

1. Introduction
An Overview on Ynamines
Alkynes represent one of the most important and versatile building blocks in organic synthesis. Heteroatom-substituted alkynes, which can be considered as subgroups of alkynes, have also been vastly utilized in developing synthetic methods. In particular, ynamines [1-amino-alkynes or N-alkynyl amines] became the most valuable subgroup of alkynes after the establishment of their practical synthesis in the 1960's. The first attempt at preparation of an ynamine was reported by Bode1,2 in 1892. While well-characterized ynamines were reported in 19583 and 1960,4 a practical synthesis was not achieved until the effort led by Viehe5 in 1963 in addition to other subsequent works. In the ensuing twenty years, the synthetic significance of ynamines in organic and organometallic chemistry was firmly established by the work of many creative synthetic chemists. These elegant pioneer works have been informatively and carefully reviewed by Viehe in 19676 and 1969;7 Ficini in 1976;8 Pitacco and Valentin9 in 1979; Collard-Motte and Janousek10 in 1986; Himbert11 in 1993; and most recently by us12,13 and Katritzky14.

The synthetic eminence of ynamines is well merited because of the predicable regioselectivity in their transformations as shown by the generalization in Scheme i, and more importantly, because they are inherently highly reactive. However, this latter attribute is also the source of the limitation that has seriously hampered the development of ynamine chemistry, thereby shortening the period of its prominence in synthesis. Ynamines are very sensitive toward hydrolysis, as protonation of the electron-rich alkynyl motif affords reactive keteniminium intermediates, which upon trapping with water leads to simple amides in a rather expensive manner (Scheme i). This hydrolytic instability has caused much difficulty in the experimental preparation and general handling of ynamines, and more detrimentally, rendered ynamine chemistry inaccessible.
Scheme i.
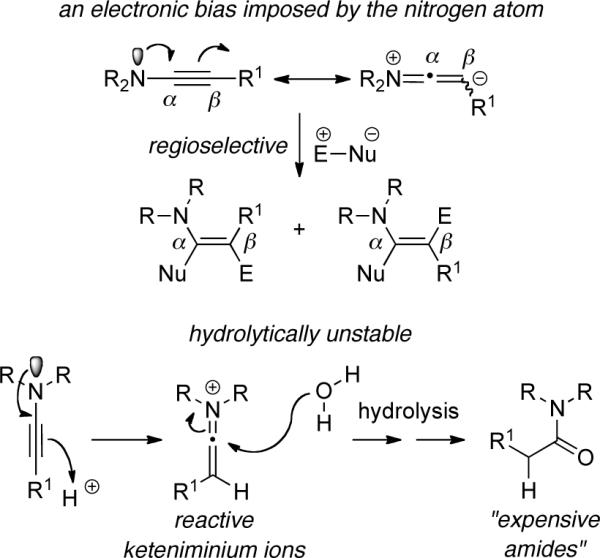
Consequently, the synthetic utility of ynamines has suffered a dramatic decline during the last thirty years.15 The most glaring limitations have been in the development of intramolecular and stereoselective reactions.7–14 The only reported intramolecular reaction of ynamines was Genet and Kahn's acid catalyzed addition of a hydroxyl group to an ynamine [i→ii in Scheme ii] in 1980,16 and although clever, it constitutes a hydrolytic process.
Scheme ii.

Besides Reinhoudt's17 sole account in 1987 reporting hetero-[4 + 2] cycloadditions of chiral ynamine iii with nitroalkenes that led to cycloadducts iv in modest de, the only other notable studies were reported ten years later by Fischer18 showcasing [2 + 2] cycloadditions of chiral ynamides v and vi with vinylidene chromium carbene complexes, and another three years later by Pericàs19 in their Pauson-Khand cycloadditions using chiral ynamines vii.
