, a transcription factor that that plays key role in regulating the cellular response to inflammation [132]. In the brain, IL-1RI is expressed primarily by non-neuronal cells including endothelial cells, glial cells, and macrophages [133]. Only a few neuronal cell groups express IL-1RI, however these groups are predominately found in areas associated with energy homeostasis and behavior including the ARC, NTS, area postrema, basolateral nucleus of the amygdala, hippocampus, and the trigeminal and hypoglossal motor nuclei [133, 134]. Expression of IL-1RI in the hypothalamus and hindbrain is increased by acute inflammation induced by single i.c.v injections of LPS [135], or IL-1β [136] and by chronic inflammation due to tumor growth [127].
The first described physiological role for IL-1β in the brain was the induction of fever, [137] by regulating the activity of thermosensitive neurons in the anterior and preoptic hypthalamus [138]. IL-1β-induced fever is attenuated by salicylates and non-steroidal anti-inflammatory drugs that prevent PG synthesis [139]. IL-1β also activates the hypothalamic-pituitary-adrenal (HPA) axis leading to elevated plasma levels of ACTH [140]. IL-1β has an increasingly recognized role in the regulation of feeding behavior and energy homeostasis. IL-1β potently suppresses food intake when injected peripherally and centrally [141, 142] and unlike IL-1β-mediated fever, the anorexic effect of IL-1β is not completely blocked by inhibition of PG synthesis [143, 144]. Further, administration of IL-1ra into the lateral ventricle abrogates sickness behavior induced by peripheral IL-1β without affecting the febrile response [142]. In the ARC, IL-1β induces cFos immunoreactivity in both POMC and NPY neurons, suggesting a role for the hypothalamic melanocortin system in mediating the feeding effects and metabolic effects of IL-1β [145].
In recently published work, our laboratory investigated the nature of the relationship between IL-1β and the central melanocortin system. Our studies demonstrate that ARC POMC and AgRP neurons both express IL-1RI (Figure 2A). Central IL-1β administration induces cFos immunoreactivity in both neuronal types in the ARC, and this activation was only modestly attenuated by PG synthesis inhibition. Brainstem POMC neurons, however, do not appear to be specifically activated by i.c.v. IL-1β. Activation of ARC POMC neurons was confirmed by whole-cell slice electrophysiology, which measured a dramatic increase in POMC neuron firing rate in response to IL-1β. We found that IL-1β also increased the release of α-MSH from hypothalamic explants in vitro, and that this effect was blunted, but not reversed by inhibiting PG synthesis [146]. IL-1β was found to significantly reduce AgRP release from hypothalami in vitro. Surprisingly, rat models of peripheral inflammation, including LPS, tumor, and chronic renal failure all increased AgRP mRNA expression above control, though i.c.v. IL-1β did not [147]. The in vitro and electrophysiology data strongly suggest that IL-1β, like leptin, increases central melanocortin tone in the ARC by increasing the ratio of α-MSH: AgRP available at the post-synaptic MC4-R. The paradoxically increased transcription of AgRP in inflammatory states may serve to prepare the animal for rebound hyperphagia, to recover energy stores lost during the acute illness. Taken together, these data demonstrate the existence of a neuroanatomic framework capable of supporting direct cytokine regulation of POMC and AgRP activity. These data conflict with a previous study, which showed worsened hypophagia following ablation of the medial ARC and suggested that the role of the ARC in cachexia may be to limit the extent of the anorexia [145]. These ablation studies killed off the entire AgRP population as well as the medial POMC population, but lateral POMC neurons, which are thought to be more important in the control of body weight and feeding, were spared [148, 149]. Therefore, the worsened anorexia reported by this group may be explained by unopposed α-MSH at the MC4-R.
Figure 2.
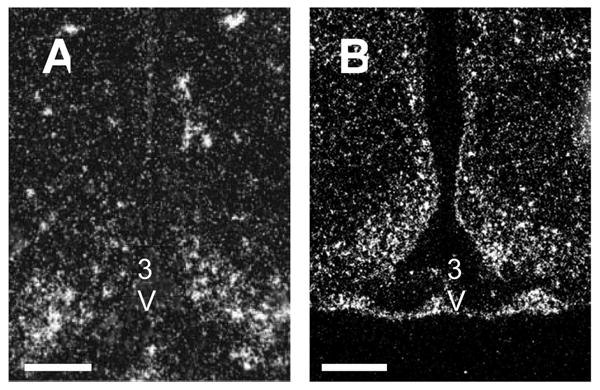
Pro-inflammatory cytokine receptors are densely expressed in the ARC. A, Photomicrograph showing IL-1RI expression in rat hypothalamus. Expression is densest in ARC. B, Photomicrograph of LIF-R expression in rat hypothalamus. Expresssion of LIF-R exactly defines the borders of the ARC. 50X. Scale bar, 100 μm. 3V, Third ventricle. (Adapted with permission from Scarlett, J.M.; Jobst, E.E.; Enriori, P.J.; Bowe, D.D.; Batra, A.K.; Grant, W.F.; Cowley, M.A.; and Marks, D.L. Regulation of central melanocortin signaling by interleukin-1 beta. Endocrinology, 2007. 148: 4217-25. Figure 7A. and Grossberg, A.J.; Scarlett, J.M.; Zhu, X.; Bowe, D.D.; Batra, A.K.; Braun, T.P.; and Marks, D.L. Arcuate Nucleus Proopiomelanocortin Neurons Mediate the Acute Anorectic Actions of Leukemia Inhibitory Factor via gp130. Endocrinology, 2010. 151. Figure 4A.)
It is well established that central administration of IL-1β initiates a cachexia-like metabolic phenotype, which is reversed by melanocortin antagonism. Though we demonstrated that IL-1β increases melanocortin signaling by acting directly on POMC and AgRP neurons, the relative contribution of this mechanism in disease states remains uncertain. However the low EC50 for IL-1β-mediated changes in neuropeptide release and the anatomic location of IL-1β expression in inflammatory states, suggest that this mechanism is likely to play a significant role. Cachexia becomes overtly maladaptive only when wasting persists as in late-stage chronic disease. Though IL-1β potently induces acute anorexia, animals rapidly desensitize to continuous i.c.v. administration of the cytokine [150]. Therefore, IL-1β may not be sufficient on its own to produce sustained catabolism. For this reason, we have begun to investigate the contribution of other pro-inflammatory cytokines, which may contribute to the persistent wasting syndrome.
LIF
LIF was initially cloned and named for its ability to induce differentiation of the murine leukemic M1 cell line and has subsequently been found to play an important role in inflammatory signaling, neuro-immune function, and development [151]. Depending on alternative splicing, LIF is synthesized as either a secreted protein or a matrix-associated protein from a single gene locus [152]. LIF is variably glycosylated, which can directly influence the bioactivity of the molecule in a cell-specific manner [153, 154]. The effects of LIF further depend on the cell type and developmental stage of the organism. In the developing nervous system, LIF is known to be neuropoietic and involved in the differentiation of autonomic nerves and astrocytes in several brain locations, suggesting that its receptors may be expressed in the adult CNS. In the adult animal, LIF is thought to be involved predominately in the inflammatory response to tissue damage or infection (reviewed in [155]).
LIF binds to a heterodimeric receptor complex consisting of the ligand-specific LIF receptor (LIF-R) and the signal transducing gp130 subunit, which is common to all IL-6 family cytokines, including IL-6, CNTF, and oncostatin-M (reviewed in [156]). Receptor binding stimulates gp130-mediated activation of the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) pathway [157], the same pathway activated by the binding of leptin to its receptor [158]. Importantly, STAT3 activation by any of these cytokines induces transcription of an autoregulatory gene product, suppressor of cytokine signaling-3 (SOCS-3), which inhibits further JAK/STAT signaling in response to ligand binding and is thought to play a role in leptin resistance [159].
Previous studies linking leptin-induced STAT3 phosphorylation to its anorectic effects suggest that IL-6 family cytokines could also be used to inhibit feeding [160]. This hypothesis is supported by the observation that activators of gp130 can acutely reduce food intake when injected in the CNS [37]. Though there is consensus on the anorectic activity of LIF and CNTF in the brain, the ability to IL-6 to induce negative energy balance remains somewhat controversial [150]. An elegant study using mice that have gp130 specifically deleted from only POMC neurons found that the anorectic effects of CNTF specifically depended on expression of gp130 on POMC neurons, indicating that this may be a common pathway for IL-6 family cytokine-induced anorexia [161]. A series of experiments exploring the potential role of chronic gp130 activation using CNS CNTF or LIF demonstrate that both of these cytokines are capable of sustaining negative energy balance in animals, in contrast with similar studies using chronic leptin administration [162]. These studies suggest that despite initiating similar signaling cascades, gp130 activation must fundamentally differ from Lep-R signaling. A recent study indicates that differential activation of phospo-tyrosine phosphatase 1B (PTP1B), an autoihibitory phosphatase that is induced in neurons by leptin, but not by gp130 activation may underlie these observed differences in sustained anorectic efficacy [163]. A role for PTP1B in cachexia is further supported by research demonstrating that its expression is diminished in tumor bearing rats and that silencing of the gene in the brain using antisense oligonucleotides can cause fatal anorexia [164].
