

Abstract
A series of new phosphatidylcholine analogues with structurally modified sn-2-substituents have been prepared. The synthetic compounds include oligo(ethyleneglycol) derivatives with chain-terminal pharmacophores that upon catalytic hydrolysis by phospholipaseA2 yielded a series of oligo(ethyleneglycol)-conjugates of the respective drugs. The approach here outlined may open a new way to employ OEG derivatives of phospholipids for therapeutic applications as secretory PLA2-targeted precursors of prodrugs.
Keywords: Oligo(ethyleneglycol) substituted phosphatidylcholines, Phospholipase A2-targeted precursor of prodrugs, Non-steroidalanti-inflammmatory drugs, Drug delivery methods
1. Introduction
Phospholipases A2 (PLA2’s, EC 3.1.1.4) comprise a large group of intracellular and secreted enzymes that catalyze the hydrolysis of the sn-2-ester bond of glycerophospholipids to yield free fatty acids such as arachidonic acid, and lysophospholipids (Burke and Dennis, 2009a, eq.1).
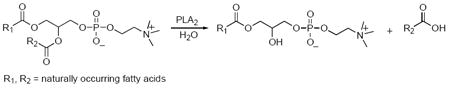 |
(1) |
Both products are precursors for signaling molecules with a wide range of biological functions (Burke and Dennis, 2009b). Specifically, arachidonic acid is converted to eicosanoids that have been shown to be involved in immune response, inflammation, pain perception, and sleep regulation (Funk, 2001, Schaloske and Dennis, 2007), while lysophospholipids are precursors of lipid mediators such as lysophosphatidic acid (LPA) and platelet activating factor (PAF). Lysophosphatidic acid has been shown to be involved in cell proliferation, survival and migration (Rivera and Chun, 2008, Zhao and Natarajan, 2009), while PAF is particularly involved in inflammatory processes (Prescott et al., 2000).
Secreted phospholipases A2 (sPLA2’s) are widespread in nature (Six and Dennis, 2000). Early studies have focused on members of the sPLA2 family isolated from insect and snake venoms; more recently sPLA2’s have been found in plants, bacteria, fungi, viruses, and mammals (Boyanovsky and Webb, 2009). The mammalian family of secreted phospholipase A2 enzymes consists of twelve different members at the present (Rouault et al., 2007). Isolated from the variety of sources, the sPLA2’s share a series of common structural features. They are small, secreted proteins (14-18 kDa), with a compact structure usually containing 5 to 8 disulfide bonds (Fuentes et al., 2002). Studies focusing on their mechanism of action have shown that an active site histidine in close proximity to a conserved aspartate are the residues involved in the catalytic reaction, with absolute dependence on Ca2+ for activation (Scott et al., 1990).
At the present there is a growing interest in elucidation of the in vivo biological functions of mammalian sPLA2’s, since they have been implicated in a series of physiological and pathophysiological functions including lipid digestion, signal transduction, prostaglandin biosynthesis, cell proliferation, neurosecretion (Burke and Dennis, 2009b), antibacterial defense (Nevalainen et al., 2008), inflammatory diseases (Nevalainen et al., 2000) and cancer (Dong et al., 2006). In this context, secretory phospholipase A2 enzymes have recently been targets for a therapeutic application, as elevated levels of one subtype, sPLA2-IIA, were observed in the microenvironment surrounding tumors in human colorectal adenocarcinomas (Abe et al., 1997, Kennedy et al., 1998, Mounier et al., 2008), and in neoplastic prostate tissue (Graff et al., 2001, Menschikowski et al., 2008). The high levels of sPLA2 in tumors prompted the design of a tumor-activated drug delivery system in the form of sPLA2 degradable liposomes, transporting conventional therapeutics that were released at the tumor site using sPLA2 as a tumor-specific trigger (Davidsen et al., 2003).
In this communication we report a new approach to develop sPLA2-directed therapeutically applicable synthetic phospholipid analogues. We have designed a series of structurally modified phosphatidylcholine analogues incorporating oligoethylene glycol-substituted sn-2-ester functions. Relying on the minimum structural requirements for catalytic hydrolysis by the enzyme (Kuipers et al., 1990), including the presence of an essential α-methylene group adjacent to the sn-2-ester carbonyl of the substrate, replacement of the hydrocarbon portion of the naturally occurring fatty acid chain with an oligoethylene glycol group appeared to be a promising modification to qualify for catalytic cleavage by sPLA2 enzymes.
2. Results and Discussion
2.1.Design of the synthetic target compounds
In designing the synthetic phospholipid analogues we sought to develop PLA2-directed precursors of oligoethylene glycol-conjugated pharmacophores to be released as prodrugs upon catalytic hydrolysis by the enzyme. Specifically, it has been shown recently, that oligoethylene modification of therapeutic agents, much like PEGylation using their high-molecular weight polymeric counterparts (Veronese et al., 2005), greatly improves the pharmacokinetic profiles and targeting of drugs (Marsac et al., 2006). These benefits include: 1) increase in solubility to achieve better bioavailability, 2) resistance to proteolytic degradation, 3) reduction in immunogenicity and antigenicity, and 4) prolonged plasma circulation time (Harris and Chess, 2003, Juillerat-Jeanneret and Schmitt, 2007, Marcus et al., 2008). Furthermore, oligomeric PEG analogues (OEG’s) are readily derivatized for site-selective targeting, and they are also amenable to a wide range of structural modification including construction of linear or branched scaffolds (Bowen et al., 2007). To explore the feasibility of the approach we used indomethacin 4, sulindac 5, and diclofenac 6, as these nonsteroidal anti-inflammatory drugs (NSAID) are subject of ongoing biochemical and pharmacological investigation (Figure 1).
Figure 1.