Emergence of Ynamides
To improve the stability of ynamines and revitalize their synthetic utility, diminishing the electron-density by substituting the nitrogen atom and/or the alkyne with electronegative elements would appear to be a logical solution. These could be classified as electron-deficient ynamines [ix–xiv in Figure i] and their reactivities and relative stabilities are known in literature.7–14 These would include beautiful recent work by Katritzky20 using benzotriazole substituted ynamines [x]; and by Kerwin21 involving imidazole-substituted ynamines [xi]. In addition, elegant chemistry has been reported during the last 30 years using so called push-pull ynamines or alkynes.7–14 Ishihara's reports22 employing N,N-dialkyl-(3,3,3-trifluoro-1-propynyl)amines xiii and Stang's push-pull ynamines xiv23 represent examples of such electron deficient ynamines.
Figure i.
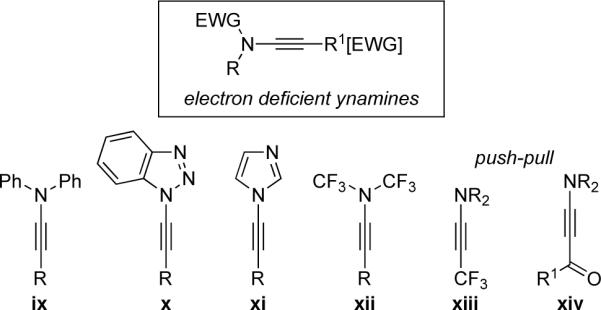
While the aforementioned electron-deficient variants of ynamines [ix–xiv] depend upon inductive effects, or delocalization of the nitrogen lone pair into an aryl or hetero-aryl system, or through the triple bond to reduce the electron density of the ynamine motif, chemistry of another class of electron-deficient ynamines has emerged in the last 15 years. By simply placing an electron-withdrawing carbonyl group on the nitrogen atom, the donating ability of the nitrogen lone pair toward the alkynyl motif is greatly diminished through resonance delocalization into the carbonyl oxygen (Figure ii). These new electron-deficient ynamines have been called ynamides, or 1-amido-alkynes [or N-alkynyl amides]. Not only do they offer superior stability to traditional ynamines, but they also have set the gold standard for balancing reactivity and stability.
Figure ii.
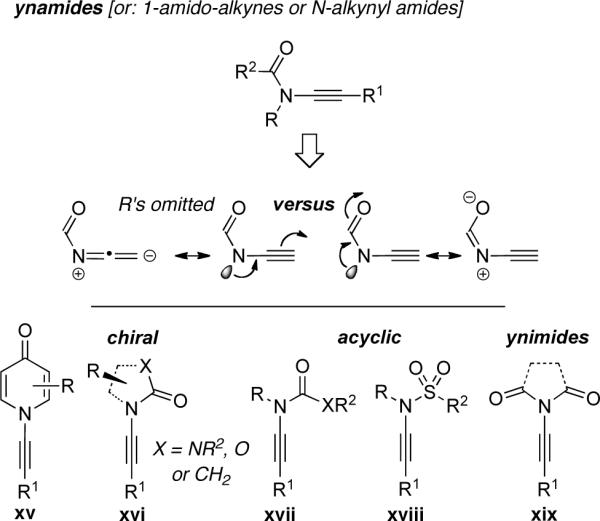
Examples of ynamides would include those in which the nitrogen atom is a member of (1) vinylogous amides or pyridones xv; (2) imidazolidinones, oxazolidinones, or lactams xvi, which can also be chiral; (3) ureas, urethanes, or simple amides xvii, and sulfonamides xviii, which represent acyclic ynamides; or (4) imides xix, which remain elusive to date. This very concept of improving thermal stability and stability toward hydrolytic conditions by using an electron-withdrawing carbonyl group should be credited to Viehe who in 1972 reported the synthesis of the first ynamide [see xvii: R = R2 = Me, R1 = Ph, X = NMe].24
While identifying stable variants of ynamines represents a pressing issue, more important is the need for the new variants to retain high levels of reactivity. In meeting both of these criteria, ynamides may lead to rich new areas of chemistry where their improved thermal and hydrolytic stability could allow access to intramolecular and stereoselective reactions that are not possible with ynamines. Most significantly, while the chemistry of other heteroatom-substituted alkynes is of high impact and value, nitrogen atom-substituted alkynes offer greater advantages. The trivalent nature of the nitrogen atom allows: (1) tethering of a chirality-inducing unit for providing asymmetric induction; (2) inclusion of the coordinating unit to provide conformational rigidity; (3) a much greater flexibility in designing intramolecular reactions or tandem processes than with oxygen- or sulfur-substituted alkynes; and last but not the least, (4) a novel entry to alkaloids if the nitrogen atom can be preserved throughout the transformations. These are remarkably attractive features for developing highly stereoselective methodologies and rapid assembly of structural complexity.