Serum LIF is elevated in chronic disease and malignancy [165-167], an observation that has been correlated to poor prognosis [168]. Hypothalamic expression of LIF is induced by acute LPS administration [32, 48, 51], and, in contrast to IL-1β or LPS, animals do not desensitize to chronic LIF-induced anorexia [171-173]. This lack of tachyphylaxis more closely resembles clinical cachexia, indicating that LIF could be an essential CNS mediator of chronic inflammation-induced anorexia. Studies by Shlomo Melmed demonstrate that LIF expression in the pituitary is necessary to drive the increased POMC mRNA expression and ACTH release by pituitary corticotrophs in response to inflammation [32, 174]. This process is dependent on gp130-mediated activation of JAK2/STAT3 signaling, which is known to underlie the induction of POMC mRNA expression by leptin, suggesting that LIF may activate hypothalamic POMC neurons in a similar manner [160, 175]. Indeed, our laboratory recently demonstrated that LIF induces anorexia by directly activating ARC POMC neurons [51]. We observed that acute i.c.v. LIF injection activates ARC POMC neurons, but brainstem POMC neurons were not activated [51], despite significant activation of neighboring cells and widespread expression of the LIF-R throughout the brainstem (including the NTS) [unpublished data]. ARC POMC neurons also express the LIF-R (Figure 2B) and hypothalamic explants increase α-MSH release in response to LIF. When the gp130 subunit is deleted specifically from POMC neurons, the ability of LIF to induce α-MSH release and inhibit food intake is completely abolished, indicating that the acute anorectic effects of LIF are mediated by direct activation of gp130 signaling in POMC neurons. Importantly, we found that though LIF and IL-6 expression were increased in the hypothalami of LPS or IL-1β treated animals, CNTF expression was actively inhibited by inflammation, suggesting that this cytokine is not involved in inflammation-induced anorexia. We did not observe any detectable IL-6 receptor mRNA expression by POMC neurons, strongly supporting a unique role for LIF in activating gp130 signaling on POMC neurons in inflammatory states. This is the first study that directly implicates inflammation-induced changes in melanocortin signaling with the anorectic component of cachexia and provides mechanistic support for therapeutic use of melanocortin antagonists. However, many questions remain regarding the mechanistic details of cytokine-induced alterations in melanocortinergic neuron activity. For example, a large number of cells in the ARC express LIF-R but not POMC. Other cell types, such as AgRP neurons and glia, may be influenced by CNS LIF signaling and contribute to its effects on energy homeostasis. Further, it is uncertain whether the acute mechanisms of cytokine action described above are important in the chronic disease process, and studies looking into this are vital to establish an effective therapeutic strategy for melanocortin antagonism.
Future Directions
Collectively, the studies presented in this review describe a common model for sickness behavior in which peripheral inflammation stimulates the transport and local production of pro-inflammatory cytokines within the hypothalamus and brainstem. These cytokines then directly and indirectly alter the activity of neuronal populations involved in regulating feeding behavior and energy expenditure (Figure 3). It has not been established, however, whether CNS inflammation may play a direct role in other salient features of the sickness response.
Figure 3.

Hypothetical model for the regulation of feeding, metabolic rate, and sickness behavior by hypothalamic cytokine signaling. In chronic disease elevated circulating cytokines stimulate the production of pro-inflammatory cytokines, including IL-1β and LIF, in the hypothalamus. These molecules then increase melanocortin signaling by directly activating POMC neurons and inhibiting AgRP neurons. This leads to decreased appetite and increased energy expenditure, primarily as basal metabolic rate, by increasing activation of MC4-R. Other elements of sickness behavior, including insulin resistance, lethargy, anhedonia, and reproductive axis failure may also result from inflammation-induced changes in melanocortin signaling
Insulin Resistance
A key feature common to cachexia induced by several disease etiologies is peripheral insulin resistance [20]. In patients with chronic heart failure, the degree of heart failure is correlated to the degree of insulin resistance [176]. Further, insulin resistance is an independent risk factor for mortality in these patients [177]. The prevalence of insulin resistance in cachectic patients suggests a common pathophysiology. Recent work has shown that intact central melanocortin signaling is necessary for normal insulin sensitivity and glucose homeostasis [178]. In obese animals, insufficient melanocortin signaling secondary to leptin resistance is often cited as the major CNS cause of insulin resistance [179]. The role of the melanocortin system in energy partitioning and utilization is further supported by a series of recent experiments that demonstrate changes in melanocortin tone alter glucose utilization and lipolysis via sympathetic nervous system [180, 181]. Therefore, it is plausible that altered melanocortin signaling secondary to cytokine influence may directly impact insulin sensitivity and energy partitioning. Given the overlap in receptor-activated signaling cascades between leptin and gp130-activating cytokines, one possible mechanism may involve cytokine induction of SOCS-3, which could inhibit leptin signaling [182]. Because insulin resistance in cachexia has been directly linked to increased mortality, further research on this mechanism is warranted.
Anhedonia
Patients suffering from major depression exhibit a constellation of symptoms that is remarkably similar to sickness behavior, including significant weight loss, sleep disturbances, psychomotor disturbances, fatigue, and anhedonia. Elevated IL-6 and acute phase proteins have been observed in plasma from depressed patients [183]. Further, non-depressed patients treated with IL-2 or interferon alpha, two pro-inflammatory cytokines, develop a depressed mood and altered cognition, which may be successfully treated with anti-depressant pharmacotherapy [184, 185]. This relationship is supported by animal studies of depression, which have demonstrated that cytokine treatment can induce anhedonia in rats, measured by decreases in rewarding self-stimulation and reduced saccharine preference [186, 187]. Notably, i.c.v. IL-1ra can attenuate learned helplessness behavior, another hallmark of animal depression models [188]. A potential role for melanocortins is suggested by the existence of an anatomical link between MC4-R-expressing neurons and nuclei implicated in mediating depressive behavior, including brainstem serotoninergic and noradrenergic centers, as well as ventral tegmental reward pathways [189-193].
Lethargy
Lethargy, presenting as decreased voluntary activity and motivation, is a commonly observed component of the sickness response. In severely cachectic patients, the etiology of lethargy may be complicated by debilitation and weakness resulting from extended inflammatory response and negative energy balance. However, acute inflammatory states consistently induce significant reductions in spontaneous activity and motivated behaviors long before an appreciable energy deficit or significant weakness can accumulate. For example, endotoxin-treated rats stop bar pressing to receive water and exhibit decreased water intake when freely available [194]. Also, both wheel running activity and spontaneous home cage movement are reduced in LPS-treated animals [195]. Recent research has linked control of locomotor activity to central melanocortin activation. MC4-R null mice are resistant to this inflammation-induced decrease in wheel running, suggesting that melanocortins may mediate this response [18]. Recently, intact leptin signaling in both POMC and AgRP neurons has been associated with controlling locomotor activity to the extent that it can significantly alter total energy homeostasis [196, 197]. These findings indicate that pro-inflmmatory cytokines may have similar actions on these neurons. Melanocortin neurons are also known to communicate with arousal and motivation centers in the hypothalamus and nucleus accumbens, providing a neuroanatomic basis for this relationship. Little research investigating these relationships in inflammatory states has been conducted, however.
Reproductive Axis Failure
Gonadal hormone release and reproductive viability are regulated by neuronal populations in the hypothalamus. Hypogonadotropic hypogonadism and reproductive failure have been observed in a variety of inflammatory conditions including lupus erythematosus, rheumatoid arthritis, type 2 diabetes mellitus, and acute infection [198-201]. Because reproduction is energetically very expensive for an organism, suppression of the hypothalamic-pituitary-gonadal axis makes teleologic sense during acute immune challenges, while the body is actively fighting off infection. However, several chronic inflammatory conditions similarly suppress the reproductive axis, leading to infertility and delayed onset of puberty. In rats, i.c.v., but not peripheral administration of IL-1β completely blocks induction of hypothalamic gonadotropin secretion, suggesting that IL-1 suppresses hypothalamic-pituitary-gonadal function by inhibiting gondadotropin releasing hormone neurons [202]. These observations indicate that pro-inflammatory cytokines in the brain may play a role in mediating the neuroendocrine response to disease, though little additional research investigating this relationship has been reported. The reproductive axis is known to be tightly associated with energy status, and leptin levels as well as signaling through the MC4-R regulate hypothalamic control of gonadotropin release [203, 204]. Taken together, these findings again suggest that pro-inflammatory cytokines may suppress the reproductive axis by inhibiting hypothalamic gonadotropin release through melanocortin-mediated pathways.
