

Abstract
Emerging data suggests that host immune cells with a suppressive phenotype represent a significant hurdle to successful therapy for metastatic cancer. Among the suppressor cells, T regulatory cells (Treg) and myeloid-derived suppressor cells (MDSC) are significantly increased in hosts with advanced malignancies. MDSC mediate the suppression of the tumor antigen-specific T-cell response through the induction of T-cell anergy and the development of Treg in tumor-bearing mice. These results provide robust evidence of an in vivo immunoregulatory function of MDSC in the establishment of tumor antigen-specific tolerance and the development of Treg in tumor-bearing hosts. To achieve effective anti-tumor immunity, tumor-induced immunosuppression must be reversed. Our preliminary results indicate that c-kit ligand (stem cell factor) expressed by tumor cells may be required for MDSC accumulation in tumor-bearing mice, and that blocking the c-kit ligand/c-kit receptor interaction can prevent the development of Treg and reverse immune tolerance induced by MDSC. Since c-kit can be readily inhibited by several small molecule inhibitors including imatinib, sunitinib and dasatinib, targeting immune suppressing cells can be readily accomplished in the clinic.
Keywords: Myeloid-derived suppressor cells, sunitinib, stem cell factor (c-kit), radiation therapy, tumor immunotherapy, T regulatory cells
1. The tumor microenvironment is immunosuppressive
Within the last two decades, oncology research has increasingly focused on the role of biologics in cancer treatment [1-11]. Many classes of these agents have already been approved for clinical use, including monoclonal antibodies to tumor-associated antigens (e.g., trastuzumab, bevacizumab or rituximab) or small molecule inhibitors to key signaling pathways (e.g., tyrosine kinase inhibitors such as imatinib, sunitinib and erlotinib). As these agents have been developed, their anti-tumor effects, both direct and indirect, have become better defined from both preclinical and clinical studies [12]. The interplay between the host immune system and the tumor has become a significant topic of interest because of evidence that they profoundly influence each other [13, 14].
There is ample evidence from the literature on the association between host immune response and prognosis in a variety of tumor types, including breast, colorectal, melanoma, ovarian, head and neck, prostate and bladder cancer [15-17]. One of the major intermediaries in this process is the tumor microenvironment, where many of these interactions occur. The tumor microenvironment is a heterogeneous mix of cellular and noncellular components. Primary tumors consist of cancer cells and stromal cells, including endothelial cells, pericytes, fibroblasts, and many other cells that are mostly of hematopoietic lineage [14]. The bone marrow-derived cells have been shown to play key roles in modulating the immune response and angiogenesis [18]. Bone marrow derived cells, particularly those of myeloid origin, play important roles in promoting tumor progression, immune escape, angiogenesis, and metastasis [19, 20]. These cellular populations are quite heterogeneous and their defining characteristics, whether by morphologic or antigenic phenotyping, are often controversial. Some of these subsets, including immature toleragenic dendritic cells and regulatory T cells, have served either as targets for modulating the immune response, or as proxies for determining whether the host immune response has been biased in favor or against an anti-tumor immune response [17, 21].
To date, the role of tumor immunotherapy remains poorly defined, and there remains a wide discrepancy between impressive anti-tumor findings in the preclinical setting and the limited clinical results. For example, although significant regressions of bulky tumors have been observed in preclinical models utilizing IL-2 and interferon-alpha, the clinical response rate in melanoma and renal cell carcinoma has been less than 10% [7, 22-24]. This failure of immunotherapy has been attributed to a suboptimal level of anti-tumor immunity and the evasion of an immune response by the tumor. In the first scenario, there may be defects in the elicitation or maintenance of an effective antitumor immune response, including (1) insufficient antigen presentation by dendritic cells; (2) poor recruitment of effector cells; or (3) poor activation of these effectors. In the latter scenario, among the tumor evasion mechanisms it is known that tumors not only have the potential to downregulate tumor-associated HLA class I antigens and downregulation of antigen processing machinery components but they may also produce immunosuppressive cytokines [13, 15, 25].