Surprisingly, despite the precedent for ynamide synthesis and documentation of its superior thermal stability by Viehe, reactions involving ynamides remained almost unknown for 25 years (Figure iii).7–14 It was not until the late 1990's that the chemistry of ynamides began to appear in literature commencing with Feldman's synthesis of chiral ynamides,25 and later, in a series of beautiful works done by Witulski, as well as those of Rainier and Chen. Meanwhile, our lab initially focused developing an atom-economical way of synthesizing ynamides. These early developments awakened the synthetic community to utilizing nitrogen-substituted alkynes in organic synthesis, and led to a Tetrahedron-Symposium-in-Print26 focused on the chemistry of electron-deficient ynamines and ynamides.
Figure iii.

In the last five years, the chemistry of ynamides has exploded (Figure iii), particularly involving ynamides xvi–xviii. It can be stated that the chemistry of ynamines has reemerged in the forms of ynamides. These efforts demonstrate that ynamides possess the right balance between reactivity and stability and can be employed in a diversified array of stereoselective and intramolecular reactions that were not possible with traditional ynamines. With such compelling evidence for the reemergence of “ynamine” chemistry, it is the purpose of this review to provide proper illustrations of elegant chemistry involving ynamides xvi–xviii that has come to pass and to illicit a greater interest from the synthetic community, leading to new ynamide chemistry in the future.
A Personal Perspective
Despite working with alkynes extensively in Professor Bill Wulff's lab, my interest in the chemistry of ynamines truly began with Professor Gilbert Stork in the summer of 1996 when a discussion of ours led to the topic of Professor Jacqueline Ficini's dihydroantirhine synthesis,27 which featured Ficini's ynamine-[2 + 2] cycloaddition to cleverly control the relative stereochemistry at C3, C15, and C20 in dihydroantirhine. It was a memorable discussion because I had not seen Professor Stork this passionate before, and I could sense that he has a profound respect for Jacqueline. I queued into phrases such as, “ynamines are not stable but clearly useful” and “it's an unsolved area and you should look into it.” After two more similar conversations, I was into it.
Six months later, I learned something invaluable from an interview trip to Palo Alto. In his office, Professor Barry Trost was quite fascinated with our proposed ynamine chemistry, asking what I had really thought of in terms of improving stability and what the future might hold. Not having found Viehe's paper, I was not sure of a good answer. However, from our conversation, I grasped that to truly revitalize interest in this functionally rich organic building block, one would need to address issues not only related to stability, but more importantly, synthetic accessibility in an atom-economical manner, which became our foremost quest.
A few weeks later in Lansing, it was Professor Robert Maleczka who pointed out that our proposed novel ynamines with chiral oxazolidinone and imidazolidinone motifs [see xvi in Figure ii] could offer the needed thermal stability given the electron-withdrawing carbonyl group. In hindsight, while having read Viehe's paper would have made my life a little easier, that particular conversation has significance of its own. It led me to focus on these novel oxazolidinone and imidazolidinone substituted ynamides xvi instead of sulfone-substituted ynamides xviii. The implication is that while the latter could be accessible using the iodonium triflate salt protocol developed by Stang and Zhdankin28 as a new method for ynamine synthesis from lithium amides, the same protocol was not useful for the former, thereby requiring the development of new methods. Without the conversation with Professor Maleczka, we might not have embarked on this adventure, which ultimately led to the development of a copper-catalyzed N-alkynylation or Ullmann-type amidative cross-coupling.