Conclusions
We have shown that cytokines liberated during the disease process activate pro-opiomelanocortin (POMC) neurons by both direct and indirect mechanisms [51, 146]. At the same time, we have shown that the release of the endogenous antagonist/inverse agonist agouti-related peptide (AgRP) is diminished by inflammation, further potentiating activation of downstream melanocortin pathways [147]. We hypothesize that inflammation alters the function of these neurons and that this will in turn produce various aspects of the illness response including anorexia, decreased movement, elevated basal metabolic rate, lethargy, and alterations in energy partitioning. We further hypothesize that this cellular mechanism represents a critical common pathway for the production of cachexia in a variety of chronic disease states, particularly those in which inflammation is known to play an important role (Figure 3). All of these constituents of the sickness response are sensitive to nutritional status, and neuronal populations responsible for maintaining these functions are known to have extensive hypothalamic connections. Given that the ARC functions as a metabolic integration point and that melanocortin antagonism has been successful in ameliorating multiple facets of the behavioral response to inflammation, it is reasonable to consider that melanocortinergic neurons may be critical in the pathogenesis of several behavioral responses to sickness. Further studies are needed to determine whether these pro-inflammatory cytokine-induced changes in melanocortin signaling are essential in mediating sickness behavior, as a means to develop integrative, targeted therapies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hart BL. The behavior of sick animals. Vet Clin North Am Small Anim Pract. 1991;21:225–37. doi: 10.1016/s0195-5616(91)50028-0. [DOI] [PubMed] [Google Scholar]
- 2.Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–37. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- 3.Kluger MJ, Kozak W, Conn CA, Leon LR, Soszynski D. The adaptive value of fever. Infect Dis Clin North Am. 1996;10:1–20. doi: 10.1016/s0891-5520(05)70282-8. [DOI] [PubMed] [Google Scholar]
- 4.Kluger MJ. The evolution and adaptive value of fever. Am Sci. 1978;66:38–43. [PubMed] [Google Scholar]
- 5.Ganz T. Iron in innate immunity: starve the invaders. Curr Opin Immunol. 2009;21:63–7. doi: 10.1016/j.coi.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murray MJ, Murray AB. Anorexia of infection as a mechanism of host defense. Am J Clin Nutr. 1979;32:593–6. doi: 10.1093/ajcn/32.3.593. [DOI] [PubMed] [Google Scholar]
- 7.Katz AM, Katz PB. Diseases of the heart in the works of Hippokrates. Br Heart J. 1962;24:257–264. doi: 10.1136/hrt.24.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tisdale MJ. Biology of cachexia. J Natl Cancer Inst. 1997;89:1763–73. doi: 10.1093/jnci/89.23.1763. [DOI] [PubMed] [Google Scholar]
- 9.Larkin M. Thwarting the dwindling progression of cachexia. Lancet. 1998;351:1336. doi: 10.1016/S0140-6736(05)79068-1. [DOI] [PubMed] [Google Scholar]
- 10.Loprinzi CL, Schaid DJ, Dose AM, Burnham NL, Jensen MD. Body-composition changes in patients who gain weight while receiving megestrol acetate. J Clin Oncol. 1993;11:152–4. doi: 10.1200/JCO.1993.11.1.152. [DOI] [PubMed] [Google Scholar]
- 11.Carroll PV. Treatment with growth hormone and insulin-like growth factor-I in critical illness. Best Pract Res Clin Endocrinol Metab. 2001;15:435–51. doi: 10.1053/beem.2001.0162. [DOI] [PubMed] [Google Scholar]
- 12.Della Cuna GR, Pellegrini A, Piazzi M. Effect of methylprednisolone sodium succinate on quality of life in preterminal cancer patients: a placebo-controlled, multicenter study. The Methylprednisolone Preterminal Cancer Study Group. Eur J Cancer Clin Oncol. 1989;25:1817–21. doi: 10.1016/0277-5379(89)90353-2. [DOI] [PubMed] [Google Scholar]
- 13.Crown AL, Cottle K, Lightman SL, Falk S, Mohamed-Ali V, Armstrong L, Millar AB, Holly JM. What is the role of the insulin-like growth factor system in the pathophysiology of cancer cachexia, and how is it regulated? Clin Endocrinol (Oxf) 2002;56:723–33. doi: 10.1046/j.1365-2265.2002.01540.x. [DOI] [PubMed] [Google Scholar]
- 14.Deboer MD, Zhu X, Levasseur PR, Inui A, Hu Z, Han G, Mitch WE, Taylor JE, Halem HA, Dong JZ, Datta R, Culler MD, Marks DL. Ghrelin treatment of chronic kidney disease: improvements in lean body mass and cytokine profile. Endocrinology. 2008;149:827–35. doi: 10.1210/en.2007-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeBoer MD, Zhu XX, Levasseur P, Meguid MM, Suzuki S, Inui A, Taylor JE, Halem HA, Dong JZ, Datta R, Culler MD, Marks DL. Ghrelin treatment causes increased food intake and retention of lean body mass in a rat model of cancer cachexia. Endocrinology. 2007;148:3004–12. doi: 10.1210/en.2007-0016. [DOI] [PubMed] [Google Scholar]
- 16.Garcia JM, Polvino WJ. Effect on body weight and safety of RC-1291, a novel, orally available ghrelin mimetic and growth hormone secretagogue: results of a phase I, randomized, placebo-controlled, multiple-dose study in healthy volunteers. Oncologist. 2007;12:594–600. doi: 10.1634/theoncologist.12-5-594. [DOI] [PubMed] [Google Scholar]
- 17.Markison S, Foster AC, Chen C, Brookhart GB, Hesse A, Hoare SR, Fleck BA, Brown BT, Marks DL. The regulation of feeding and metabolic rate and the prevention of murine cancer cachexia with a small-molecule melanocortin-4 receptor antagonist. Endocrinology. 2005;146:2766–73. doi: 10.1210/en.2005-0142. [DOI] [PubMed] [Google Scholar]
- 18.Marks DL, Ling N, Cone RD. Role of the central melanocortin system in cachexia. Cancer Res. 2001;61:1432–8. [PubMed] [Google Scholar]
- 19.Wisse BE, Frayo RS, Schwartz MW, Cummings DE. Reversal of cancer anorexia by blockade of central melanocortin receptors in rats. Endocrinology. 2001;142:3292–301. doi: 10.1210/endo.142.8.8324. [DOI] [PubMed] [Google Scholar]
- 20.Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H, Mantovani G, Marks D, Mitch WE, Muscaritoli M, Najand A, Ponikowski P, Rossi Fanelli F, Schambelan M, Schols A, Schuster M, Thomas D, Wolfe R, Anker SD. Cachexia: a new definition. Clin Nutr. 2008;27:793–9. doi: 10.1016/j.clnu.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Cavaillon JM, Haeffner-Cavaillon N. Signals involved in interleukin 1 synthesis and release by lipopolysaccharide-stimulated monocytes/macrophages. Cytokine. 1990;2:313–29. doi: 10.1016/1043-4666(90)90061-w. [DOI] [PubMed] [Google Scholar]
- 22.Hillhouse EW, Mosley K. Peripheral endotoxin induces hypothalamic immunoreactive interleukin-1 beta in the rat. Br J Pharmacol. 1993;109:289–90. doi: 10.1111/j.1476-5381.1993.tb13567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Dam AM, Bauer J, Tilders FJ, Berkenbosch F. Endotoxin-induced appearance of immunoreactive interleukin-1 beta in ramified microglia in rat brain: a light and electron microscopic study. Neuroscience. 1995;65:815–26. doi: 10.1016/0306-4522(94)00549-k. [DOI] [PubMed] [Google Scholar]
- 24.van Dam AM, Poole S, Schultzberg M, Zavala F, Tilders FJ. Effects of peripheral administration of LPS on the expression of immunoreactive interleukin-1 alpha, beta, and receptor antagonist in rat brain. Ann N Y Acad Sci. 1998;840:128–38. doi: 10.1111/j.1749-6632.1998.tb09557.x. [DOI] [PubMed] [Google Scholar]
- 25.Baile CA, Naylor J, McLaughlin CL, Catanzaro CA. Endotoxin-elicited fever and anorexia and elfazepam-stimulated feeding in sheep. Physiol Behav. 1981;27:271–7. doi: 10.1016/0031-9384(81)90269-9. [DOI] [PubMed] [Google Scholar]
- 26.Plata-Salamán CR, Ilyin SE, Gayle D, Flynn MC. Gram-negative and gram-positive bacterial products induce differential cytokine profiles in the brain: analysis using an integrative molecular-behavioral in vivo model. Int J Mol Med. 1998;1:387–97. doi: 10.3892/ijmm.1.2.387. [DOI] [PubMed] [Google Scholar]
- 27.Anker SD, Ponikowski PP, Clark AL, Leyva F, Rauchhaus M, Kemp M, Teixeira MM, Hellewell PG, Hooper J, Poole-Wilson PA, Coats AJ. Cytokines and neurohormones relating to body composition alterations in the wasting syndrome of chronic heart failure. Eur Heart J. 1999;20:683–93. doi: 10.1053/euhj.1998.1446. [DOI] [PubMed] [Google Scholar]