2. Myeloid-derived suppressor cells are associated with immunosuppression
The presence of several cell subsets including T regulatory cells (Treg), type 2 tumor associated macrophages and myeloid derived suppressor cells (MDSC) have also been shown to contribute to an immunosuppressive tumor microenvironment. There has been a high level of recent interest in MDSC, a heterogeneous population of immature myeloid cells that are defined as CD11b+Gr1+ in mice and CD11b+CD14-CD33+ or Lin-HLA-DR-CD33+ in humans [13, 26]. Approximately 5% of total cells in experimental tumors are comprised of MDSC [19]. MDSC are characterized by their potent ability to suppress T and NK cell function via increased expression of arginase I and inducible nitric oxide synthase, and increased production of reactive oxygen species [19]. Additionally, MDSC promote the development of forkhead box P3 (FoxP3)+ regulatory T cells and modulate cytokine production by macrophages.
In mice, granulocytic MDSC (CD11b+LY6G+LY6Clow with high side scatter) and monocytic MDSC (CD11b+LY6G-LY6Chi) have been identified [27]. The terminally differentiated granulocytic MDSC represent 70-80% of MDSC and generate reactive oxygen species [27, 28]. Monocytic MDSC, accounting for 20-30% of MDSC, retain the ability to differentiate into mature dendritic cells and macrophages, and produce reactive nitrogen species [27, 28]. In healthy individuals, MDSC readily differentiate into mature granulocytes, macrophages or dendritic cells [30]. In pathological states, such as cancer, MDSC accumulate in blood, bone marrow and spleen of cancer patients and tumor-bearing mice [12]. As a result, while MDSC constitute <1% of peripheral blood mononuclear cells in healthy individuals, they increase by a factor of 4 to 10-fold depending on tumor type and stage [29, 30]. MDSC expansion is associated with changes in the expression of COX-2, prostaglandins, stem-cell factor (c-kit), M-CSF, IL-6, GM-CSF, BV8, S100A9, VEGF and STAT3 [14, 31-34]. (See Figure 1) Increased levels of MDSC are associated with poor prognosis in multiple tumor types [29, 35]. Furthermore, MDSC and tumor-associated macrophage crosstalk increased production of IL-10 and decreased IL-2, biasing towards a less favorable Th2 type immune response [36].
Figure 1.
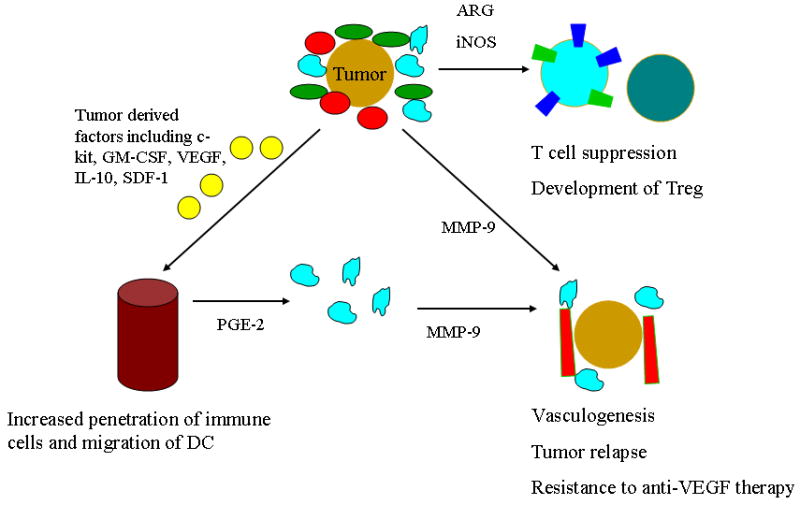
Role of MDSC in regulating immune response, vasculogenesis, and tumor progression.