It is noteworthy that when one digs deep enough into history, one can always find a precedent. In this case, an earlier account related to copper-promoted N-alkynylations was reported by Balsamo and Domiano et al. in 198529 using CuCl/O2, although the resulting ynamide was a by-product. Even earlier in 1968, Peterson30 at Dow-Midland reported the use of Cu(OAc)2/O2 in the synthesis of ynamines from terminal acetylenes and secondary amines, which is as economical as it gets. I should also note that while we were exploring various possible synthetic approaches toward ynamides, it was Chris Douglas, a college sophomore at the time [now at Minnesota], who suggested the possibility of pursing a Buchwald-Hartwig-Migita type palladium-catalyzed amination to make ynamides.
Lastly, to advance any area or field of interest, it requires creativity from as many people as possible. This perspective remains true here, as the chemistry of ynamides would not be what it is today without these collective innovations. On that note, while we tried our very best to be comprehensive, it is likely that we have inadvertently missed some beautiful work for which we express our regret here in advance. Overall, this review is intended to highlight ynamide chemistry in the past 10–15 years with the hope that work in the next decade and beyond will truly render ynamides a modern functional group for the new millennium.
2. Synthesis of Ynamides
The first ynamine synthesis was reported more than 50 years ago, however the very first example of ynamide preparation was not disclosed until 14 years later. One major obstacle for the broader synthetic application of this versatile building block lies in the fact that until recently, a highly practical, efficient ynamide preparation procedure, which also possesses general substrate scope, remained elusive. While the emergence of alkynyliodonium chemistry greatly helped the development of ynamide chemistry, the more recent discovery of metal-catalyzed ynamide formation provided significant fuel for the explosion of this area since 2003–2004.
2.1. Isomerization
In 1958, Zaugg3 described the first example of ynamine synthesis via isomerization. Twenty years later several more syntheses were reported by Galy31,32 and Katritzky33 (Scheme 1). Galy initially reported a phase-transfer condition which involved propargylation of acridones 1 followed by in situ isomerization to give ynamides 2 in 55 – 80% yield. The same group later also reported conditions for selective preparation of ynamides 2 and allenamides 3. Katritzky found that it was more efficient to follow a two-step procedure in which the intermediate 4 was isolated and then rearranged to the desired ynamides.
Scheme 1.
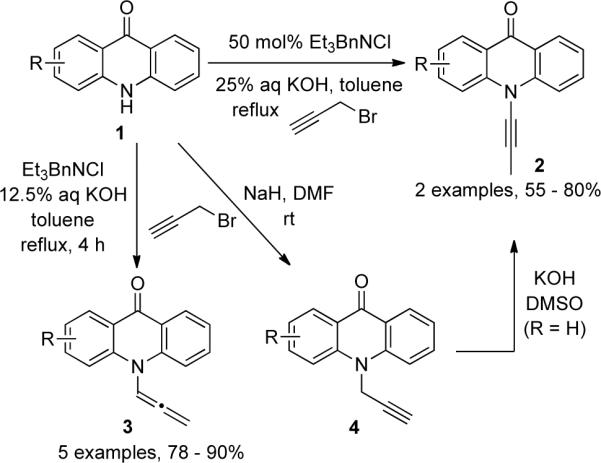
According to Majumdar's34 study, propargylation of acridones 1 with propargyl bromide 5 followed by isomerization and elimination under phase-transfer conditions gave various ynamides 8. Alternatively 8 could also be accessed by a one-pot procedure using propargyl chlorides 7 (Scheme 2).
Scheme 2.