- 28.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 29.Deans C, Wigmore SJ. Systemic inflammation, cachexia and prognosis in patients with cancer. Curr Opin Clin Nutr Metab Care. 2005;8:265–9. doi: 10.1097/01.mco.0000165004.93707.88. [DOI] [PubMed] [Google Scholar]
- 30.Scott HR, McMillan DC, Crilly A, McArdle CS, Milroy R. The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br J Cancer. 1996;73:1560–2. doi: 10.1038/bjc.1996.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plata-Salaman CR, Sonti G, Borkoski JP, Wilson CD, Ffrench-Mullen JMH. Anorexia induced by chronic central administration of cytokines at estimated pathophysiological concentrations. Phys Behav. 1996;60:867–875. [PubMed] [Google Scholar]
- 32.Chesnokova V, Melmed S. Leukemia inhibitory factor mediates the hypothalamic pituitary adrenal axis response to inflammation. Endocrinology. 2000;141:4032–40. doi: 10.1210/endo.141.11.7778. [DOI] [PubMed] [Google Scholar]
- 33.Bendtzen K, Baek L, Berild D, Hasselbach H, Dinarello CA, Wolff SM. Demonstration of circulating leukocytic pyrogen/interleukin-1 during fever. N Engl J Med. 1984;310:596. doi: 10.1056/NEJM198403013100915. [DOI] [PubMed] [Google Scholar]
- 34.Krueger JM, Fang J, Taishi P, Chen Z, Kushikata T, Gardi J. Sleep. A physiologic role for IL-1 beta and TNF-alpha. Ann N Y Acad Sci. 1998;856:148–59. doi: 10.1111/j.1749-6632.1998.tb08323.x. [DOI] [PubMed] [Google Scholar]
- 35.Irwin MR, Miller AH. Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun. 2007;21:374–83. doi: 10.1016/j.bbi.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Sonti G, Ilyin SE, Plata-Salaman CR. Anorexia induced by cytokine interactions at pathophysiological concentrations. Am J Physiol. 1996;270:R1394–402. doi: 10.1152/ajpregu.1996.270.6.R1394. [DOI] [PubMed] [Google Scholar]
- 37.Plata-Salaman CR. Anorexia induced by activators of the signal transducer gp 130. Neuroreport. 1996;7:841–4. doi: 10.1097/00001756-199602290-00038. [DOI] [PubMed] [Google Scholar]
- 38.Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol. 2003;5:87–90. doi: 10.1038/ncb0203-87. [DOI] [PubMed] [Google Scholar]
- 39.Langen RC, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-kappaB. Faseb J. 2001;15:1169–80. doi: 10.1096/fj.00-0463. [DOI] [PubMed] [Google Scholar]
- 40.Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–98. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 41.Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS., Jr NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–6. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- 42.Baracos V, Rodemann HP, Dinarello CA, Goldberg AL. Stimulation of muscle protein degradation and prostaglandin E2 release by leukocytic pyrogen (interleukin-1). A mechanism for the increased degradation of muscle proteins during fever. N Engl J Med. 1983;308:553–8. doi: 10.1056/NEJM198303103081002. [DOI] [PubMed] [Google Scholar]
- 43.Chida D, Osaka T, Hashimoto O, Iwakura Y. Combined interleukin-6 and interleukin-1 deficiency causes obesity in young mice. Diabetes. 2006;55:971–7. doi: 10.2337/diabetes.55.04.06.db05-1250. [DOI] [PubMed] [Google Scholar]
- 44.Garcia MC, Wernstedt I, Berndtsson A, Enge M, Bell M, Hultgren O, Horn M, Ahren B, Enerback S, Ohlsson C, Wallenius V, Jansson JO. Mature-onset obesity in interleukin-1 receptor I knockout mice. Diabetes. 2006;55:1205–13. doi: 10.2337/db05-1304. [DOI] [PubMed] [Google Scholar]
- 45.Somm E, Henrichot E, Pernin A, Juge-Aubry CE, Muzzin P, Dayer JM, Nicklin MJ, Meier CA. Decreased fat mass in interleukin-1 receptor antagonist-deficient mice: impact on adipogenesis, food intake, and energy expenditure. Diabetes. 2005;54:3503–9. doi: 10.2337/diabetes.54.12.3503. [DOI] [PubMed] [Google Scholar]
- 46.Wallenius V, Wallenius K, Ahren B, Rudling M, Carlsten H, Dickson SL, Ohlsson C, Jansson JO. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8:75–9. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]
- 47.Plata-Salaman CR. Immunoregulators in the nervous system. Neurosci Biobehav Rev. 1991;15:185–215. doi: 10.1016/s0149-7634(05)80001-6. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, Ren SG, Melmed S. Hypothalamic and pituitary leukemia inhibitory factor gene expression in vivo: a novel endotoxin-inducible neuro-endocrine interface. Endocrinology. 1996;137:2947–53. doi: 10.1210/endo.137.7.8770918. [DOI] [PubMed] [Google Scholar]
- 49.Dinarello CA. The proinflammatory cytokines interleukin-1 and tumor necrosis factor and treatment of the septic shock syndrome. J Infect Dis. 1991;163:1177–84. doi: 10.1093/infdis/163.6.1177. [DOI] [PubMed] [Google Scholar]
- 50.Turrin NP, Plata-Salaman CR. Cytokine-cytokine interactions and the brain. Brain Res Bull. 2000;51:3–9. doi: 10.1016/s0361-9230(99)00203-8. [DOI] [PubMed] [Google Scholar]
- 51.Grossberg AJ, Scarlett JM, Zhu X, Bowe DD, Batra AK, Braun TP, Marks DL. Arcuate Nucleus Proopiomelanocortin Neurons Mediate the Acute Anorectic Actions of Leukemia Inhibitory Factor via gp130. Endocrinology. 2009 doi: 10.1210/en.2009-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wisse BE, Ogimoto K, Tang J, Harris MK, Jr, Raines EW, Schwartz MW. Evidence that lipopolysaccharide-induced anorexia depends upon central, rather than peripheral, inflammatory signals. Endocrinology. 2007;148:5230–7. doi: 10.1210/en.2007-0394. [DOI] [PubMed] [Google Scholar]
- 53.Broback JR, Tepperman J, Long CNH. Experimental hypothalamic hyperphagia in the albino rat. Yale Journal of Biological Medicine. 1943;15:831–853. [PMC free article] [PubMed] [Google Scholar]
- 54.Hotheriogtoa AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Anatomical Record. 1940;78:149–172. [Google Scholar]
- 55.Laflamme N, Rivest S. Effects of systemic immunogenic insults and circulating proinflammatory cytokines on the transcription of the inhibitory factor kappaB alpha within specific cellular populations of the rat brain. J Neurochem. 1999;73:309–21. doi: 10.1046/j.1471-4159.1999.0730309.x. [DOI] [PubMed] [Google Scholar]
- 56.Eriksson C, Nobel S, Winblad B, Schultzberg M. Expression of interleukin 1 alpha and beta, and interleukin 1 receptor antagonist mRNA in the rat central nervous system after peripheral administration of lipopolysaccharides. Cytokine. 2000;12:423–431. doi: 10.1006/cyto.1999.0582. [DOI] [PubMed] [Google Scholar]
- 57.Gayle D, Ilyin SE, Flynn MC, Plata-Salaman CR. Lipopolysaccharide (LPS)- and muramyl dipeptide (MDP)-induced anorexia during refeeding following acute fasting: Characterization of brain cytokine and neuropeptide systems mRNAs. Brain Res. 1998;795:77–86. doi: 10.1016/s0006-8993(98)00280-7. [DOI] [PubMed] [Google Scholar]
- 58.Laye S, Gheusi G, Cremona S, Combe C, Kelly K, Dantzer R, Parnet P. Endogenous brain IL-1 mediates the response to peripheral LPS. Am J Phys Reg Integr Comp Physiol. 2000;279:R93–R98. doi: 10.1152/ajpregu.2000.279.1.R93. [DOI] [PubMed] [Google Scholar]
- 59.Takao T, Hashimoto K, De Souza EB. Modulation of interleukin-1 receptors in the neuro-endocrine-immune axis. Int J Dev Neurosci. 1995;13:167–178. doi: 10.1016/0736-5748(95)00015-9. [DOI] [PubMed] [Google Scholar]
- 60.Ericsson A, L C, Hart RP, Sawchenko PE. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol. 1995;361:681–698. doi: 10.1002/cne.903610410. [DOI] [PubMed] [Google Scholar]
- 61.Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 1999;93:1449–64. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- 62.Utsuyama M, Hirokawa K. Differential expression of various cytokine receptors in the brain after stimulation with LPS in young and old mice. Exp Gerontol. 2002;37:411–20. doi: 10.1016/s0531-5565(01)00208-x. [DOI] [PubMed] [Google Scholar]
- 63.Yamakuni H, Minami M, Satoh M. Localization of mRNA for leukemia inhibitory factor receptor in the adult rat brain. J Neuroimmunol. 1996;70:45–53. doi: 10.1016/s0165-5728(96)00097-5. [DOI] [PubMed] [Google Scholar]