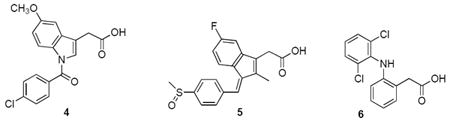
The structures of NSAID’s used in the syntheses: indomethacin 4, sulindac 5, and diclofenac 6.
Specifically, in addition to their well-established inhibitory profiles against cyclooxygenases (Prusakiewicz et al., 2004, Felts et al., 2007, Blobaum and Marnett, 2007) nonsteroidal anti-inflammatory drugs have been found both in epidemiological and clinical studies to reduce the prevalence and severity of Alzheimer’s disease (Veld et al., 2001, Gasparini et al., 2004), as inflammation exacerbates the pathology of that disease and other neurodegenerative diseases (Klegeris and McGeer 2005). However, long-term use of NSAID’s is limited by gastrointestinal and renal toxicity of these drugs. Attempts to mitigate the toxic side effects include development of improved delivery systems such as sustained release strategies with the use of prodrugs (Dahan and Hoffman, 2007).
2.2. Syntheses
The synthesis of the target compounds shown in Scheme 1 relied on the following key elements: 1) introduction of the oligoethylene glycol ester at the sn-2-position incorporating a chain-terminal protected amino group, 2) activation of the NSAID’s by preparation of the corresponding p-nitrophenyl esters, and 3) deprotection of the amino group, followed by attachment of the desired anti-inflammatory drug. In implementing the synthesis we used a two-prong approach: of the commercially available orthogonally functionalized oligoethylene glycol building blocks we selected OEG- carboxylic acids using both acid-labile t-BOC vs. base-labile FMOC protected amino groups to develop the best way for preparation of the target phospholipids.
Scheme 1.

Reagents and conditions: t-BOC-NH(CH2CH2O)3COOH/DCC/DMAP, CHCl3, 25°C; (b) FMOC-NH(CH2CH2O)2COOH/DCC/DMAP, CHCl3, 25°C; (c) (i) 1.0 M HCl/dioxane, (ii) Et3N/p-nitrophenyl indomethacin, CHCl3, rt, 3 h; (d) (i) 1.0 M HCl/dioxane, (ii) Et3N, CHCl3, (iii) p-nitrophenyl sulindac/DMAP, rt, 3 h; (e) (i) DBU/CHCl3, rt, 1 h, (ii) dansyl chloride/Et3N, CHCl3, rt, 1h; (f) (i) DBU/CHCl3, rt, 1 h, (ii) p-nitrophenyl dichlofenac/DMAP, 5 h, rt.
For acylation of sn-1-palmitoyl lysophosphatidylcholine 7 we used the method that we recently developed for acylation of lysophospholipids: 1) increasing the surface of the reaction vessel, where the reaction is believed to take place, by addition of glass beads, and 2) preventing intramolecular acyl migration by keeping the temperature at 25°C. Thus, we used sonication rather than stirring the reaction mixture and maintained the temperature at 25°C by cooling the sonicated vessel in a circulatory water bath (Rosseto and Hajdu, 2005). Under these conditions the reactions reached completion in 6 – 8 h and the products were obtained in good yield, with high regioselectivity. Specifically, reaction of compound 7 with t-BOC protected 11-amino-3,6,9-trioxaundecanoic acid with dicyclohexylcarbodiimide (DCC), in the presence of 4-dimethylaminopyridine (DMAP) as catalyst, produced the target compound 8 in 59% yield after purification on silica gel chromatography, while a similar reaction using the base-labile FMOC-protected 8-amino-3,6-dioxaoctanoic acid as acyl donor resulted in formation of compound 11 in 75% yield.
Acid-catalyzed deprotection of the chain-terminal amino group of compound 8 was carried out using 1.0 M anhydrous HCl solution in dioxane at room temperature for 2.5 h, and the resulting amine hydrochloride was isolated from the reaction mixture by freeze-drying. Deprotonation with anhydrous triethylamine, followed by triethylamine catalyzed introduction of the desired pharmacophore via the corresponding p-nitrophenyl active-ester yielded the NSAID substituted phosphatidylcholines of indomethacin 9 and sulindac 10 in 92% and 97% yields, respectively.
In the second series, the FMOC protecting group of compound 11 was removed by DBU-catalyzed elimination reaction in chloroform at room temperature for 1 h, followed by acylation of the amino group in the same pot, without isolating the intermediate, using the p-nitrophenyl ester of diclofenac in the presence of DMAP as catalyst, to obtain the desired phosphatidylcholine 12 in 51% overall yield.
Finally, we have explored the use of the synthetic method here developed for preparation an oligoethylene glycol-substituted phosphatidylcholine analogue 13, carrying a spectroscopically active reporter group at the sn-2-chain-terminal, shown in Scheme 1. Incorporation of fluorescent reporter groups into phospholipid analogues have been shown to be useful for both kinetic studies of lipolytic enzymes, as well as for providing a convenient way for in vivo tracking the fate of the carboxylic acid produced on the catalytic hydrolysis by PLA2 enzymes (Feng et al., 2002).
2.3. Enzymatic hydrolysis
Catalytic hydrolysis of the synthetic phosphatidylcholine analogues 8-13 was carried out with bee-venom phospholipase A2, a widely used, readily available representative of the low-molecular weight secretory PLA2 enzymes (Valentin et al., 2000). In an assay system (Roodsari et al., 1999) containing Triton X-100-phospholipid mixed micelles, in the presence of catalytically essential Ca2+, each one of the synthetic phosphatidylcholine compounds was completely hydrolyzed by the enzyme, yielding the corresponding lysophosphatidylcholine 7 and the series of oligoethylene substituted long-chain fatty acid analogues, including OEG-conjugated prodrugs of the NSAID’s indomethacin, sulindac, and diclofenac (Figure 2).
Figure 2.