3. CD11b+Gr1+ bone marrow derived cells augment tumor blood vessel development
CD11b+Gr1+ bone marrow derived cells have also been implicated in angiogenesis and vasculogenesis, another mechanism by which they promote tumor progression (Figure 1). Murine tumors that were refractory to anti-VEGF therapy were able to prime and recruit CD11b+Gr1+ bone marrow mononuclear cells [37]. Subsequent experiments suggest that tumor implantation results in Bv8 upregulation in CD11b+Gr1+ cells [38]. Bv8 or G-CSF expression is important in mobilizing CD11b+Gr1+ cells from the bone marrow and MDSC-dependent angiogenesis [38, 39].
Furthermore, MDSC secretion of MMP9 has been associated with increased tumor growth and vasculogenesis and Gr1+CD11b+ cells appear to be incorporated into neovasculature [19]. Gr1+CD11b+ cells are noted in the invasive front of tumors and are associated with increased secretion of TGF-β1 and metastasis. A preclinical model of tumor recurrence following high-dose radiation that is lethal to tumor endothelial cells demonstrates that bone marrow derived CD11b+ cells expressing MMP9 are responsible for vasculogenesis in tumors transplanted in irradiated tissue [40]. Taken together, these data suggest that targeting CD11b+Gr1+ bone marrow derived cells is an attractive strategy to overcome resistance to immune therapy, angiogenesis inhibition and conventional cytotoxic cancer therapy.
4. Limiting MDSC accumulation enhances antitumor immunity
One preclinical model for studying immune suppression in bulky tumors that are resistant to immune therapy was recently described. A combination of adenovirus-mediated gene delivery of mIL-12 plus systemic administration of an agonistic monoclonal antibody (mAb) against the co-stimulatory molecule 4-1BB had a synergistic effect on inducing potent anti-tumor responses and tumor eradication in mice bearing small (5×5-7×7 mm2) MCA26 colon tumors [41]. However, the survival rate of IL-12 + anti-4-1BB mAb treated animals was significantly decreased in animals with large (≥9×9 mm2) tumors. Furthermore, the anti-4-1BB-induced CTL activity of tumor infiltrating leukocytes (TILs) decreased dramatically in mice bearing large tumors, consistent with immune suppression. Recent work in our laboratory demonstrated that tumor growth induces accumulation of both Gr-1+CD11b+ MDSC and T regulatory cell development in tumor-bearing mice [42]. MDSC can suppress T cell immune response and induce tumor specific Treg development and T cell tolerance during tumor progression. Gene expression profile analysis of multiple tumor types identified SCF (c-kit ligand) as a candidate tumor factor involved in MDSC accumulation. It was hypothesized that SCF secreted by tumor cells may regulate the accumulation of MDSC by simultaneously enhancing myelopoiesis and attenuating monocyte/granulocyte/DC differentiation. The siRNA knockdown of SCF expression in tumor cells significantly reduced MDSC accumulation and reverted immune tolerance of the tumor microenvironment. More importantly, blockade of SCF receptor (c-kit)-SCF interaction by the use of anti-c-kit in tumor-bearing mice prevented tumor antigen-specific T cell anergy, Treg development, and tumor angiogenesis [33]. Furthermore, the prevention of MDSC accumulation in conjunction with immune activation therapy showed synergistic therapeutic effect when treating mice with large tumor burdens [33].