- 64.Wan W, Janz L, Vriend CY, Sorensen CM, Greenberg AH, Nance DM. Differential induction of c-Fos immunoreactivity in hypothalamus and brain stem nuclei following central and peripheral administration of endotoxin. Brain Res Bull. 1993;32:581–7. doi: 10.1016/0361-9230(93)90158-8. [DOI] [PubMed] [Google Scholar]
- 65.Herkenham M, Lee HY, Baker RA. Temporal and spatial patterns of c-fos mRNA induced by intravenous interleukin-1: a cascade of non-neuronal cellular activation at the blood-brain barrier. J Comp Neurol. 1998;400:175–96. doi: 10.1002/(sici)1096-9861(19981019)400:2<175::aid-cne2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 66.Elmquist JK, Scammell TE, Jacobsen CD, Saper CB. Distribution of Fos-like immunoreactivity in the rat brain following intravenous lipopolysaccharide administration. J Comp Neurol. 1996;371:85–103. doi: 10.1002/(SICI)1096-9861(19960715)371:1<85::AID-CNE5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 67.Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–32. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 68.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–8. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 69.Broberger C, Johansen J, Johansson C, Schalling M, Hökfelt T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci USA. 1998;95:15043–8. doi: 10.1073/pnas.95.25.15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benoit S, Schwartz M, Baskin D, Woods SC, Seeley RJ. CNS melanocortin system involvement in the regulation of food intake. Horm Behav. 2000;37:299–305. doi: 10.1006/hbeh.2000.1588. [DOI] [PubMed] [Google Scholar]
- 71.Jegou S, Boutelet I, Vaudry H. Melanocortin-3 receptor mRNA expression in pro-opiomelanocortin neurones of the rat arcuate nucleus. J Neuroendocrinol. 2000 Jun;12(6):501–505. doi: 10.1046/j.1365-2826.2000.00477.x. [DOI] [PubMed] [Google Scholar]
- 72.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Molecular Endocrinology. 1994;8:1298–308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 73.Xia Y, Wikberg JES. Postnatal expression of melanocortin-3 receptor in rat diencephalon and mesencephalon. Neuropharmacology. 1997;36:217–224. doi: 10.1016/s0028-3908(96)00151-7. [DOI] [PubMed] [Google Scholar]
- 74.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–8. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 75.Garfield AS, Lam DD, Marston OJ, Przydzial MJ, Heisler LK. Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol Metab. 2009;20:203–15. doi: 10.1016/j.tem.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 76.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 77.Hagan MM, Rushing PA, Pritchard LM, Schwartz MW, Strack AM, Van Der Ploeg LH, Woods SC, Seeley RJ. Long-term orexigenic effects of AgRP-(83-132) involve mechanisms other than melanocortin receptor blockade. Am J Physiol Regul Integr Comp Physiol. 2000;279:R47–R52. doi: 10.1152/ajpregu.2000.279.1.R47. [DOI] [PubMed] [Google Scholar]
- 78.Woods SC, Seeley RJ, Porte D, Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280:1378–1382. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- 79.Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, Chen A, Camacho RE, Shearman LP, Gopal-Truter S, MacNeil DJ, Van Der Ploeg LH, Marsh DJ. Neither Agouti-Related Protein nor Neuropeptide Y Is Critically Required for the Regulation of Energy Homeostasis in Mice. Mol Cell Biol. 2002;22:5027–35. doi: 10.1128/MCB.22.14.5027-5035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheung W, Yu PX, Little BM, Cone RD, Marks DL, Mak RH. Role of leptin and melanocortin signaling in uremia-associated cachexia. J Clin Invest. 2005;115:1659–65. doi: 10.1172/JCI22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marks DL, Butler AA, Turner R, Brookhart G, Cone RD. Differential role of melanocortin receptor subtypes in cachexia. Endocrinology. 2003;144:1513–23. doi: 10.1210/en.2002-221099. [DOI] [PubMed] [Google Scholar]
- 82.Sartin JL, Marks DL, McMahon CD, Daniel JA, Levasseur P, Wagner CG, Whitlock BK, Steele BP. Central role of the melanocortin-4 receptors in appetite regulation after endotoxin. J Anim Sci. 2008;86:2557–67. doi: 10.2527/jas.2008-0916. [DOI] [PubMed] [Google Scholar]
- 83.Inui A. Neuropeptide Y: a key molecule in anorexia and cachexia in wasting disorders? Mol Med Today. 1999;5:79–85. doi: 10.1016/s1357-4310(98)01395-1. [DOI] [PubMed] [Google Scholar]
- 84.McCarthy HD, Crowder RE, Dryden S, Williams G. Megestrol acetate stimulates food and water intake in the rat: effects on regional hypothalamic neuropeptide Y concentrations. Eur J Pharmacol. 1994;265:99–102. doi: 10.1016/0014-2999(94)90229-1. [DOI] [PubMed] [Google Scholar]
- 85.Chance WT, Balasubramaniam A, Thompson H, Mohapatra B, Ramo J, Fischer JE. Assessment of feeding response of tumor-bearing rats to hypothalamic injection and infusion of neuropeptide Y. Peptides. 1996;17:797–801. doi: 10.1016/0196-9781(96)00108-8. [DOI] [PubMed] [Google Scholar]
- 86.Anand BK, Brobeck JR. Localization of a “feeding center” in the hypothalamus of the rat. Proc Soc Exp Biol Med. 1951;77:323–4. doi: 10.3181/00379727-77-18766. [DOI] [PubMed] [Google Scholar]
- 87.Hetherington AW, R S. Hypothalamic lesions and adiposity in the rat. Anat Rec. 1940;78:149–172. [Google Scholar]
- 88.Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 1973;9:294–298. doi: 10.1007/BF01221857. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 90.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Woolf EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 91.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinol. 1997;138:4489–4492. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 92.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Morgan PJ, Trayhurn P. Coexpression of leptin receptor and preproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. J Neuroendocrinol. 1996;8:733–5. doi: 10.1046/j.1365-2826.1996.05161.x. [DOI] [PubMed] [Google Scholar]
- 93.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;22:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 94.Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med. 1996;2:589–93. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 95.Ciofi P, Garret M, Lapirot O, Lafon P, Loyens A, Prévot V, Levine J. Brain-Endocrine Interactions: A Microvascular Route in the Mediobasal Hypothalamus. Endocrinology. 2009 doi: 10.1210/en.2009-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Banks WA. Blood-brain barrier and energy balance. Obesity (Silver Spring) 2006;14 5:234S–237S. doi: 10.1038/oby.2006.315. [DOI] [PubMed] [Google Scholar]
- 97.Banks WA, Kastin AJ, Gutierrez EG. Penetration of interleukin-6 across the murine blood-brain barrier. Neurosci Lett. 1994;179:53–6. doi: 10.1016/0304-3940(94)90933-4. [DOI] [PubMed] [Google Scholar]
- 98.Banks WA, Ortiz L, Plotkin SR, Kastin AJ. Human interleukin (IL) 1 alpha, murine IL-1 alpha and murine IL-1 beta are transported from blood to brain in the mouse by a shared saturable mechanism. J Pharmacol Exp Ther. 1991;259:988–96. [PubMed] [Google Scholar]
- 99.Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol. 1993;47:169–76. doi: 10.1016/0165-5728(93)90027-v. [DOI] [PubMed] [Google Scholar]
- 100.Pan W, Yu C, Hsuchou H, Zhang Y, Kastin AJ. Neuroinflammation facilitates LIF entry into brain: role of TNF. Am J Physiol, Cell Physiol. 2008;294:C1436–42. doi: 10.1152/ajpcell.00489.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lugarini F, Hrupka BJ, Schwartz GJ, Plata-Salaman CR, Langhans W. A role for cyclooxygenase-2 in lipopolysaccharide-induced anorexia in rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R862–8. doi: 10.1152/ajpregu.00200.2002. [DOI] [PubMed] [Google Scholar]
- 102.Tizard I. Sickness behavior, its mechanisms and significance. Anim Health Res Rev. 2008;9:87–99. doi: 10.1017/S1466252308001448. [DOI] [PubMed] [Google Scholar]
- 103.Engblom D, Saha S, Engström L, Westman M, Audoly LP, Jakobsson PJ, Blomqvist A. Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat Neurosci. 2003;6:1137–8. doi: 10.1038/nn1137. [DOI] [PubMed] [Google Scholar]
