

Abstract
Background
Two novel genetic polymorphisms on chromosome 6q23 are associated with susceptibility to rheumatoid arthritis (RA). Both polymorphisms (rs6920220 and rs10499194) reside in a region close to the gene encoding tumour necrosis factor α-induced protein 3 (TNFAIP3). TNFAIP3 is a negative regulator of NF-κB and is involved in inhibiting TNF-receptor-mediated signalling effects. Interestingly, the initial associations were detected in patients with longstanding RA. However, no association was found for rs10499194 in a Swedish cohort with early arthritis. This might be caused by over-representation of patients with severe disease in cohorts with longstanding RA.
Objective
To analyse the effect of the 6q23 region on the rate of joint destruction.
Methods
Five single nucleotide polymorphisms in 6q23 were genotyped in 324 Dutch patients with early RA. Genotypes were correlated with progression of radiographic joint damage for a follow-up time of 5 years.
Results
Two polymorphisms (rs675520 and rs9376293) were associated with severity of radiographic joint damage in patients positive for anti-citrullinated protein/peptide antibodies (ACPA). Importantly, the effects were present after correction for confounding factors such as secular trends in treatment.
Conclusions
These data associate the 6q23 region with the rate of joint destruction in ACPA+ RA.
Introduction
Recent whole-genome association scans have disclosed novel genetic polymorphisms associated with susceptibility to anti-citrullinated protein/peptide antibody-positive (ACPA+) rheumatoid arthritis (RA).1 2 Among those, two single nucleotide polymorphisms (SNPs), rs6920220 (A allele) and rs10499194 (C allele), were found to be independently associated with ACPA+ disease. Both SNPs map to a single linkage disequilibrium block spanning ~60 kb in a region on chromosome 6q23 that lacks known genes or transcripts. The closest genes are oligodendrocyte lineage transcription factor 3 (OLIG3) and tumour necrosis factor α-induced protein 3 (TNFAIP3). The latter is of potential importance to RA pathogenesis, as the protein TNFAIP3 acts as a negative regulator of NF-κB.3 So far, however, functional relevance of the reported polymorphisms is unknown.
rs6920220 was initially identified in ACPA+ patients with RA (minor allele OR=1.38) originating from the United Kingdom (UK).1 It was further replicated in an extended UK-based case–control study.4 rs10499194 was initially identified in North American ACPA+ patients (the Brigham Rheumatoid Arthritis Sequential Study, BRASS; minor allele OR=0.67).2 Replication was successful in two additional US cohorts selected from the North American Rheumatoid Arthritis Consortium (NARAC). Replication failed, however, in ACPA+ patients of a Swedish population-based inception cohort (the Epidemiological Investigation of Rheumatoid Arthritis cohort, EIRA).2 This last finding is of interest, as BRASS and NARAC are cohorts of patients with longstanding RA (mean disease duration BRASS: 15.4±12.8 years5; NARAC: 14.3±11.1 years6). The EIRA study, however, was designed to identify incident cases of RA as soon as possible after disease onset, resulting in an estimated mean disease duration at inclusion of only 10 months.7
Association of a genetic polymorphism in cohorts of patients with longstanding disease but absence of this association in an early arthritis cohort led us to hypothesise that the 6q23 region would be associated with disease severity in ACPA+ patients. Very little information is currently available on the effects of genetic variation on outcome measures in RA.8 Therefore, we genotyped five SNPs in a Dutch early arthritis cohort (the Leiden Early Arthritis Clinic, EAC) and correlated genotyping data to progression of radiographic joint damage for a maximum follow-up of 5 years.
Patients and methods
Patients
The Leiden EAC is a population-based inception cohort that includes patients with self-reported symptom duration of ≤2 years.9 DNA samples of 324 patients consecutively included between 1993 and 2003 were used for analysis. For further details see online supplementary file 1.
SNP selection and genotyping
Five SNPs (rs1878658, rs675520, rs9376293, rs10499194 and rs6920220) were selected based on a haplotype analysis across the 6q23 locus published previously.2 All SNPs are in imperfect linkage disequilibrium with one another (supplementary table 1). Genotyping was performed using predesigned TaqMan allelic discrimination probes (Applied Biosystems, Foster City, California, USA).Each 384-well plate contained 10 ng sample DNA per well and at least eight negative and six positive controls. Genotype calls and clusters were manually checked for discrepancies and doubtful calls were rejected. No SNP deviated from Hardy–Weinberg equilibrium. Genotyping call rates were 96.5% (rs1878658), 98% (rs675520), 95% (rs9376293), 94% (rs10499194) and 98.1% (rs6920220).
Table 1.