The concept of using PLA2-targeted phospholipids as precursors of prodrugs
2.4. Discussion
The synthesis of the phospholipid analogues here reported provides a new way of employing oligoethylene glycol-substituted phosphatidylcholines as secretory phospholipase A2 targeted precursors of NSAID prodrugs. Specifically, polyethylene glycol modification (PEGylation) of biomolecules such as proteins, peptides as well as other small-molecular therapeutic agents continues to be a method of growing interest for development of new strategies in drug delivery, diagnostics, and for modification of the physicochemical properties of the compounds. While it has been well established, that PEGylation of a therapeutic agent extends its half-life in circulation, decreases its immunogenicity and antigenicity, and alters the pattern of drug distribution, with the availability of functionalized short-chain oligomers of ethylene glycol (OPE’s) new design strategies have become possible. Specifically, OPE’s have been shown to be useful not only for improving drug delivery (Warnecke and Kratz, 2003), but also for preparation of membrane biosensors (Chen et al., 2000), and for modulation of the physicochemical properties of membrane-targeted lipoproteins (Malolanarasimhan et al., 2007), opening the way toward the design of biomolecule analogues with targeted physical behaviors (Grogan et al., 2005). In addition, branched-chain OPE conjugates of peptides were used to incorporate fluorescent reporter groups to study receptor-ligand interactions, showing retention of the biological potency of the ligand (Bowen et al., 2007).
Preparation and phospholipase A2 hydrolysis of compounds present a new approach to design of PLA2 targeted oligoethylene glycol conjugates of pharmacophores and other biomolecules. In contrast to earlier applications of PEGylated phospholipid derivatives with polymeric ethylene glycol substituents introduced at the polar headgroup (Zalipsky et al 1999), the use of structurally well-defined oligoethylene glycol-substituted sn-2-chain here reported, offers improved targeting, flexibility in design of oligomeric linkers, and applicability for introduction of various different therapeutically active pharmacophores as well. Specifically, while NSAID’s have been shown to reduce the prevalence and severity of Alzheimer’s disease, as well as cyclooxygenase dependent anti-inflammatory and neuroprotecting effects, their long-term use has been limited by the gastrointestinal and renal toxicity exhibited by the drugs (Dahan and Hoffman, 2007). Availability of compounds such as 9, 10, and 12 may lead to development of drug delivery systems relying on sPLA2-targeted release of the respective OEG-prodrug conjugates to circumvent the problem.
Finally, the approach of sPLA2-directed drug delivery systems here illustrated may lead to design of precursors of OEG-prodrugs targeted at tissues with high levels of sPLA2 enzymes. Significantly, elevated levels of secretory phospholipase A2’s have been found in a number of tissues under pathological conditions such as prostate and colorectal cancers (Menschikowski, et al., 2008, Mounier et al., 2008), atherosclerosis (Bostrom et al., 2007, Kimura-Matsumoto et al., 2008), rheumatoid arthritis, and coronary heart disease (Khuseyinova et al., 2005, Nijmeijer,et al., 2008). Thus, the approach here presented may open the way to employ new strategies involving PLA2-aided tissue specific drug delivery for the development of improved treatments of specific pathological conditions.
3. Experimental procedures
1-Palmitoyl-2-(11’-N-t.BOC-amino-3,6,9-trioxaundecanoyl)-sn-glycerophosphocholine (8)
To a suspension of 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (0.4987 g, 1 mmol) in 25 mL of CHCl3 were added 11-N-t.BOC-amino-3,6,9-trioxaundecanoic acid (0.7011 g, 1.43 mmol), DCC (0.5218 g, 2.5 mmol), DMAP (0.3120 g, 2.5 mmol) and approximately 1g of glass beads. The reaction was sonicated for 8 h at 25 °C. The solvent was then evaporated and the residue was purified on silica gel chromatography with CHCl3/MeOH (4:1) to elute the impurities, followed by CHCl3/MeOH/H2O (65:25:4) to isolate the phospholipid. The fractions corresponding to the product were combined and freeze-dried from benzene to give 8 (0.5721 g, 59 %) as white solid. IR (CHCl3): 3342, 1741 br, 1248 cm-1. 1H NMR (CDCl3, 200 MHz) δ 0.84 (br t, 3H), 1.22 (br s, 24H), 1.40 (s, 9H), 1.56 (m, 2H), 2.24 (t, 2H, J = 7.4 Hz), 3.27 (m, 2H), 3.33 (br s, 9H), 3.42 (t, 2H, J = 9.9 Hz), 3.65 (s, 4H), 3.67 (m, 4H), 3.75 (m, 2H), 3.88 (m, 2H), 4.13 (m, 2H), 4.30 (m, 4H), 5.03-5.30 (br m, 2H). 13C NMR (CDCl3, 50 MHz) δ 14.03, 22.57, 24.76, 28.35, 29.08, 29.25, 29.44, 31.82, 33.94, 40.24, 54.26, 59.43, 62.51, 63.56, 66.21, 68.26, 70.09, 70.37, 70.69, 71.36, 80.73, 155.91, 169.93, 173.38. 31P NMR (CDCl3, 160 MHz, pyrophosphate ref. ext.) δ -1.28. Rf (CHCl3/MeOH/H2O 65:25:4) 0.41. [α]D20 +4.7° (c 0.98, CHCl3/MeOH 4:1). Anal. Calcd for C37H73N2O13P.H2O.⅓.CHCl3: C, 52.09; H, 9.06; N, 3.25; Found: C, 52.00; H, 8.82; N, 3.39. MS MH+ C37H73N2O13PH Calcd: 785.4923, Found: 785.4916.