5. Sunitinib inhibits MDSC accumulation and reverses immune tolerance
Based on these data, we hypothesized that clinically available inhibitors of c-kit could reverse MDSC-mediated immune suppression and modulate the tumor microenvironment. Sunitinib is a small molecule tyrosine kinase inhibitor with potency against vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), c-kit, FLT3, colony-stimulating factor 1 (CSF-1) and RET. Treatment with sunitinib decreased the number of MDSC and Treg in advanced tumor-bearing animals [12]. Furthermore, sunitinib prevented tumor antigen-specific T cell anergy and Treg development. Analysis of isolated TIL from control or sunitinib treated mice demonstrates that treatment with sunitinib skewed the immune response towards a Th1 type response, reducing the expression of IL-10, TGF-β, and Foxp3 but enhancing the expression of IFN-gamma. Sunitinib increased the percentage of CD8 and CD4 cells in treated mice while decreasing CD4+Foxp3+ gated Treg. MDSC was significantly reduced from 53.9% in PBS-treated mice to 39% in sunitinib-treated mice. Additionally, the number of plasmacytoid dendritic cells (pDC) was also significantly reduced by sunitinib treatment, from 34.8% in PBS treated to 22.5% in sunitinib treated mice [12]. This effect was not seen in c-kit mutant mice (Wv/Wv). Significantly, when given in combination with an immune therapy protocol with IL-12 and 4-1BB activation, sunitinib significantly improved the long-term survival of large tumor-bearing mice [12]. Together, these data suggest that sunitinib can overcome tumor-mediated immune tolerance and enhance tumor immunotherapy via a c-kit dependent mechanism.
6. Radiation therapy influences tumor immunity
Recently, there has been increasing interest in combining immune therapy with conventional anti-cancer therapy. Although some chemotherapy agents also augment immune responses (e.g., gemcitabine), we will focus on ionizing radiation as the prototype cytotoxic agent [43]. Of note, other drug regimens such as doxorubicin and cyclophosphamide increase MDSC accumulation in peripheral blood [35]. While total body irradiation is immunosuppressive, numerous in vivo studies have demonstrated that tumor-directed RT enhances the effectiveness of different forms of immunotherapy, including dendritic cell vaccines with tumor associated antigens, cytokine-based viral gene therapy, and adoptive transfer of cytotoxic T cells [21]. For instance, in one preclinical model, the combination of adoptive transfer of activated T cells and RT eradicated tumors in the majority of immune competent mice, whereas tumors regrew in mice given either treatment alone. The enhancement of anti-tumor responses following RT was attributed to the ability of RT to alter the tumor microenvironment and enhance cross priming by stromal cells [44]. Recently, regression in non-irradiated metastases after extracranial stereotactic radiotherapy was reported, clearly demonstrating the ability of RT to achieve an abscopal effect on renal cell carcinoma [45]. The observed effect on cells outside of the radiation field was hypothesized to reflect a potentiation of tumor antigen-specific immunity by RT. Some possible mechanisms underlying this observation include an increased uptake of tumor cells treated with RT, the limitation of immune suppressing Treg and MDSC, inhibition of tumor angiogenesis, and enhanced penetration of immune effector cells due to RT-induced alterations in the tumor microenvironment [21, 46]. When these observations are translated to the clinical setting, the potentiation of tumor immunity by RT represents a mechanism by which localized RT to a tumor site may lead to the augmentation of tumor antigen-specific immunity systemically. This would allow for the eradication of microscopic systemic disease in a manner that is more tumor antigen-specific than that offered by systemic chemotherapy. It remains to be seen whether the effectiveness of these mechanisms can be demonstrated clinically, and whether the resultant anti-tumor immunity can improve tumor control both locally and systemically.
Some preclinical studies have investigated the optimization of RT schedule for the induction of an effective anti-tumor response. For example, a recent study suggests that B16 melanoma responds to high dose RT (20 Gy × 1) but not to fractionated RT (5 Gy × 4) [47]. In this model, high dose RT resulted in the maturation and priming of dendritic cells and the induction of tumor antigen-specific cytolytic T cell responses, resulting in tumor rejection. This effect appeared to be blunted with concurrent chemotherapy, which suggests that chemotherapy may limit the ability of one or more subsets of immune cells in the coordination of an effective anti-tumor response. Taken together, these observations suggest that focal RT can elicit anti-tumor immunity, which may be via a combination of factors including (i) enhancing trafficking of antigen presenting cells to the tumor site; (ii) augmenting antigen uptake of irradiated tumor cells; (iii) increasing the maturation of antigen presenting cells to elicit an effective immune response; (iv) inducing the maturation of immune effector cells to generate a robust immune response; and/or (v) limiting the immunomodulatory effects of suppressor cells.