Median Sharp van der Heijde scores (M) and interquartile ranges (IQR; 25%–75% centiles) per genotype for patients with ACPA+ RA (n = number of patients). Genotypes were combined for rs1878658 (G), rs10499194 (T) and rs6920220 (A), as the frequency of patients homozygous for the respective minor allele was ≤5% for these single nucleotide polymorphisms
| Year | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | |||||||||||||
| ACPA+ | M | IQR | n | M | IQR | n | M | IQR | n | M | IQR | n | M | IQR | n | M | IQR | n |
| rs1878658 | ||||||||||||||||||
| AA | 6 | 2–13 | 126 | 14 | 6–24 | 111 | 18 | 10–32 | 113 | 22.5 | 11–42 | 98 | 26.5 | 14–51.5 | 86 | 32 | 14–51.5 | 89 |
| AG/GG | 3 | 1–7 | 40 | 12 | 5.5–20.5 | 40 | 17 | 6–32 | 39 | 30 | 11.5–56.5 | 34 | 32 | 16–69 | 31 | 27 | 17–74 | 31 |
| rs675520 | ||||||||||||||||||
| AA | 9 | 4.5–13 | 33 | 14 | 6.5–21 | 32 | 15 | 8–27.5 | 33 | 19 | 9–34 | 27 | 22 | 13.5–39.5 | 24 | 20 | 12.5–47.5 | 21 |
| AG | 5 | 1–10 | 83 | 12 | 5–21 | 73 | 18 | 9.5–32.5 | 78 | 25 | 11.5–54 | 68 | 32.5 | 14–61 | 58 | 33 | 17–62.5 | 66 |
| GG | 5 | 2–11 | 51 | 14 | 7–27 | 48 | 18 | 8–37 | 43 | 26 | 14–48 | 39 | 28 | 16–63 | 37 | 32 | 20–64 | 35 |
| AG/GG | 5 | 2–10 | 134 | 13 | 6–24.5 | 121 | 18 | 9.5–33 | 121 | 26 | 12–53 | 107 | 32 | 15–61 | 95 | 32 | 18.5–61 | 101 |
| rs9376293 | ||||||||||||||||||
| CC | 6.5 | 3–13.5 | 28 | 15 | 9–28 | 27 | 27 | 12–49.5 | 25 | 33 | 15.5–58 | 24 | 35 | 16.5–62 | 24 | 35 | 16.5–56 | 21 |
| CT | 4 | 1–9 | 84 | 12 | 5–21 | 79 | 17 | 7–32.5 | 78 | 21 | 10.5–54 | 66 | 32 | 14–68 | 57 | 36 | 20–68 | 61 |
| TT | 8 | 3–13 | 54 | 14 | 6–21 | 46 | 16 | 10.5–25.5 | 50 | 21 | 12–30.5 | 44 | 23 | 13.5–38 | 38 | 23 | 13–47 | 40 |
| CC/CT | 4 | 2–10 | 112 | 13 | 6–26.5 | 106 | 18 | 8–36 | 103 | 28 | 11–54.5 | 90 | 34 | 15–63 | 81 | 36 | 19–67.5 | 82 |
| rs10499194 | ||||||||||||||||||
| CC | 7 | 2–12.5 | 90 | 14 | 6–21.5 | 77 | 18 | 9.5–28 | 82 | 21 | 12–35 | 71 | 25.5 | 14–45 | 62 | 29 | 14–48 | 62 |
| CT/TT | 4 | 2–7.5 | 65 | 12 | 6–24.5 | 64 | 17 | 7–37 | 61 | 31 | 11–55 | 55 | 29.5 | 14.5–67 | 50 | 32 | 15–68 | 51 |
| rs6920220 | ||||||||||||||||||
| AA/AG | 7 | 2–12 | 58 | 14 | 6–21 | 53 | 20 | 10–32.5 | 57 | 24 | 12–48 | 47 | 32 | 18–56 | 35 | 35.5 | 18–65 | 42 |
| GG | 4.5 | 2–10.5 | 108 | 13 | 6–25 | 99 | 16.5 | 8–30.5 | 96 | 22 | 11–48 | 86 | 25 | 14–61 | 84 | 24 | 14–51 | 79 |
ACPA, anti-citrullinated protein/peptide antibodies; RA, rheumatoid arthritis.
Serology and radiographs
Serum samples were tested for citrulline-specific IgG antibodies using a commercially available ELISA kit (Immunoscan Mark2; Eurodiagnostica, The Netherlands). Radiographs were scored according to the Sharp van der Heijde method10 with known time order by one blinded, independent trained reader (supplementary file 1).
Statistical analysis
Association between genotypes and radiographic scoring data was analysed using SPSS version 16.0 (SPSS, Chicago, Illinois, USA). p Values <0.05 were considered significant. All p values reported are two-sided.