1-Palmitoyl-2-(11’-N-indomethacincarboxylamino-3,6,9-trioxaundecanoyl)-sn-glycerophosphocholine (9)
(i) Indomethacin p-nitrophenyl ester. To a solution of indomethacin (0.5011 g, 1.4 mmol) in 30 mL CHCl3 were added p-nitrophenol (0.2102 g, 1.54 mmol), DCC (0.3203 g, 1.54 mmol) and DMAP (40 mg, 0.3 mmol). The reaction mixture was stirred for 1 h at room temperature. The DCC-urea was then filtered and the filtrate was evaporated to give the p-nitrophenyl ester of the drug; Rf (CHCl3) 0.43, which was suitable for use in the subsequent acylation step without further purification. (ii) Deprotection of the amino group of compound 2. To a solution of 2 (0.2351 g, 0.24 mmol) in 20 mL 1,4-dioxane was added 4 M HCl (6 mL) in dioxane, and the mixture was stirred for 2.5 h at room temperature. A positive ninhydrin test confirmed the presence of the free amino group in the product near the origin on a TLC plate. The amine hydrochloride was freeze-dried from benzene to give a white solid. (iii) Coupling of the indomethacin active-ester with the chain-terminal amino group. The amine hydrochloride just obtained was dissolved in 10 mL CHCl3 and the mixture was treated with excess triethylamine (0.3 mL, 2.1 mmol) to adjust the pH of the solution to about 9, followed by addition of indomethacin p-nitrophenyl ester (0.3 g, 0.63 mmol). The mixture was kept at room temperature for 3 h, when a negative ninhydrin test indicated that the reaction reached completion. The solution was loaded onto a silica gel column and chromatographed using first CHCl3/MeOH (4:1), followed by CHCl3/MeOH/H2O (65:25:4). The fractions corresponding to the product were combined, evaporated, dispersed in benzene and freeze-dried to give 9 (0.2231 g, 92%) as pale-yellow solid. IR (CHCl3): 3347, 1740 br, 1690 cm-1; 1H NMR (CDCl3, 200 MHz) δ 0.82 (br t, 3H), 1.19 (br s, 24H), 1.50 (m, 2H), 2.21 (t, 2H, J = 7.4 Hz), 2.31 (s, 3H), 3.28 (br s, 11H), 3.43 (br s, 6H), 3.54 (m, 6H), 3.76 (br s, 5H), 3.94 (m, 2H), 3.98 (m, 3H), 4.53 (m, 3H), 4.81 (br m, 1H), 5.27 (m, 1H), 6.62-6.92 (m, 3H), 7.42 (d, 2H, 8.4 Hz), 7.60 (d, 2H, 8.4 Hz). 13C NMR (CDCl3, 50 MHz) δ 13.18, 13.93, 22.48, 24.64, 28.95, 29.11, 29.14, 29.31, 29.45, 29.49, 31.70, 31.84, 33.81, 39.06, 54.11, 55.56, 59.22, 62.37, 63.29, 65.99, 68.14, 69.86, 70.13, 70.51, 71.19, 71.32, 101.25, 111.53, 113.22, 114.74, 128.97, 130.46, 130.69, 130.99, 133.56, 135.90, 139.14, 155.91, 168.07, 169.77, 169.95, 173.26. 31P NMR (CDCl3, 160 MHz, pyrophosphate ref. ext.) δ - 0.68. Rf (CHCl3/MeOH/H2O 65:25:4) 0.54. Anal. Calcd for C51H79ClN3O14P.⅓.H2O: C, 59.43; H, 7.79; N, 4.08; Found: C, 59.26; H, 7.80; N, 4.07. MS MH+ C51H79ClN3O14PH Calcd: 1024.5061, Found: 1024.5075. [α]D20 +5.8° (c 1.09, CHCl3/MeOH 4:1).
1-Palmitoyl-2-(11’-N-sulindaccarboxylamino-3,6,9-trioxaundecanoyl)-sn-glycerophosphocholine (10)
This compound was prepared following the same activation/deprotection/acylation protocol outlined for compound 9. The product 10 was obtained from the t-BOC protected precursor 8 in an overall yield of 97%. IR (CHCl3): 3355, 1739 br cm-1; 1H NMR (CDCl3, 200 MHz) δ 0.86 (br t, 3H), 1.23 (br s, 24H), 1.54 (m, 2H), 2.24 (m, 5H), 2.80 (s, 3H), 3.34 (br s, 11H), 3.51 (br s, 8H), 3.65 (m, 6H), 3.83 (m, 2H), 4.04 (m, 2H), 4.17 (m, 2H), 4.34 (m, 2H), 5.28 (m, 1H), 6.51 (br t, 1H), 6.97-7.27 (m, 3H), 7.69 (dd, 4H, J = 5.5 Hz and J = 8.9 Hz). 13C NMR (CDCl3, 50 MHz) δ 10.54, 14.06, 22.61, 24.76, 29.08, 29.24, 29.28, 29.45, 29.59, 29.63, 31.84, 33.48, 33.93, 39.30, 43.81, 54.37, 59.52, 62.35, 63.76, 66.23, 68.27, 69.59, 69.99, 70.29, 70.66, 71.19, 105.89, 106.36, 110.46, 110.91, 123.47, 123.79, 128.23, 129.62, 130.18, 133.06, 138.40, 139.55, 141.58, 145.42, 146.90, 160.77, 165.66, 169.47, 169.94, 173.40. 31P NMR (CDCl3, 160 MHz, pyrophosphate ref. ext.) δ -1.47. Rf (CHCl3/MeOH/H2O 65:25:4) 0.56. Anal. Calcd for C52H80FN2O13P.3/2.H2O.⅓CHCl3: C, 56.81; H, 7.58; N, 2.52; Found: C, 56.92; H, 7.86; N, 2.34. MS MH+ C52H80FN2O13PSH Calcd: 1023.5176, Found: 1023.5147. [α]D20 +5.7 (c 1.14, CHCl3/MeOH 4:1).