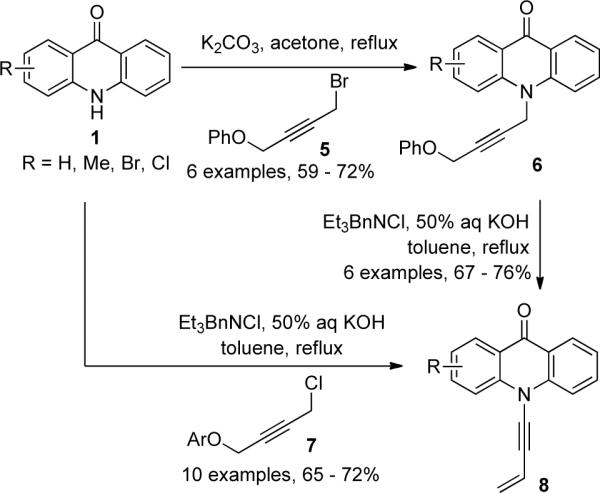
Attempts to extend the scope of isomerization to chiral ynamides were initially met with difficulty. In 2001, Hsung35a described that isomerization of 9 failed to give the desired ynamides 10 (Scheme 3). It was later found that the isomerization of propargyl urethanes 11a and 11b gave exclusively allenamides 12, however, the attempted rearrangement of 12 to ynamides 13 was not successful.
Scheme 3.
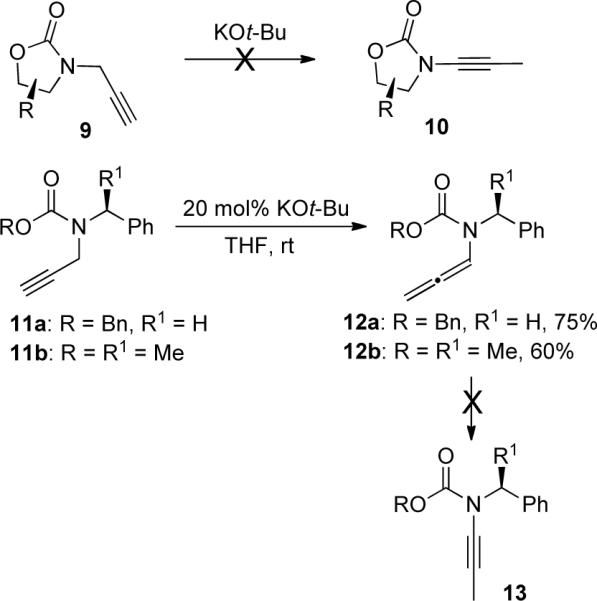
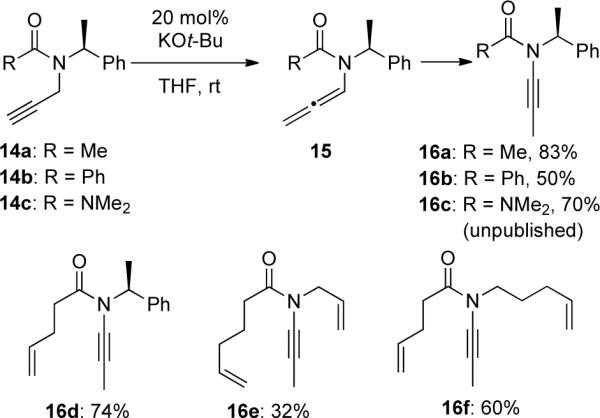
However, Hsung35b discovered that propargyl amides 14 underwent complete isomerization to ynamides 16 in the presence of a catalytic amount of KOt-Bu (Scheme 4). The chiral ynamides were then used in ring-closing metathesis (Section 3.3.3).
Scheme 4.