7. Improved clinical responses are associated with immune changes after treatment with sunitinib and radiation therapy
Given the promising preclinical data, we investigated whether sunitinib can favorably impact the immune profile of patients with advanced malignancies. At our institution, an ongoing phase I/II study is investigating the efficacy of concurrent sunitinib and focal image guided radiation therapy for patients with 1 to 5 distant metastases from solid tumors [11]. Sunitinib (25-50 mg) is administered on days 1-28 followed by a 2 week rest period. Radiation (40-50 Gy in 10 fractions) is administered on days 8-19. Maintenance sunitinib was allowed but was not required. Peripheral blood was collected on days 0, 8 and 19. Preliminary analysis suggest that the effect of 7 days of sunitinib in peripheral blood on MDSC, T reg, pDC and CD8+ T cells are similar to those seen in mice. While analysis of additional patients is ongoing, sunitinib clearly decreases the percentage of monocytes and neutrophils without affecting total lymphocytes, as detected by clinical complete blood counts [48]. There was a strong correlation between a decrease in monocytes within 7 days of starting sunitinib and freedom from relapse [48]. At a median follow-up of 10 months, 9 of 21 patients remain free from progression [11]. Figure 2 demonstrates the durable clinical, biochemical and radiographic response of a patient with metastatic pancreatic adenocarcinoma more than 24 months after sunitinib and RT. Although peripheral blood for flow cytometry was not collected for this patient, absolute monocyte counts decreased from 600/μL prior to treatment to 100/μL within 7 days of sunitinib.
Figure 2.

Figure 2a. Patient with a history of pancreatic adenocarcinoma status post surgery, adjuvant gemcitabine, and chemoradiation who presents with three biopsy-proven liver metastases. Two enhancing liver metastases are seen in the dome of the liver on pretreatment CT.
Figure 2b. Image-guided radiation therapy plan encompassing 2 liver metastases. Note radio-opaque fiducial markers placed adjacent to tumors to allow for daily kV image guidance. The third metastasis was resected with negative margins.
Figure 2c. Follow-up CT of the abdomen and pelvis at 24 months after radiation and concurrent sunitinib demonstrates no evidence of disease.
Figure 2d. Correlation of longitudinal CA19-9 values to therapeutic interventions. Interestingly, the CA19-9 rose abruptly during radiation before returning to normal limits, which may indicate the release of tumor antigens.
8. Other candidates for pharmacological inhibition of MDSC
Several classes of agents have been studied for their ability to inhibit MDSC proliferation, survival, or function. Agents that promote myeloid-cell differentiation include all-trans retinoic acid and Vitamin D [49, 50]. Agents that inhibit MDSC function by decreasing arginase and/or iNOS include cyclooxygenase 2 inhibitors (celecoxib), phosphodiesterase 5 inhibitors (sildenafil, tadalafil) or reactive oxygen species inhibitors (nitroaspirin). These agents appear effective in vivo and several of these agents are FDA approved for non-cancer related indications [13]. Potentially, selected cytotoxic agents such as ionizing radiation or gemcitabine may selectively deplete MDSC. However, perhaps the most promising approach is inhibiting MDSC expansion by blocking tumor-derived factors such as SCF (c-kit) or VEGF, since there are several FDA approved anti-cancer agents of this class that are currently available.
8.1. Inhibition of c-kit
Targeting immune suppressor cells via small molecule inhibitors of c-kit is a strategy that should explored in well-designed clinical and translational studies. Unlike sunitinib and dasatinib, which affect multiple kinases, imatinib is relatively specific for c-kit and BCR-ABL [54]. Although imatinib reduces MDSC expansion, published data cautions that there may also be some impairment in T and B cell proliferation and function [52-42]. However, other studies suggest that, following imatinib treatment, there is increased T cell production of TNF-alpha, restoration of plasmacytoid dendritic cell function, enhancement of antigen-presenting cell function, reversion of tumor-induced CD4 T cell tolerance, and inhibition of Treg function 55-58]. These conflicting reports highlight the complexity of studying immune response to pharmacological inhibitors.