- 104.Pecchi E, Dallaporta M, Thirion S, Salvat C, Berenbaum F, Jean A, Troadec JD. Involvement of central microsomal prostaglandin E synthase-1 in IL-1beta-induced anorexia. Physiol Genomics. 2006;25:485–92. doi: 10.1152/physiolgenomics.00306.2005. [DOI] [PubMed] [Google Scholar]
- 105.Jakobsson PJ, Thorén S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci USA. 1999;96:7220–5. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–92. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 107.Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J Neurosci. 2001;21:2669–77. doi: 10.1523/JNEUROSCI.21-08-02669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dallaporta M, Pecchi E, Jacques C, Berenbaum F, Jean A, Thirion S, Troadec JD. c-Fos immunoreactivity induced by intraperitoneal LPS administration is reduced in the brain of mice lacking the microsomal prostaglandin E synthase-1 (mPGES-1) Brain Behav Immun. 2007;21:1109–21. doi: 10.1016/j.bbi.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 109.Pecchi E, Dallaporta M, Jean A, Thirion S, Troadec JD. mPGES-1 knock-out mice are resistant to cancer-induced anorexia despite the absence of central mPGES-1 up-regulation in wild-type anorexic mice. J Neuroimmunol. 2008;199:104–14. doi: 10.1016/j.jneuroim.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 110.Serrats J, Schiltz JC, García-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron. 2010;65:94–106. doi: 10.1016/j.neuron.2009.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.García-Bueno B, Serrats J, Sawchenko PE. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. J Neurosci. 2009;29:12970–81. doi: 10.1523/JNEUROSCI.2373-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ching S, Zhang H, Belevych N, He L, Lai W, Pu Xa, Jaeger LB, Chen Q, Quan N. Endothelial-specific knockdown of interleukin-1 (IL-1) type 1 receptor differentially alters CNS responses to IL-1 depending on its route of administration. J Neurosci. 2007;27:10476–86. doi: 10.1523/JNEUROSCI.3357-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hansen MK, Taishi P, Chen Z, Krueger JM. Vagotomy blocks the induction of interleukin-1beta (IL-1beta) mRNA in the brain of rats in response to systemic IL-1beta. J Neurosci. 1998;18:2247–53. doi: 10.1523/JNEUROSCI.18-06-02247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Layé S, Bluthé RM, Kent S, Combe C, Médina C, Parnet P, Kelley K, Dantzer R. Subdiaphragmatic vagotomy blocks induction of IL-1 beta mRNA in mice brain in response to peripheral LPS. Am J Physiol. 1995;268:R1327–31. doi: 10.1152/ajpregu.1995.268.5.R1327. [DOI] [PubMed] [Google Scholar]
- 115.Konsman JP, Luheshi GN, Bluthé RM, Dantzer R. The vagus nerve mediates behavioural depression, but not fever, in response to peripheral immune signals; a functional anatomical analysis. Eur J Neurosci. 2000;12:4434–46. doi: 10.1046/j.0953-816x.2000.01319.x. [DOI] [PubMed] [Google Scholar]
- 116.Luheshi GN, Bluthé RM, Rushforth D, Mulcahy N, Konsman JP, Goldbach M, Dantzer R. Vagotomy attenuates the behavioural but not the pyrogenic effects of interleukin-1 in rats. Autonomic neuroscience : basic & clinical. 2000;85:127–32. doi: 10.1016/S1566-0702(00)00231-9. [DOI] [PubMed] [Google Scholar]
- 117.Bret-Dibat JL, Bluthé RM, Kent S, Kelley KW, Dantzer R. Lipopolysaccharide and interleukin-1 depress food-motivated behavior in mice by a vagal-mediated mechanism. Brain Behav Immun. 1995;9:242–6. doi: 10.1006/brbi.1995.1023. [DOI] [PubMed] [Google Scholar]
- 118.Porter MH, Hrupka BJ, Langhans W, Schwartz GJ. Vagal and splanchnic afferents are not necessary for the anorexia produced by peripheral IL-1beta, LPS, and MDP. Am J Physiol. 1998;275:R384–9. doi: 10.1152/ajpregu.1998.275.2.R384. [DOI] [PubMed] [Google Scholar]
- 119.Ericsson A, Kovács KJ, Sawchenko PE. A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons. J Neurosci. 1994;14:897–913. doi: 10.1523/JNEUROSCI.14-02-00897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sawchenko PE, Swanson LW. The organization of noradrenergic pathways from the brainstem to the paraventricular and supraoptic nuclei in the rat. Brain Res. 1982;257:275–325. doi: 10.1016/0165-0173(82)90010-8. [DOI] [PubMed] [Google Scholar]
- 121.Sergeyev V, Broberger C, Hokfelt T. Effect of LPS administration on the expression of POMC, NPY, galanin, CART and MCH mRNAs in the rat hypothalamus. Brain Res Mol Brain Res. 2001;90:93–100. doi: 10.1016/s0169-328x(01)00088-2. [DOI] [PubMed] [Google Scholar]
- 122.Endo M, Masaki T, Seike M, Yoshimatsu H. Involvement of stomach ghrelin and hypothalamic neuropeptides in tumor necrosis factor-alpha-induced hypophagia in mice. Regul Pept. 2007;140:94–100. doi: 10.1016/j.regpep.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 123.Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines: neuromodulators in normal brain? J Neurochem. 2000;74:457–71. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- 124.Giulian D, Tapscott MJ. Immunoregulation of cells within the central nervous system. Brain Behav Immun. 1988;2:352–8. doi: 10.1016/0889-1591(88)90040-2. [DOI] [PubMed] [Google Scholar]
- 125.Higgins GA, Olschowka JA. Induction of interleukin-1 beta mRNA in adult rat brain. Brain Res Mol Brain Res. 1991;9:143–8. doi: 10.1016/0169-328x(91)90139-o. [DOI] [PubMed] [Google Scholar]
- 126.Stalder AK, Campbell IL. Simultaneous analysis of multiple cytokine receptor mRNAs by RNase protection assay in LPS-induced endotoxemia. Lymphokine Cytokine Res. 1994;13:107–12. [PubMed] [Google Scholar]
- 127.Turrin NP, Ilyin SE, Gayle DA, Plata-Salaman CR, Ramos EJ, Laviano A, Das UN, Inui A, Meguid MM. Interleukin-1beta system in anorectic catabolic tumor-bearing rats. Curr Opin Clin Nutr Metab Care. 2004;7:419–26. doi: 10.1097/01.mco.0000134373.16557.92. [DOI] [PubMed] [Google Scholar]
- 128.Van Dam AM, Bauer J, Tilders FJ, Berkenbosch F. Endotoxin-induced appearance of immunoreactive interleukin-1 beta in ramified microglia in rat brain: a light and electron microscopic study. Neuroscience. 1995;65:815–826. doi: 10.1016/0306-4522(94)00549-k. [DOI] [PubMed] [Google Scholar]
- 129.Webb AC, Collins KL, Auron PE, Eddy RL, Nakai H, Byers MG, Haley LL, Henry WM, Shows TB. Interleukin-1 gene (IL1) assigned to long arm of human chromosome 2. Lymphokine Res. 1986;5:77–85. [PubMed] [Google Scholar]
- 130.Thornberry NA, Molineaux SM. Interleukin-1 beta converting enzyme: a novel cysteine protease required for IL-1 beta production and implicated in programmed cell death. Protein Sci. 1995;4:3–12. doi: 10.1002/pro.5560040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hirsch E, Irikura VM, Paul SM, Hirsh D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc Natl Acad Sci U S A. 1996;93:11008–13. doi: 10.1073/pnas.93.20.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Boraschi D, Tagliabue A. The interleukin-1 receptor family. Vitam Horm. 2006;74:229–54. doi: 10.1016/S0083-6729(06)74009-2. [DOI] [PubMed] [Google Scholar]
- 133.Ericsson A, Liu C, Hart RP, Sawchenko PE. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol. 1995;361:681–98. doi: 10.1002/cne.903610410. [DOI] [PubMed] [Google Scholar]
- 134.Parnet P, Kelley KW, Bluthe RM, Dantzer R. Expression and regulation of interleukin-1 receptors in the brain. Role in cytokines-induced sickness behavior. J Neuroimmunol. 2002;125:5–14. doi: 10.1016/s0165-5728(02)00022-x. [DOI] [PubMed] [Google Scholar]
- 135.Ilyin SE, Gayle D, Flynn MC, Plata-Salaman CR. Interleukin-1beta system (ligand, receptor type I, receptor accessory protein and receptor antagonist), TNF-alpha, TGF-beta1 and neuropeptide Y mRNAs in specific brain regions during bacterial LPS-induced anorexia. Brain Res Bull. 1998;45:507–15. doi: 10.1016/s0361-9230(97)00437-1. [DOI] [PubMed] [Google Scholar]
- 136.Gayle D, Ilyin SE, Plata-Salaman CR. Central nervous system IL-1 beta system and neuropeptide Y mRNAs during IL-1 beta-induced anorexia in rats. Brain Res Bull. 1997;44:311–7. doi: 10.1016/s0361-9230(97)00159-7. [DOI] [PubMed] [Google Scholar]