2.2. Elimination
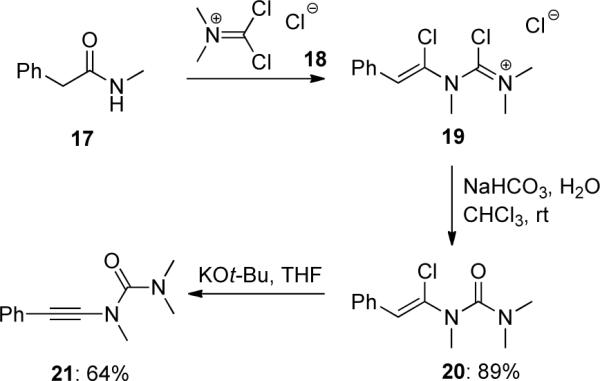
The first example of an elimination protocol was reported by Viehe36 in 1972 (Scheme 5). A secondary acetamide 17 reacted with phosgeneimmonium chloride 18 gave a chloroformamidinium salt 19. Upon hydrolysis 19 afforded urea 20, which underwent elimination in the presence of base to produce ynamide 21.
Scheme 5.
Zemlicka37 prepared several ynamines/ynamides derived from nucleic acid bases (Scheme 6). Chloroenamine 22 and 24 were prepared in ~20% yield from respective deprotonated 2-pyrimidinones and tetrachloroethylene. Treatment of 22 and 24 with n-BuLi at −70 °C afforded the respective ynamides 23 and 25 in 34–51% yields.
Scheme 6.
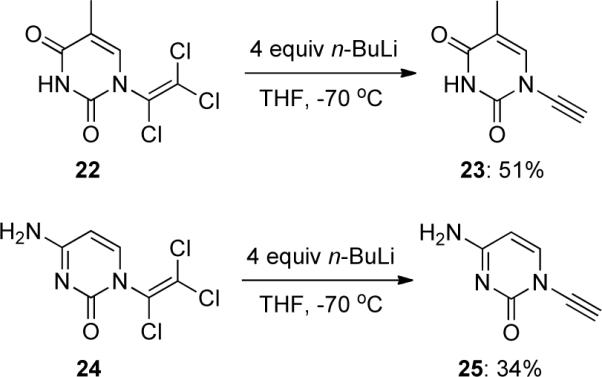
Brückner38 published a facile elimination protocol via formamides (Scheme 7). Intermediate formamides 27 could be accessed from tosyl amides 26 via coupling reactions with formyl benzotriazole 29. Alternatively, 27 could be prepared from formamides 28 and alkyl mesylates. Treatment of 27 with CCl4 and PPh3 leads to β,β-dichloroenamides 30, and subsequent elimination with n-BuLi afforded various N-alkyl, phenyl ynamides 31 in generally excellent yields over two steps in a homologous Corey-Fuchs manner. Brückner also described that the reactions of 27 with CBr4/PPh3 were not satisfactory as the corresponding β,β-dichloroenamides gave mixtures of desired ynamides 31 and starting tosylamides 26.
Scheme 7.
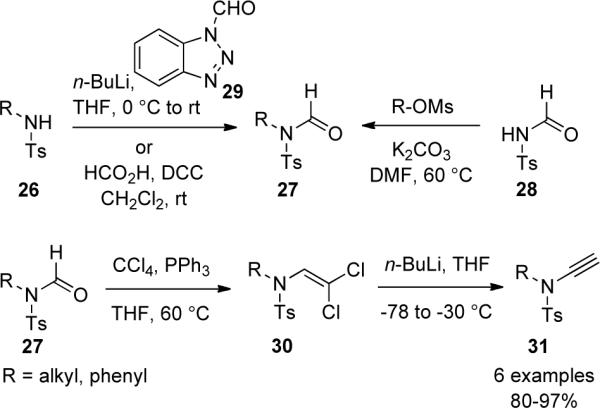
Hsung35 described an attempt to prepare ynamides via the elimination of α,β-dicholoenamides 32 (Scheme 8), however the only isolated product was 34, which presumably resulted from the attack of KOt-Bu on chloroketenimine 33.
Scheme 8.