1-Palmitoyl-2-(8’-N-FMOC-amino-3,6-dioxaoctanoyl)-sn-glycerophosphocholine (11)
To a suspension of 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (0.4012 g, 0.8 mmol) in 25 mL CHCl3 were added 8-N-FMOC-amino-3,6-dioxaoctanoic acid (0.3305 g, 0.86 mmol), DCC (0.2482 g, 1.2 mmol), DMAP (0.1504 g, 1.2 mmol) and 1g of glass beads. The reaction was sonicated for 6 h at 25 °C. The solvent was then evaporated and the residue was purified by silica gel chromatography, first with CHCl3/MeOH (4:1), followed by CHCl3/MeOH/H2O (65:25:4). The fractions of the product were combined and freeze-dried from benzene to yield compound 11 (0.5134 g, 75 %) as white solid. IR (CHCl3): 3330, 1732 br, 1690 cm-1. 1H NMR (CDCl3, 200 MHz) δ 0.87 (br t, 3H), 1.24 (br s, 24H), 1.54 (m, 2H), 2.24 (t, 2H, J = 7.4 Hz), 3.30 (br s, 11H), 3.55-3.75 (br m, 6H), 3.98 (m, 2H), 4.15 (m, 4H), 4.24-4.50 (br m, 6H), 4.67 (m, 1H), 5.28 (m, 1H), 5.86 (m, 1H), 7.25-7.37 (m, 4H), 7.60 (d, 2H, J = 7.4 Hz), 7.73 (d, 2H, J = 7.4 Hz).13C NMR (CDCl3, 50 MHz) δ 13.93, 22.48, 24.63, 28.95, 29.11, 29.15, 29.32, 29.46, 31.71, 33.81, 40.60, 47.02, 54.03, 59.27, 62.33, 63.45, 65.97, 66.42, 68.15, 69.79, 70.59, 71.35, 119.75, 124.98, 126.87, 127.48, 141.04, 143.79, 156.42, 169.85, 173.26. Anal. Calcd for C45H71N2O12P.¼ CHCl3: C, 60.87; H, 8.04; N, 3.14; Found: C, 60.64; H, 8.26; N, 3.03. MS MH+ C45H71N2O12PH Calcd: 863.4817, Found: 863.4836. 31P NMR (CDCl3, 160 MHz, pyrophosphate ref. ext.) δ -0.85. Rf (CHCl3/MeOH/H2O 65:25:4) 0.57. [α]D20 +5.2° (c 0.97, CHCl3/MeOH 4:1).
1-Palmitoyl-2-(8’-N-diclofenaccarboxylamino-3,6-dioxaoctanoyl)-sn-glycerophosphocholine (12)
(i)Diclofenac p-nitrophenyl ester. To a suspension of diclofenac sodium salt (0.6362 g, 2 mmol) in 50 mL acetone was added Dowex-H+ (25 mL). The mixture was stirred for 10 min, and then it was filtered and the filtrate was evaporated to give the corresponding free acid as a white solid. This acid was dissolved in 50 mL CHCl3 and to the solution were added p-nitrophenol (0.2546 g, 1.83 mmol), DCC (0.4126 g, 2 mmol) and DMAP (52 mg, 0.43 mmol). The reaction mixture was stirred for 1 h at room temperature. The DCC-urea was removed by filtration, and the solvent was evaporated to give the p-nitrophenyl active ester of diclofenac as a yellow solid; Rf (CHCl3/EtOAc 5:1) 0.92. It was suitable to be used for the subsequent acylation step without further purification. (ii) Elimination of the FMOC protecting group of compound 11, and N-acylation at the chain-terminal. To a solution of compound 11 (0.2107 g, 0.24 mmol) in 5 mL CHCl3 was added DBU (0.1852 g, 1.2 mmol) and the mixture was kept at room temperature for 1 h. To this solution were added diclofenac p-nitrophenyl ester (0.4 g, 0.96 mmol), DMAP (42 mg, 0.34 mmol). The reaction was stopped after 5 h, when a ninhydrin test gave negative result on the spot near the origin on the TLC plate. The reaction mixture was loaded directly onto a silica gel column, eluted first with CHCl3/MeOH (4:1) to remove the impurities, followed by CHCl3/MeOH/H2O (65:25:4) to elute the pure phospholipid. The fractions corresponding to the product were combined, evaporated, dispersed in benzene and freeze-dried to give 12 (0.1125 g, 51%) as pale yellow solid. IR (CHCl3): 3330, 1737 br, 1532 cm-1; 1H NMR (CDCl3, 200 MHz) δ 0.86 (br t, 3H), 1.23 (br s, 24H), 1.54 (m, 2H), 2.25 (t, 2H, J = 7.4 Hz), 3.24 (br s, 11H), 3.42 (m, 2H), 3.54 (br m, 6H), 3.73 (m, 4H), 4.04-4.17 (br m, 4H), 4.29 (m, 2H), 4.62 (m, 1H), 5.44 (m, 1H), 6.46 (d, 1H, J = 7.5 Hz), 6.85-6.98 (m, 3H), 7.24-7.32 (m, 3H), 8.08 (br s, 1H). 13C NMR (CDCl3, 50 MHz) δ 14.05, 22.61, 24.77, 29.07, 29.27, 29.44, 29.62, 31.84, 33.95, 39.33, 40.37, 54.24, 59.37, 62.41, 63.77, 66.11, 68.31, 69.52, 70.01, 70.71, 71.48, 117.19, 121.06, 123.86, 125.49, 127.45, 128.78, 129.75, 130.67, 137.76, 143.24, 170.20, 172.44, 173.45. 31P NMR (CDCl3, 160 MHz, pyrophosphate ref. ext.) δ -1.28. Rf (CHCl3/MeOH/H2O 65:25:4) 0.56. Anal. Calcd for C44H70Cl2N3O11P.⅓.CHCl3: C, 55.54; H, 7.39; N, 4.38; Found: C, 55.58; H, 7.61; N, 4.40. MS MH+ C44H70Cl2N3O11PH Calcd: 918.4198, Found: 918.4232. [α]D20 +4.3° (c 0.87, CHCl3/MeOH 4:1).