Two approaches were chosen for statistical analysis. First, the average increase in Sharp van der Heijde scores during the follow-up period was estimated for each person by regression analysis. Subsequently, the average increase (slope) of scores for each genotype was compared non-parametrically using the Mann–Whitney rank-sum test.
We observed an influence of the time of inclusion (1993–2003) on the progression of radiographic joint damage, probably reflecting an improvement of treatment intensity during this 10-year time period. To account for this effect, we performed, as a second approach, a mixed model analysis described in detail in supplementary file 1.
Results
Radiographic scores of 324 Dutch patients with RA (181 ACPA+, 143 ACPA−) were available for analysis. At least five radiographic follow-up observations were available in 57% of patients. A dominant model was chosen for analysis, as the frequency of patients homozygous for the minor allele of rs1878658 (G), rs10499194 (T) and rs6920220 (A) was ≤5%.Figure 1 depicts the influence of genotypes on radiographic joint damage. ACPA+ and ACPA− subgroups were analysed separately. Median scores and interquartile ranges (IQR) are provided for ACPA+ patients in table 1 (for ACPA− patients, see supplementary table 2).
Figure 1.
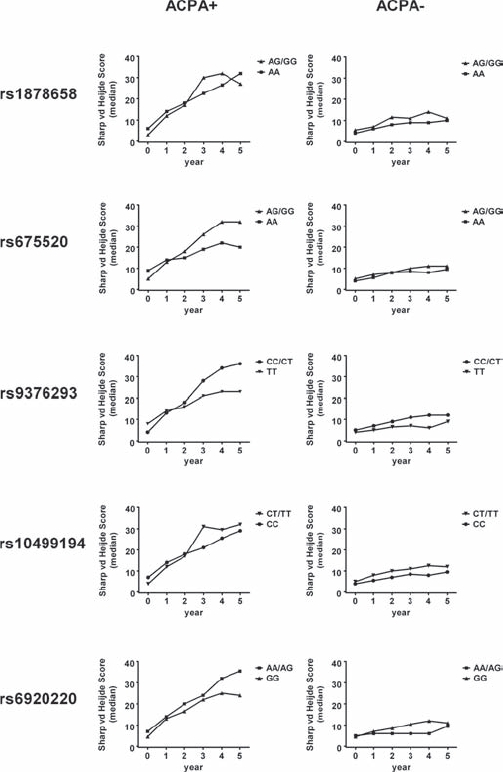
Development of median Sharp van der Heijde scores plotted according to genotype/allele in ACPA+ (left column) and ACPA− (right column) patients with RA. Year 0 equals baseline-values. Regression analysis was performed in order to estimate the average increase (slope) in Sharp van der Heijde scores over time. Slopes were subsequently compared using the non-parametric Mann–Whitney test (for the ACPA+ subgroup: p=0.37 (rs1878658); p=0.007 (rs675520); p=0.021 (rs9376293); p=0.05 (rs10499194); p=0.76 (rs6920220)).
No influence of genotypes on radiographic joint damage was seen in ACPA− patients (fig 1). In ACPA+ patients, however, two polymorphisms showed reproducible association with disease progression over time. Presence of the G allele of rs675520 was found to be associated with increased Sharp van der Heijde scores, as a significant difference was observed when the average increase (slope) in radiographic scores over time was compared with G as the dominant allele (median slope AG/GG=4.6, AA=2.3; Mann–Whitney p=0.007). In order to account for an effect of improving treatment strategies on radiographic progression during the 10-year period in which patients were included in the study, we next performed a mixed-model analysis. This analysis identified the year of inclusion as a significant variable influencing the extent of radiographic joint damage (p=0.005). After correcting for the year of inclusion, however, we still observed a significant influence of the G allele of rs675520 (AG/GG vs AA, p=0.026).
Similar to the G allele of rs675520, we noted an influence of the C allele of rs9376293 on progression of radiographic joint damage (fig 1). The average increase (slope) in Sharp van der Heijde scores over time was significantly higher for C allele carriers than for T homozygotes (median slope CC/CT=4.5, median slope TT=3.0, Mann–Whitney p=0.021). After correcting for the year of inclusion as described above a trend effect of the C allele remained (p=0.097).
For rs1878658, rs10499194 and rs6920220, no significant influence of individual genotypes on radiographic joint damage was noted.
Discussion
The 6q23 region has recently been associated with disease susceptibility in RA. This region contains no known transcripts. The closest genes with known function are OLIG3 and TNFAIP3. TNFAIP3 encodes protein A20, a TNFα-induced negative regulator of NF-κB.3 11 Decreased levels of A20 lead to uncontrolled NF-κB activity, resulting in increased inflammation. This observation makes TNFAIP3/A20 and the 6q23 region interesting candidates which may modulate inflammation also in RA.