While the effect of dasatinib on MDSC is currently unknown, multiple preclinical and clinical studies suggest that sunitinib decreases immune suppressing MDSC and Treg in solid tumors via mechanisms involving c-kit, Stat3 and possibly VEGF inhibition [12, 23, 59, 60]. Further sunitinib promoted maturation of CD1c dendritic cells, an effect not seen in patients treated with cediranib, a VEGF receptor tyrosine kinase inhibitor [61, 62]. Interestingly, sunitinib only partly reversed VEGF-mediated inhibition of dendritic cell maturation [63]. Taken together, these data suggest that sunitinib modulates immune cells through a VEGF-independent mechanism that involves c-kit inhibition. Although the clinical significance of these effects is not completely characterized, the potent inhibitory effect of sunitinib on MDSC and Treg suggests a potential role of bone marrow derived cells in achieving tumor control. Sunitinib offers significant promise to overcome immune suppression associated with advanced solid tumors. Preclinical and clinical research with the goal of rationally exploiting the immune modulatory effects of sunitinib and other multitargeted tyrosine kinase inhibitors promises to be a fruitful avenue of investigation in the coming years.
8.2. Inhibition of VEGF signaling
The efficacy of various strategies to inhibit VEGF signaling (bevacizumab, sorafenib, VEGF-trap) on MDSC remains incompletely understood. Most published data suggests that sorafenib has immune suppressing function [64-68]. For instance, sorafenib inhibits CD8+ T cell-mediated immune response and dendritic cell function via a MAPK-independent mechanism [65, 67]. However, in addition to inhibiting CD4+ and CD8+ T cells, sorafenib also inhibits immunosuppressive Treg [66, 68]. While VEGF-trap had no effect on MDSC in a cohort of patients with advanced solid tumors, bevacizumab decreased MDSC in patients with renal cell carcinoma [69, 70]. Currently, it is unknown whether strategies targeting both c-kit and VEGF are more effective than either alone.
9. Future directions: Alternative approaches to combination therapy?
There is significant interest in harnessing the immune system for the rejection of established tumors bearing tumor antigens. While specific therapeutic cancer vaccines are an active area of development, there is great potential for exploiting the immune modulating effects of existing cancer therapies. For instance, the effect of combined inhibition of c-kit and VEGF via sunitinib appears to reverse MDSC-mediated immune suppression. Whether this biologic effect translates into improved cancer control, with or without focal radiation and/or immune therapy, will be rigorously tested in future studies.
Acknowledgments
This work was supported by funding from Pfizer (IIR 2005-1082, to J.K.), DOD Physician Research Training Program (PC041206, to J.K.), William Harris Research Fellowship (J.K.), Katz Foundation Research Grant (J.K.) and the National Cancer Institute (R01 CA127483 and R01 CA109322, to S.-H.C.).