1-Palmitoyl-2-(8’-N-[5”-dimethylaminonaphthalene-1”-sulfonyl]-amino-3,6- dioxaoctanoyl)-sn-glycerophosphocholine (13)
To a solution of compound 11 (0.2115 g, 0.24 mmol) in 5 mL CHCl3 was added DBU (0.1922 g, 1.2 mmol). The reaction mixture was kept at room temperature for 1 h. To this solution were then added dansyl chloride (0.1295 g, 0.48 mmol) and NEt3 (0.17 mL, 1.2 mmol), followed by 30 min later a second portion of dansyl chloride (0.1287 g, 0.48 mmol). The reaction was over after 1 h, as shown by a negative ninhydrin test on a TLC plate. The mixture was purified directly by silica gel chromatography using CHCl3/MeOH (4:1), to remove the impurities, followed by CHCl3/MeOH/H2O (65:25:4) to elute the target phospholipid. The fractions corresponding to the product were combined, evaporated, dispersed in benzene and freeze-dried to give compound 13 (0.1028 g, 49%) as a white solid. IR (CHCl3): 3340, 1742 br cm-1; 1H NMR (CDCl3, 200 MHz) δ 0.87 (br t, 3H), 1.24 (br s, 24H), 1.54 (m, 2H), 2.25 (t, 2H, J = 7.4 Hz), 2.85 (s, 6H), 2.97 (m, 2H), 3.32 (br s, 11H), 3.42 (m, 4H), 3.57 (m, 2H), 3.83 (m, 2H), 4.15 (br m, 4H), 4.30 (m, 2H), 4.67 (m, 1H), 5.32 (m, 1H), 7.15 (d, 1H, J = 7.4 Hz), 7.49-7.69 (m, 3H), 8.20 (d, 1H, 7.4 Hz), 8.38 (d, 1H, J = 7.4 Hz). 13C NMR (CDCl3, 50 MHz) δ 14.08, 22.64, 24.77, 29.10, 29.27, 29.31, 29.47, 29.66, 31.87, 33.95, 42.73, 45.38, 54.38, 59.65, 62.33, 64.06, 66.21, 68.40, 69.41, 69.99, 70.67, 71.50, 115.23, 119.41, 123.21, 128.26, 128.78, 129.65, 129.94, 135.59, 151.64, 170.24, 173.38. 31P NMR (CDCl3, 160 MHz, pyrophosphate ref. ext.) δ -1.72. Rf (CHCl3/MeOH/H2O 65:25:4) 0.40. Anal. Calcd for C42H72N3O12PS.CHCl3.C6H6: C, 54.92; H, 7.43; N, 3.93; Found: C, 55.27; H, 7.71; N, 3.83. MS MH+ C42H72N3O12PSH Calcd: 874.4667, Found: 874.4602. [α]D20 +5.7° (c 1.04, CHCl3/MeOH 4:1).
Enzymatic hydrolysis of the synthetic phosphatidylcholine analogues
In a typical experiment, to a sample of phosphatidylcholine 9 (5.0 mg, 5 μmole) was added a solution of 4.1 mL of 0.05 M Tris (pH 8.5) containing 10 mM Triton X-100 and 50mM CaCl2. The mixture was vortexed thoroughly, followed by incubation of the resulting dispersion at 40°C in a water bath for 10 min. The mixture was then once again vortexed and used for the phospholipase assay directly. The reaction was initiated by addition of bee-venom phospholipase A2 (8μg in 45μl buffer). The reaction was run at 40°C for 60 min using a constant temperature water bath, and formation of the products was analyzed by thin layer chromatography. The compounds were visualized on the silica gel plates by UV-absorption, iodine adsorption, and molybdic acid spray. TLC analysis (chloroform/methanol/water 65:25:4) showed complete hydrolysis of phosphatidylcholine 9 (Rf 0.56) yielding 1-palmitoyl-2-lysophosphatidylcholine 7 (Rf 0.17) and the 3,6,9- trioxaundecanylamide of indomethacin (Rf 0.90) both identified by respective standards. Under similar assay conditions phosphatidylcholines 10, 12, 13 were also completely hydrolyzed by the enzyme.
Acknowledgments
We are grateful to the National Institutes of Health, grant 2S06 GM/HD48680 for financial support.