- 137.Kluger MJ. Fever: role of pyrogens and cryogens. Physiol Rev. 1991;71:93–127. doi: 10.1152/physrev.1991.71.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hori T, Shibata M, Nakashima T, Yamasaki M, Asami A, Asami T, Koga H. Effects of interleukin-1 and arachidonate on the preoptic and anterior hypothalamic neurons. Brain Res Bull. 1988;20:75–82. doi: 10.1016/0361-9230(88)90010-x. [DOI] [PubMed] [Google Scholar]
- 139.Li S, Ballou LR, Morham SG, Blatteis CM. Cyclooxygenase-2 mediates the febrile response of mice to interleukin-1beta. Brain Res. 2001;910:163–73. doi: 10.1016/s0006-8993(01)02707-x. [DOI] [PubMed] [Google Scholar]
- 140.Katsuura G, Gottschall PE, Dahl RR, Arimura A. Adrenocorticotropin release induced by intracerebroventricular injection of recombinant human interleukin-1 in rats: possible involvement of prostaglandin. Endocrinology. 1988;122:1773–9. doi: 10.1210/endo-122-5-1773. [DOI] [PubMed] [Google Scholar]
- 141.Dantzer R. Cytokine-induced sickness behavior: where do we stand? Brain Behav Immun. 2001;15:7–24. doi: 10.1006/brbi.2000.0613. [DOI] [PubMed] [Google Scholar]
- 142.Kent S, Bluthe RM, Dantzer R, Hardwick AJ, Kelley KW, Rothwell NJ, Vannice JL. Different receptor mechanisms mediate the pyrogenic and behavioral effects of interleukin 1. Proc Natl Acad Sci U S A. 1992;89:9117–20. doi: 10.1073/pnas.89.19.9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shimizu H, Uehara Y, Shimomura Y, Kobayashi I. Central administration of ibuprofen failed to block the anorexia induced by interleukin-1. Eur J Pharmacol. 1991;195:281–4. doi: 10.1016/0014-2999(91)90547-4. [DOI] [PubMed] [Google Scholar]
- 144.Shimomura Y, Inukai T, Kuwabara S, Shimizu H, Takahashi M, Sato N, Uehara Y, Tanaka Y, Kobayashi I. Both cyclooxygenase and lipoxygenase inhibitor partially restore the anorexia by interleukin-1 beta. Life Sci. 1992;51:1419–26. doi: 10.1016/0024-3205(92)90536-x. [DOI] [PubMed] [Google Scholar]
- 145.Reyes TM, Sawchenko PE. Involvement of the arcuate nucleus of the hypothalamus in interleukin-1-induced anorexia. J Neurosci. 2002;22:5091–9. doi: 10.1523/JNEUROSCI.22-12-05091.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Scarlett JM, Jobst EE, Enriori PJ, Bowe DD, Batra AK, Grant WF, Cowley MA, Marks DL. Regulation of central melanocortin signaling by interleukin-1 beta. Endocrinology. 2007;148:4217–25. doi: 10.1210/en.2007-0017. [DOI] [PubMed] [Google Scholar]
- 147.Scarlett JM, Zhu X, Enriori PJ, Bowe DD, Batra AK, Levasseur PR, Grant WF, Meguid MM, Cowley MA, Marks DL. Regulation of agouti-related protein messenger ribonucleic acid transcription and peptide secretion by acute and chronic inflammation. Endocrinology. 2008;149:4837–45. doi: 10.1210/en.2007-1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bergen HT, Mizuno TM, Taylor J, Mobbs CV. Hyperphagia and weight gain after gold-thio-glucose: relation to hypothalamic neuropeptide Y and proopiomelanocortin. :1–22. doi: 10.1210/endo.139.11.6324. [DOI] [PubMed] [Google Scholar]
- 149.Broberger C, Johansson C, Schalling M, Hokfelt T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci USA. 1998;95:15043–15048. doi: 10.1073/pnas.95.25.15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hill AG, Jacobson L, Gonzalez J, Rounds J, Majzoub JA, Wilmore DW. Chronic central nervous system exposure to interleukin-1 beta causes catabolism in the rat. Am J Physiol. 1996;271:R1142–8. doi: 10.1152/ajpregu.1996.271.5.R1142. [DOI] [PubMed] [Google Scholar]
- 151.Gearing DP, Gough NM, King JA, Hilton DJ, Nicola NA, Simpson RJ, Nice EC, Kelso A, Metcalf D. Molecular cloning and expression of cDNA encoding a murine myeloid leukaemia inhibitory factor (LIF) EMBO J. 1987;6:3995–4002. doi: 10.1002/j.1460-2075.1987.tb02742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Owczarek CM, Layton MJ, Robb LG, Nicola NA, Begley CG. Molecular basis of the soluble and membrane-bound forms of the murine leukemia inhibitory factor receptor alpha-chain. Expression in normal, gestating, and leukemia inhibitory factor nullizygous mice. J Biol Chem. 1996;271:5495–504. doi: 10.1074/jbc.271.10.5495. [DOI] [PubMed] [Google Scholar]
- 153.Aikawa J, Sato E, Kyuwa S, Sato E, Sasai K, Shiota K, Ogawa T. Asparagine-linked glycosylation of the rat leukemia inhibitory factor expressed by simian COS7 cells. Biosci Biotechnol Biochem. 1998;62:1318–25. doi: 10.1271/bbb.62.1318. [DOI] [PubMed] [Google Scholar]
- 154.Sasai K, Aikawa Ji, Saburi S, Tojo H, Tanaka S, Ogawa T, Shiota K. Functions of the N-glycans of rat leukemia inhibitory factor expressed in Chinese hamster ovary cells. J Biochem. 1998;124:999–1003. doi: 10.1093/oxfordjournals.jbchem.a022219. [DOI] [PubMed] [Google Scholar]
- 155.Auernhammer CJ, Melmed S. Leukemia-inhibitory factor-neuroimmune modulator of endocrine function. Endocr Rev. 2000;21:313–45. doi: 10.1210/edrv.21.3.0400. [DOI] [PubMed] [Google Scholar]
- 156.Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 157.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Febbraio MA. gp130 receptor ligands as potential therapeutic targets for obesity. J Clin Invest. 2007;117:841–9. doi: 10.1172/JCI30453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006;4:123–32. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 160.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AWK, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–9. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 161.Janoschek R, Plum L, Koch L, Münzberg H, Diano S, Shanabrough M, Müller W, Horvath TL, Brüning JC. gp130 signaling in proopiomelanocortin neurons mediates the acute anorectic response to centrally applied ciliary neurotrophic factor. Proc Natl Acad Sci USA. 2006;103:10707–12. doi: 10.1073/pnas.0600425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Prima V, Tennant M, Gorbatyuk OS, Muzyczka N, Scarpace PJ, Zolotukhin S. Differential modulation of energy balance by leptin, ciliary neurotrophic factor, and leukemia inhibitory factor gene delivery: microarray deoxyribonucleic acid-chip analysis of gene expression. Endocrinology. 2004;145:2035–45. doi: 10.1210/en.2003-1376. [DOI] [PubMed] [Google Scholar]
- 163.Benomar Y, Berthou F, Vacher CM, Bailleux V, Gertler A, Djiane J, Taouis M. Leptin but not ciliary neurotrophic factor (CNTF) induces phosphotyrosine phosphatase-1B expression in human neuronal cells (SH-SY5Y): putative explanation of CNTF efficacy in leptin-resistant state. Endocrinology. 2009;150:1182–91. doi: 10.1210/en.2008-1097. [DOI] [PubMed] [Google Scholar]
- 164.Ropelle ER, Rocha GZ, Cintra DE, Prada PO, Pauli JR, Picardi PK, KG Z, Velloso LA, Saad MJA, Carvalheira JBC. Key phosphatase in cancer-induced anorexia. 5th Cachexia Conference; Barcelona, Spain. 2009. [Google Scholar]
- 165.Ren SG, Seliktar J, Li X, Braunstein GD, Melmed S. Measurement of leukemia inhibitory factor in biological fluids by radioimmunoassay. J Clin Endocrinol Metab. 1998;83:1275–83. doi: 10.1210/jcem.83.4.4702. [DOI] [PubMed] [Google Scholar]
- 166.Gouin F, Heymann D, Raher S, De Groote D, Passuti N, Daculsi G, Godard A. Increased levels of leukaemia inhibitory factor (LIF) in urine and tissue culture supernatant from human primary bone tumours. Cytokine. 1998;10:110–4. doi: 10.1006/cyto.1997.0264. [DOI] [PubMed] [Google Scholar]
- 167.Lorgeot V, Praloran V, Turlure P, Denizot Y. Concentrations of serum leukemia inhibitory factor (LIF) in patients with hematologic malignancies. Leukemia. 1997;11:311–2. doi: 10.1038/sj.leu.2400570. [DOI] [PubMed] [Google Scholar]
- 168.Villers D, Dao T, Nguyen JM, Bironneau E, Godard A, Moreau M, De Groote D, Nicolas F, Soulillou JP, Anegon I. Increased plasma levels of human interleukin for DA1.a cells/leukemia inhibitory factor in sepsis correlate with shock and poor prognosis. J Infect Dis. 1995;171:232–6. doi: 10.1093/infdis/171.1.232. [DOI] [PubMed] [Google Scholar]
- 169.Carlson CD, Bai Y, Jonakait GM, Hart RP. Interleukin-1 beta increases leukemia inhibitory factor mRNA levels through transient stimulation of transcription rate. Glia. 1996;18:141–51. doi: 10.1002/(SICI)1098-1136(199610)18:2<141::AID-GLIA6>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 170.Gayle D, Ilyin SE, Flynn MC, Plata-Salamán CR. Lipopolysaccharide (LPS)- and muramyl dipeptide (MDP)-induced anorexia during refeeding following acute fasting: characterization of brain cytokine and neuropeptide systems mRNAs. Brain Res. 1998;795:77–86. doi: 10.1016/s0006-8993(98)00280-7. [DOI] [PubMed] [Google Scholar]