Biography
Vitae: Dr. Johnny Kao is an Assistant Professor of Radiation Oncology and Director of Head and Neck Oncology Clinical Research at the Mount Sinai School of Medicine. Dr. Kao has a strong interest in developing novel strategies to enhance the efficacy of radiation therapy for patients with locally advanced head and neck cancer and oligometastases. Dr. Kao also has a strong interest in exploring the potential interaction between conformal radiation therapy and anti-tumor immunity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Demetri GD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien SG, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 3.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 4.Smyth MJ. Imatinib mesylate--uncovering a fast track to adaptive immunity. N Engl J Med. 2006;354(21):2282–4. doi: 10.1056/NEJMcibr061878. [DOI] [PubMed] [Google Scholar]
- 5.Talpaz M, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 6.Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 7.Motzer RJ, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 8.Escudier B, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–34. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 9.Miller K, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–76. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 10.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 11.Kao J, et al. Phase 1 study of concurrent sunitinib and image-guided radiotherapy followed by maintenance sunitinib for patients with oligometastases: acute toxicity and preliminary response. Cancer. 2009;115(15):3571–80. doi: 10.1002/cncr.24412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozao-Choy J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69(6):2514–22. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunn GP, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 16.Pages F, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–66. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 17.Finn OJ. Cancer immunology. N Engl J Med. 2008;358(25):2704–15. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 18.Shojaei F, Ferrara N. Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res. 2008;68(14):5501–4. doi: 10.1158/0008-5472.CAN-08-0925. [DOI] [PubMed] [Google Scholar]
- 19.Yang L, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 20.Yang L, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demaria S, et al. Combining radiotherapy and immunotherapy: a revived partnership. Int J Radiat Oncol Biol Phys. 2005;63(3):655–66. doi: 10.1016/j.ijrobp.2005.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sparano JA, et al. Randomized phase III trial of treatment with high-dose interleukin-2 either alone or in combination with interferon alfa-2a in patients with advanced melanoma. J Clin Oncol. 1993;11(10):1969–77. doi: 10.1200/JCO.1993.11.10.1969. [DOI] [PubMed] [Google Scholar]
- 23.Ko JS, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–57. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 24.Sabatino M, et al. Serum vascular endothelial growth factor and fibronectin predict clinical response to high-dose interleukin-2 therapy. J Clin Oncol. 2009;27(16):2645–52. doi: 10.1200/JCO.2008.19.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koebel CM, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450(7171):903–7. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 26.Murdoch C, et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–31. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 27.Youn JI, et al. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Movahedi K, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111(8):4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 29.Almand B, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166(1):678–89. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 30.Almand B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6(5):1755–66. [PubMed] [Google Scholar]
- 31.Nefedova Y, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004;172(1):464–74. doi: 10.4049/jimmunol.172.1.464. [DOI] [PubMed] [Google Scholar]
- 32.LeCouter J, et al. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc Natl Acad Sci U S A. 2004;101(48):16813–8. doi: 10.1073/pnas.0407697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan PY, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111(1):219–28. doi: 10.1182/blood-2007-04-086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng P, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205(10):2235–49. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diaz-Montero CM, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinha P, et al. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179(2):977–83. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 37.Shojaei F, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 38.Shojaei F, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450(7171):825–31. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 39.Shojaei F, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13(3):193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinet O, et al. Immunomodulatory gene therapy with interleukin 12 and 4-1BB ligand: long- term remission of liver metastases in a mouse model. J Natl Cancer Inst. 2000;92(11):931–6. doi: 10.1093/jnci/92.11.931. [DOI] [PubMed] [Google Scholar]