We were intrigued by recent differential findings for rs10499194, a SNP on chromosome 6q23 close to TNFAIP3, in cohorts with differing disease duration. The major allele (C) was found to be associated with disease susceptibility in ACPA+ patients with RA in three cohorts with longstanding disease, but not in an early arthritis cohort.2 This indicated a potential impact of the 6q23 region on disease severity. In order to test for such an impact, five SNPs were genotyped in a cohort of Dutch patients with early RA. These SNPs had previously been shown to identify common haplotypes in 6q23.2 We identified two SNPs for which the presence of alleles was associated with increased joint destruction in ACPA+ patients. Carriers of the G allele of rs675520 developed increased Sharp van der Heijde scores over time. A similar effect, although weaker, was found for the C allele of rs9376293. Interestingly, no association was found for any of the SNPs in ACPA− subjects. Although this does not exclude the possibility of a contribution of the 6q23 region to disease severity in ACPA− disease, the latter observation is in line with recent reports detecting an association of the 6q23 region with disease susceptibility in ACPA+ patients only.4
No effect on disease severity was observed for rs10499194 and rs6920220. Based on our data we cannot rule out the possibility that either SNP exerts a weak effect that requires larger sample numbers for detection or that cannot be observed during the first years of disease. Interestingly, we observed nominally higher scores for the risk-conferring A allele of rs6920220 without reaching statistical significance. The discrepancy between SNPs associating with susceptibility and radiographic progression also indicates that the causal variant at this locus has not yet been identified. Given the large area of linkage disequilibrium surrounding these SNPs, further fine-mapping and functional characterisation will have to be performed.
Data linking newly identified genetic polymorphisms to disease outcome in RA are only beginning to emerge. Our data are unique, as they cover a long period of radiographic follow-up and have been scrutinised for artefacts such as secular trends in treatment intensity. Albeit based on relatively small patient numbers, our data indicate a contribution of the 6q23 region to the rate of joint destruction in ACPA+ RA, thereby further refining our understanding of the effects exerted by this locus. Replication of our findings in other cohorts is needed. Nonetheless, this is the first study demonstrating such an effect for genetic polymorphisms located outside the HLA-region in ACPA+ patients with RA.
Acknowledgments
We are grateful to Robert M. Plenge for sharing genotyping data on polymorphisms in 6q23.
Footnotes
Funding: Dutch Arthritis Foundation; European Community FP6 funding project 018661 Autocure and FP7 funding project Masterswitch; Pfizer Articulum Fellowship; Centre for Medical Systems Biology (CMSB) within the framework of the Netherlands Genomics Initiative (NGI); The Netherlands Organization for Health Research and Development.
Competing interests: None.
Ethics approval: This study was conducted with the approval of the Institutional Review Board of Leiden University Medical Centre, Leiden , The Netherlands.
Contributors: HUS designed the study, performed genotyping, analysed and interpreted data and wrote the manuscript; MPMvdL performed x-ray scoring; GSR performed genotyping; SlC performed statistical analysis; FASK, TWJH, AHvdH and REMT designed the study, interpreted data and critically drafted and revised the manuscript. All authors approved the submitted version.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plenge RM, Cotsapas C, Davies L, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet 2007;39:1477–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wertz IE, O'Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004;430:694–9 [DOI] [PubMed] [Google Scholar]
- 4.Thomson W, Barton A, Ke X, et al. Rheumatoid arthritis association at 6q23. Nat Genet 2007;39:1431–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karlson EW, Chibnik LB, Cui J, et al. Associations between human leukocyte antigen, PTPN22, CTLA4 genotypes and rheumatoid arthritis phenotypes of autoantibody status, age at diagnosis and erosions in a large cohort study. Ann Rheum Dis 2008;67:358–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jawaheer D, Lum RF, Amos CI, et al. Clustering of disease features within 512 multicase rheumatoid arthritis families. Arthritis Rheum 2004;50:736–41 [DOI] [PubMed] [Google Scholar]
- 7.Stolt P, Bengtsson C, Nordmark B, et al. ; EIRA study group. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis 2003;62:835–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Helm-van Mil AH, Padyukov L, Toes RE, et al. Genome-wide single-nucleotide polymorphism studies in rheumatology: Hype or hope? Arthritis Rheum 2008;58:2591–7 [DOI] [PubMed] [Google Scholar]
- 9.van Aken J, van Bilsen JH, Allaart CF, et al. The Leiden Early Arthritis Clinic. Clin Exp Rheumatol 2003;21(5 Suppl 31):S100–5 [PubMed] [Google Scholar]
- 10.van der Heijde DM. Plain X-rays in rheumatoid arthritis: overview of scoring methods, their reliability and applicability. Baillieres Clin Rheumatol 1996;10:435–53 [DOI] [PubMed] [Google Scholar]
- 11.Opipari AW, Jr, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem 1990;265:14705–8 [PubMed] [Google Scholar]