References
- Abe T, Sakamoto K, Kamahara H, Kuwahara N, Ogawa N. Group II phospholipase A2 is increased in peritoneal and pleural effusions in patient with various types of cancer. Int J Cancer. 1997;74:245–250. doi: 10.1002/(sici)1097-0215(19970620)74:3<245::aid-ijc2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J Med Chem. 2007;50:1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- Bostrom MA, Boyanovski BB, Jordan CT, Wadsworth MP, Taatjes DJ, DeBeer FC, Webb NR. Group V secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Atheroscler Thromb Vasc Biol. 2007;27:600–606. doi: 10.1161/01.ATV.0000257133.60884.44. [DOI] [PubMed] [Google Scholar]
- Bowen ME, Monguchi Y, Sankaranarayanan R, Vagner J, Begay LJ, Xu L, Bhumasamudram J, Hruby VJ, Gillies RJ, Marsh EA. Design, synthesis and validation of a branched, flexible linker for bioactive peptides. J Org Chem. 2007;72:1675–1680. doi: 10.1021/jo062276g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyanovski BB, Webb NR. Biology of secretory phospholipase A2. Cardiovasc Drugs Ther. 2009;23:61–72. doi: 10.1007/s10557-008-6134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JL, Dennis EA. Phospholipase A2 biochemistry. Cardiovasc Drugs Ther. 2009a;23:49–59. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism and signaling. J Lipid Res. 2009b;50:5237–5242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Wong KF, Fenske DB, Palmer LR, Cullis PR. Fluorescent-labeled poly(ethylene glycol) lipid conjugates with distal cationic headgroups. Bioconjugate Chem. 2000;11:433–437. doi: 10.1021/bc990171x. [DOI] [PubMed] [Google Scholar]
- Dahan A, Hoffman A. Mode of administration-dependent brain uptake of indomethacin: sustained systemic input increases brain influx. ASPET Drug Metab Dispos. 2007;35:321–324. doi: 10.1124/dmd.106.011817. [DOI] [PubMed] [Google Scholar]
- Davidsen J, Jorgensen K, Andersen TL, Mouritsen OG. Secreted phospholipase A2 as a new enzymatic trigger mechanism for localized liposomal drug release and absorption in diseased tissue. Biochim Biophys Acta. 2003;1609:95–101. doi: 10.1016/s0005-2736(02)00659-4. [DOI] [PubMed] [Google Scholar]
- Dong O, Patel M, Scott KF, Graham GG, Russel PJ, Sved P. Oncogenic action of phospholipase A2 in prostate cancer. Cancer Lett. 2006;240:9–16. doi: 10.1016/j.canlet.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Felts AS, Ji C, Stafford JB, Crews BC, Kingsley PJ, Rouzer CA, Washington MK, Subbaramaiah K, Siegel BS, Young SM, Dannenberg AJ, Marnett LJ. Desmethyl derivatives of indomethacin and sulindac as probes for cyclooxygenase-dependent biology. ACS Chem Biol. 2007;2:479–483. doi: 10.1021/cb700077z. [DOI] [PubMed] [Google Scholar]
- Feng L, Manabe K, Shope JC, Widmer S, DeWald DB, Prestwich GD. A real- time fluorogenic phospholipase A2 assay for biochemical and cellular activity measurements. Chem Biol. 2002;9:795–803. doi: 10.1016/s1074-5521(02)00168-0. [DOI] [PubMed] [Google Scholar]
- Fuentes L, Hernandez M, Nieto ML, Crespo MS. Biological effects of a group IIA secreted phospholipase A2. FEBS Lett. 2002;531:7–11. doi: 10.1016/s0014-5793(02)03401-4. [DOI] [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Ongini E, Wenk G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of action. J Neurochem. 2004;91:521–536. doi: 10.1111/j.1471-4159.2004.02743.x. [DOI] [PubMed] [Google Scholar]
- Graff JR, Konicek BW, Deddens JA, Chedid M, Hurst BM, Colligan B, Neubaer BL, Carter HW, Carter JH. Expression of IIa secretory phospholipase A2 increases with prostate tumor grade. Clin Cancer Res. 2001;7:3857–3861. [PubMed] [Google Scholar]
- Grogan MJ, Kaizuka Y, Conrad RM, Groves JT, Bertozzi CR. Synthesis of lipidated green fluorescent protein and its incorporation in supported lipid bilayers. J Am Chem Soc. 2005;127:14383–14387. doi: 10.1021/ja052407f. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Chess RB. Effect of PEGylation on pharmaceuticals. Nat Rev Drug Discovery. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- Juillerat-Jeanneret L, Schmitt F. Chemical modification of therapeutic drugs or drug vector systems to achieve targeted therapy: looking for the grail. Med Res Rev. 2007;27:574–590. doi: 10.1002/med.20086. [DOI] [PubMed] [Google Scholar]
- Kennedy BP, Soravia C, Moffat J, Xia I, Hiruki T, Collins S, Gallinger S, Bapat B. Overexpression of nonpancreatic secretory group II PLA2 messenger RNA and protein in colorectal adenomas from familial adenomatous polyposis patients. Cancer Res. 1998;58:500–503. [PubMed] [Google Scholar]
- Khuseyinova N, Imhof A, Rothenbacher D, Trischler G, Kuelb S, Scharnagl H, Maerz W, Brenner H, Koenig W. Association between Lp-PLA2 and coronary artery disease: focus on its relationship with lipoproteins and markers of inflammation and hemostasis. Atherosclerosis. 2005;182:181–188. doi: 10.1016/j.atherosclerosis.2004.10.046. [DOI] [PubMed] [Google Scholar]
- Kimura-Matsumoto M, Ishikawa Y, Komiyama K, Tsurata T, Murakami M, Masuda S, Akasaka Y, Ito K, Ishiguro S, Ishii T. Expression of secretory phospholipase A2’s in human atherosclerosis development. Atherosclerosis. 2008;196:81–91. doi: 10.1016/j.atherosclerosis.2006.08.062. [DOI] [PubMed] [Google Scholar]
- Klegeris A, McGeer PL. Non-steroidal anti-inflammatory drugs (NSAIDs) and other anti-inflammatory agents in the treatment of neurodegenerative disease. Curr Alzheimer Res. 2005;2:355–365. doi: 10.2174/1567205054367883. [DOI] [PubMed] [Google Scholar]
- Kuipers OP, Dekker N, Verheij HM, DeHaas GH. Activities of native and tyrosine- 69 mutant phospholipase A2 on phospholipid analogues. A reevaluation of minimal substrate requirements. Biochemistry. 1990;29:6094–6102. doi: 10.1021/bi00477a029. [DOI] [PubMed] [Google Scholar]