- 171.Metcalf D, Gearing DP. Fatal syndrome in mice engrafted with cells producing high levels of the leukemia inhibitory factor. Proc Natl Acad Sci USA. 1989;86:5948–52. doi: 10.1073/pnas.86.15.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Beretta E, Dhillon H, Kalra PS, Kalra SP. Central LIF gene therapy suppresses food intake, body weight, serum leptin and insulin for extended periods. Peptides. 2002;23:975–84. doi: 10.1016/s0196-9781(02)00021-9. [DOI] [PubMed] [Google Scholar]
- 173.Mori M, Yamaguchi K, Honda S, Nagasaki K, Ueda M, Abe O, Abe K. Cancer cachexia syndrome developed in nude mice bearing melanoma cells producing leukemia-inhibitory factor. Cancer Res. 1991;51:6656–9. [PubMed] [Google Scholar]
- 174.Ray DW, Ren SG, Melmed S. Leukemia inhibitory factor regulates proopiomelanocortin transcription. Ann N Y Acad Sci. 1998;840:162–73. doi: 10.1111/j.1749-6632.1998.tb09560.x. [DOI] [PubMed] [Google Scholar]
- 175.Ray DW, Ren SG, Melmed S. Leukemia inhibitory factor (LIF) stimulates proopiomelanocortin (POMC) expression in a corticotroph cell line. Role of STAT pathway. Journal of Clinical Investigation. 1996;97:1852–9. doi: 10.1172/JCI118615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Swan JW, Anker SD, Walton C, Godsland IF, Clark AL, Leyva F, Stevenson JC, Coats AJ. Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol. 1997;30:527–32. doi: 10.1016/s0735-1097(97)00185-x. [DOI] [PubMed] [Google Scholar]
- 177.Doehner W, Rauchhaus M, Ponikowski P, Godsland IF, von Haehling S, Okonko DO, Leyva F, Proudler AJ, Coats AJS, Anker SD. Impaired insulin sensitivity as an independent risk factor for mortality in patients with stable chronic heart failure. J Am Coll Cardiol. 2005;46:1019–26. doi: 10.1016/j.jacc.2005.02.093. [DOI] [PubMed] [Google Scholar]
- 178.Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–32. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- 179.Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC, Lowell BB, Elmquist JK. The hypothalamic arcuate nucleus: a key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 180.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferré P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–74. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 181.Nogueiras R, Wiedmer P, Perez-Tilve D, Veyrat-Durebex C, Keogh JM, Sutton GM, Pfluger PT, Castaneda TR, Neschen S, Hofmann SM, Howles PN, Morgan DA, Benoit SC, Szanto I, Schrott B, Schürmann A, Joost HG, Hammond C, Hui DY, Woods SC, Rahmouni K, Butler AA, Farooqi IS, O'Rahilly S, Rohner-Jeanrenaud F, Tschöp MH. The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest. 2007;117:3475–88. doi: 10.1172/JCI31743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjørbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–8. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 183.Maes M, Smith R, Scharpe S. The monocyte-T-lymphocyte hypothesis of major depression. Psychoneuroendocrinology. 1995;20:111–6. doi: 10.1016/0306-4530(94)00066-j. [DOI] [PubMed] [Google Scholar]
- 184.Capuron L, Ravaud A, Dantzer R. Early depressive symptoms in cancer patients receiving interleukin 2 and/or interferon alfa-2b therapy. J Clin Oncol. 2000;18:2143–51. doi: 10.1200/JCO.2000.18.10.2143. [DOI] [PubMed] [Google Scholar]
- 185.Myint AM, Schwarz MJ, Steinbusch HWM, Leonard BE. Neuropsychiatric disorders related to interferon and interleukins treatment. Metab Brain Dis. 2009;24:55–68. doi: 10.1007/s11011-008-9114-5. [DOI] [PubMed] [Google Scholar]
- 186.Anisman H, Merali Z. Anhedonic and anxiogenic effects of cytokine exposure. Adv Exp Med Biol. 1999;461:199–233. doi: 10.1007/978-0-585-37970-8_12. [DOI] [PubMed] [Google Scholar]
- 187.Yirmiya R. Endotoxin produces a depressive-like episode in rats. Brain Res. 1996;711:163–74. doi: 10.1016/0006-8993(95)01415-2. [DOI] [PubMed] [Google Scholar]
- 188.Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695:279–82. doi: 10.1016/0006-8993(95)00930-o. [DOI] [PubMed] [Google Scholar]
- 189.Hsu R, Taylor JR, Newton SS, Alvaro JD, Haile C, Han G, Hruby VJ, Nestler EJ, Duman RS. Blockade of melanocortin transmission inhibits cocaine reward. Eur J Neurosci. 2005;21:2233–42. doi: 10.1111/j.1460-9568.2005.04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lindblom J, Kask A, Hägg E, Härmark L, Bergström L, Wikberg J. Chronic infusion of a melanocortin receptor agonist modulates dopamine receptor binding in the rat brain. Pharmacol Res. 2002;45:119–24. doi: 10.1006/phrs.2001.0913. [DOI] [PubMed] [Google Scholar]
- 191.Lindblom J, Opmane B, Mutulis F, Mutule I, Petrovska R, Klusa V, Bergström L, Wikberg JE. The MC4 receptor mediates alpha-MSH induced release of nucleus accumbens dopamine. Neuroreport. 2001;12:2155–8. doi: 10.1097/00001756-200107200-00022. [DOI] [PubMed] [Google Scholar]
- 192.Tatro JB, Entwistle ML. Distribution of melanocortin receptors in the lower brainstem of the rat. Ann N Y Acad Sci. 1994;739:311–4. doi: 10.1111/j.1749-6632.1994.tb19833.x. [DOI] [PubMed] [Google Scholar]
- 193.Kawashima N, Chaki S, Okuyama S. Electrophysiological effects of melanocortin receptor ligands on neuronal activities of monoaminergic neurons in rats. Neurosci Lett. 2003;353:119–22. doi: 10.1016/j.neulet.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 194.Holmes JE, Miller NE. Effects of Bacterial Endotoxin on Water Intake, Food Intake, and Body Temperature in the Albino Rat. J Exp Med. 1963;118:649–58. doi: 10.1084/jem.118.4.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kozak W, Conn CA, Kluger MJ. Lipopolysaccharide induces fever and depresses locomotor activity in unrestrained mice. Am J Physiol. 1994;266:R125–35. doi: 10.1152/ajpregu.1994.266.1.R125. [DOI] [PubMed] [Google Scholar]
- 196.Mesaros A, Koralov SB, Rother E, Wunderlich FT, Ernst MB, Barsh GS, Rajewsky K, Brüning JC. Activation of Stat3 signaling in AgRP neurons promotes locomotor activity. Cell Metab. 2008;7:236–48. doi: 10.1016/j.cmet.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 197.Huo L, Gamber K, Greeley S, Silva J, Huntoon N, Leng XH, Bjørbaek C. Leptin-dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab. 2009;9:537–47. doi: 10.1016/j.cmet.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Dandona P, Dhindsa S, Chaudhuri A, Bhatia V, Topiwala S, Mohanty P. Hypogonadotrophic hypogonadism in type 2 diabetes, obesity and the metabolic syndrome. Curr Mol Med. 2008;8:816–28. doi: 10.2174/156652408786733658. [DOI] [PubMed] [Google Scholar]
- 199.Shabanova SS, Ananieva LP, Alekberova ZS, Guzov II. Ovarian function and disease activity in patients with systemic lupus erythematosus. Clin Exp Rheumatol. 2008;26:436–41. [PubMed] [Google Scholar]
- 200.Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol. 2009;36:887–92. doi: 10.3899/jrheum.080558. [DOI] [PubMed] [Google Scholar]
- 201.Reynoso R, Ponzo O, Cardoso N, Szwarcfarb B, Carbone S, Moguilevsky J, Scacchi P. Effect of bacterial lipopolysaccharide on the reproductive axis of prepubertal and peripubertal female rats. Ontogenic changes in the immune-neuroendocrine interactions. Neuroimmunomodulation. 2008;15:125–30. doi: 10.1159/000148195. [DOI] [PubMed] [Google Scholar]
- 202.Kalra PS, Sahu A, Kalra SP. Interleukin-1 inhibits the ovarian steroid-induced luteinizing hormone surge and release of hypothalamic luteinizing hormone-releasing hormone in rats. Endocrinology. 1990;126:2145–52. doi: 10.1210/endo-126-4-2145. [DOI] [PubMed] [Google Scholar]
- 203.Schiöth HB, Watanobe H. Melanocortins and reproduction. Brain Res Brain Res Rev. 2002;38:340–50. doi: 10.1016/s0165-0173(01)00159-x. [DOI] [PubMed] [Google Scholar]
- 204.Backholer K, Smith J, Clarke I. Melanocortins May Stimulate Reproduction by Activating Orexin Neurons in the Dorsomedial Hypothalamus and Kisspeptin Neurons in the Preoptic Area of the Ewe. Endocrinology. 2009 doi: 10.1210/en.2009-0604. [DOI] [PubMed] [Google Scholar]