- 42.Huang B, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki E, et al. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–21. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 44.Zhang B, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204(1):49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wersall PJ, et al. Regression of non-irradiated metastases after extracranial stereotactic radiotherapy in metastatic renal cell carcinoma. Acta Oncol. 2006;45(4):493–7. doi: 10.1080/02841860600604611. [DOI] [PubMed] [Google Scholar]
- 46.Demaria S, Formenti SC. Sensors of ionizing radiation effects on the immunological microenvironment of cancer. Int J Radiat Biol. 2007:1–7. doi: 10.1080/09553000701481816. [DOI] [PubMed] [Google Scholar]
- 47.Lee Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589–95. doi: 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kao J, et al. Phase I Study of Concurrent Sunitinib and Image Guided Radiotherapy Followed by Maintenance Sunitinib for Patients with Oligometastases. Proc AACR. 2009:3616a. doi: 10.1002/cncr.24412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lathers DM, et al. Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunol Immunother. 2004;53(5):422–30. doi: 10.1007/s00262-003-0459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mirza N, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66(18):9299–307. doi: 10.1158/0008-5472.CAN-06-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karaman MW, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26(1):127–32. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 52.Mumprecht S, et al. Imatinib mesylate selectively impairs expansion of memory cytotoxic T cells without affecting the control of primary viral infections. Blood. 2006;108(10):3406–13. doi: 10.1182/blood-2006-04-018705. [DOI] [PubMed] [Google Scholar]
- 53.Santachiara R, et al. Development of hypogammaglobulinemia in patients treated with imatinib for chronic myeloid leukemia or gastrointestinal stromal tumor. Haematologica. 2008;93(8):1252–5. doi: 10.3324/haematol.12642. [DOI] [PubMed] [Google Scholar]
- 54.Cristofanilli M, et al. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: clinical activity and biological correlations. Ann Oncol. 2008;19(10):1713–9. doi: 10.1093/annonc/mdn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohty M, et al. Imatinib and plasmacytoid dendritic cell function in patients with chronic myeloid leukemia. Blood. 2004;103(12):4666–8. doi: 10.1182/blood-2003-09-3220. [DOI] [PubMed] [Google Scholar]
- 56.Wang H, et al. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood. 2005;105(3):1135–43. doi: 10.1182/blood-2004-01-0027. [DOI] [PubMed] [Google Scholar]
- 57.Chen CI, Maecker HT, Lee PP. Development and dynamics of robust T-cell responses to CML under imatinib treatment. Blood. 2008;111(11):5342–9. doi: 10.1182/blood-2007-12-128397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larmonier N, et al. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol. 2008;181(10):6955–63. doi: 10.4049/jimmunol.181.10.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finke JH, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14(20):6674–82. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 60.Xin H, et al. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69(6):2506–13. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Cruijsen H, et al. Defective differentiation of myeloid and plasmacytoid dendritic cells in advanced cancer patients is not normalized by tyrosine kinase inhibition of the vascular endothelial growth factor receptor. Clin Dev Immunol. 2007;2007:17315. doi: 10.1155/2007/17315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Cruijsen H, et al. Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: CD1c+ dendritic cell frequency predicts progression-free survival. Clin Cancer Res. 2008;14(18):5884–92. doi: 10.1158/1078-0432.CCR-08-0656. [DOI] [PubMed] [Google Scholar]
- 63.Alfaro C, et al. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br J Cancer. 2009;100(7):1111–9. doi: 10.1038/sj.bjc.6604965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Escudier B, et al. Phase I trial of sorafenib in combination with IFN alpha-2a in patients with unresectable and/or metastatic renal cell carcinoma or malignant melanoma. Clin Cancer Res. 2007;13(6):1801–9. doi: 10.1158/1078-0432.CCR-06-1432. [DOI] [PubMed] [Google Scholar]
- 65.Hipp MM, et al. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood. 2008;111(12):5610–20. doi: 10.1182/blood-2007-02-075945. [DOI] [PubMed] [Google Scholar]
- 66.Zhao W, et al. Sorafenib inhibits activation of human peripheral blood T cells by targeting LCK phosphorylation. Leukemia. 2008;22(6):1226–33. doi: 10.1038/leu.2008.58. [DOI] [PubMed] [Google Scholar]
- 67.Houben R, et al. MAPK-independent impairment of T-cell responses by the multikinase inhibitor sorafenib. Mol Cancer Ther. 2009;8(2):433–40. doi: 10.1158/1535-7163.MCT-08-1051. [DOI] [PubMed] [Google Scholar]
- 68.Molhoek KR, et al. Apoptosis of CD4(+)CD25(high) T cells in response to Sirolimus requires activation of T cell receptor and is modulated by IL-2. Cancer Immunol Immunother. 2009;58(6):867–76. doi: 10.1007/s00262-008-0602-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fricke I, et al. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin Cancer Res. 2007;13(16):4840–8. doi: 10.1158/1078-0432.CCR-07-0409. [DOI] [PubMed] [Google Scholar]
- 70.Kusmartsev S, et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol. 2008;181(1):346–53. doi: 10.4049/jimmunol.181.1.346. [DOI] [PubMed] [Google Scholar]