- Malolanarasimhan K, Kedei N, Sigano D, Kelley JA, Lai CC, Lewin NE, Surawski RJ, Pavlyukovets VA, Garfield SH, Wincovitch S, Blumberg PM, Marquez VE. Conformationally constrained analogues of diacylglycerol (DAG). 27. Modulation of membrane translocation of protein kinase C (PKC) isozymes α and δ by diacylglycerol lactones (DAG-lactones) containing rigid-rod acyl groups. J Med Chem. 2007;50:962–978. doi: 10.1021/jm061289j. [DOI] [PubMed] [Google Scholar]
- Marcus Y, Sasson K, Fridkin M, Shechter Y. Turning low molecular weight drugs into prolonged acting prodrugs by reversible pegylation: a study with gentanamicin. J Med Chem. 2008;51:4300–4305. doi: 10.1021/jm8002558. [DOI] [PubMed] [Google Scholar]
- Marsac Y, Cramer J, Olschewsky D, Alexandrov K, Becker CFW. Site-specific attachment of polyethylene glycol-like oligomers to proteins and peptides. Bioconjugate Chem. 2006;17:1492–1498. doi: 10.1021/bc0601931. [DOI] [PubMed] [Google Scholar]
- Menschikowski M, Hagelgans A, Gussakovski E, Kostka H, Paley EL, Siegert G. Differential expression of phospholipase A2 in normal and malignant prostate cell lines: regulation by cytokines, cell signaling pathways, and epigenic mechanisms. Neoplasia. 2008;10:279–286. doi: 10.1593/neo.07965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier CM, Wendum D, Greenspan E, Flejou J-F, Rosenberg DW, Lambeau G. Distinct expression of the full set of secreted phospholipase A2 in human colorectal adenocarcinomas: sPLA2-III as a biomarker candidate. Br J Cancer. 2008;98:587–595. doi: 10.1038/sj.bjc.6604184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevalainen TJ, Haapamaki MM, Gronroos JM. Roles of secretory phospholipase A2 in inflammatory diseases and trauma. Biochim Biophys Acta. 2000;1488:83–90. doi: 10.1016/s1388-1981(00)00112-8. [DOI] [PubMed] [Google Scholar]
- Nevalainen TJ, Graham CG, Scott KF. Antibacterial actions of secreted phospholipase A2. Biochim Biophys Acta. 2008;1781:1–9. doi: 10.1016/j.bbalip.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Nijmeijer R, Meuwissen M, Krijnen PAJ, Van Der Wal A, Piek JJ, Visser CA, Hack CE, Niessen HWM. Secretory type II phospholipase A2 in culprit coronary lesions is associated with myocardial infraction. Eur J Clin Invest. 2008;38:205–210. doi: 10.1111/j.1365-2362.2008.01933.x. [DOI] [PubMed] [Google Scholar]
- Prescott SM, Zimmerman GA, Stafforini DM, McIntire TM. Platelet-activating factor and related lipid mediators. Annu Rev Biochem. 2000;69:419–445. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- Prusakiewicz JJ, Felts AS, Mackenzie BS, Marnett LJ. Molecular basis of the time-dependent inhibition of cyclooxygenases by indomethacin. Biochemistry. 2004;43:15439–15445. doi: 10.1021/bi048534q. [DOI] [PubMed] [Google Scholar]
- Rivera R, Chun J. Biological effects of lysophospholipids. Rev Physiol Biochem Pharmacol. 2008;160:25–46. doi: 10.1007/112_0507. [DOI] [PubMed] [Google Scholar]
- Roodsari FS, Wu D, Pum GS, Hajdu J. A new approach to the synthesis of phospholipids. The use of L-glyceric acid for preparation of diacylglycerols, phosphatidylcholines and related derivatives. J Org Chem. 1999;64:7727–7737. [Google Scholar]
- Rosseto R, Hajdu J. A rapid and efficient method for migration-free acylation of lysophospholipids: synthesis of phosphatidylcholines with sn-2-chain-terminal reporter groups. Tetrahedron Lett. 2005;46:2941–2944. [Google Scholar]
- Rouault M, LeCalvez C, Boilard E, Surrel F, Singer A, Ghomashchi F, Bezzine S, Scarzello S, Bollinger J, Gelb MH. Recombinant production and properties of binding of the full set of mouse secreted phospholipase A2 to the mouse M-type receptor. Biochemistry. 2007;46:1647–1662. doi: 10.1021/bi062119b. [DOI] [PubMed] [Google Scholar]
- Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Scott DL, White SP, Otwinowski Z, Yuan W, Gelb MH, Sigler PB. Interfacial catalysis: the mechanism of phospholipase A. Science. 1990;250:1541–1546. doi: 10.1126/science.2274785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six DA, Dennis EA. The expanding superfamily of phospholipase A2 enzymes, classification and characterization. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- Valentin E, Ghomashchi F, Gelb MH, Ladzunski M, Lambeau G. Novel human secreted phospholipase A2 with homology to the group III bee venom enzyme. J Biol Chem. 2000;275:7492–7496. doi: 10.1074/jbc.275.11.7492. [DOI] [PubMed] [Google Scholar]
- Veld BA, Ruitenberg A, Hoffman A, Launer LJ, van Duin CM, Stijnen T, Breteler MMB, Stricker BHC. Nonsteroidal anti-inflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Veronese FM, Pasut G. PEGylation, a successful approach to drug delivery. Drug Discov Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- Warnecke A, Kratz F. Maleimide-oligo(ethylene glycol) derivatives of camptothecin as albumin-binding prodrugs: synthesis and antitumor efficacy. Bioconjugate Chem. 2003;14:377–387. doi: 10.1021/bc0256289. [DOI] [PubMed] [Google Scholar]
- Zalipsky S, Qazen M, Walker JA, Mullah N, Quinn YP, Huang SK. New detachable poly(ethylene glycol) conjugates: cysteine-cleavable lipopolymers regenerating natural phospholipid, diacyl phosphatidylethanolamine. Bioconjugate Chem. 1999;10:703–707. doi: 10.1021/bc990031n. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Natarajan V. Lysophosphatidic acid signaling in airway epithelium: role in airway inflammation and remodeling. Cellular Signaling. 2009;21:367–377. doi: 10.1016/j.cellsig.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]