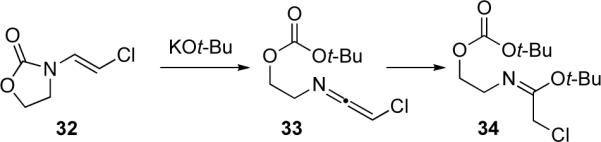
In the same article, Hsung disclosed the successful preparation of ynamides 37 via elimination (Scheme 9). It was found that by either refluxing enamides 35 (R = alkyl) with bromine or refluxing enamides 35 (R = aryl) with NBS in 1,2-dichloroethane (DCE), the desired β-bromoenamides 36 could be isolated in good yields. Subsequent treatment of the Z-bromoenamides with KOt-Bu afforded ynamides 37 in moderate to excellent yield. The ynamides were found to be quite stable to hydrolysis and could survive silica gel chromatography. It should be noted that while the brominations of 35 typically gave 36 with various E/Z ratios (1:8 to 1.5:1), efforts to prepare ynamides from E-36 were not successful.
Scheme 9.
An improved and extended protocol for the elimination process was reported by Saá39 (Scheme 10). After the treatment of β,β-dichloroenamides 38 with n-BuLi, transmetallation with ZnBr2 led to the intermediate zinc acetylide 39, and a subsequent one-pot Negishi coupling of 39 with various aryl iodides gave the desired ynamides 40 in moderate to excellent overall yields.
Scheme 10.
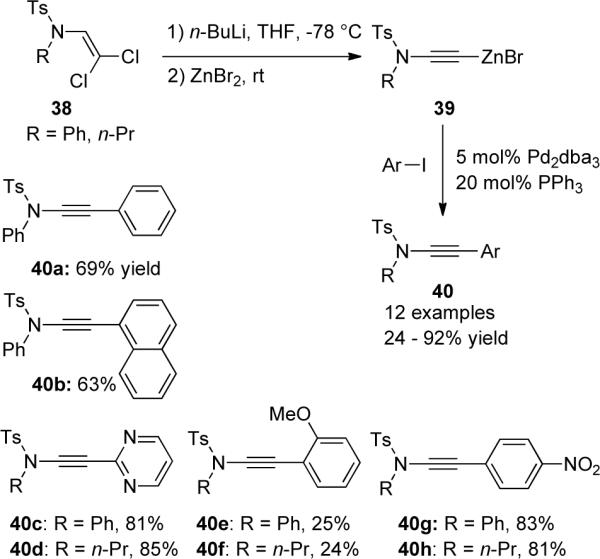
The same group later reported a different method for the synthesis of disubstituted ynamides40 (Scheme 11). It was found that after the n-BuLi mediated elimination of dichloroenamides 38, the corresponding lithium acetylide could be quenched with various electrophiles to afford ynamides 41 and 42.
Scheme 11.
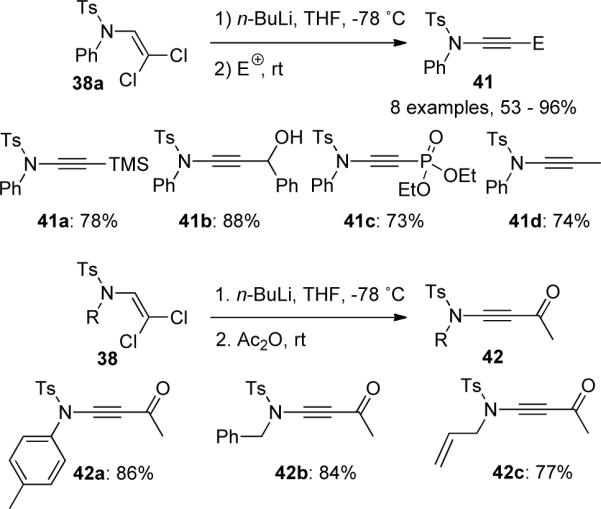
An alternative construction of the key C-C bond in ynamides was published by Cossy.41 N-formamidesc 43 were transformed into β,β-dichloroenamides 44 following Brückner's procedure (Scheme 12). Instead of direct elimination of 44 as done by Saá, Cossy reported that Suzuki-Miyaura coupling reactions of 44 with various boronic acids gave (E)-β-chloroenamides 45, which underwent elimination in the presence of LHMDS or NaOH/n-Bu4NHSO4 to yield ynamides 46.
Scheme 12.
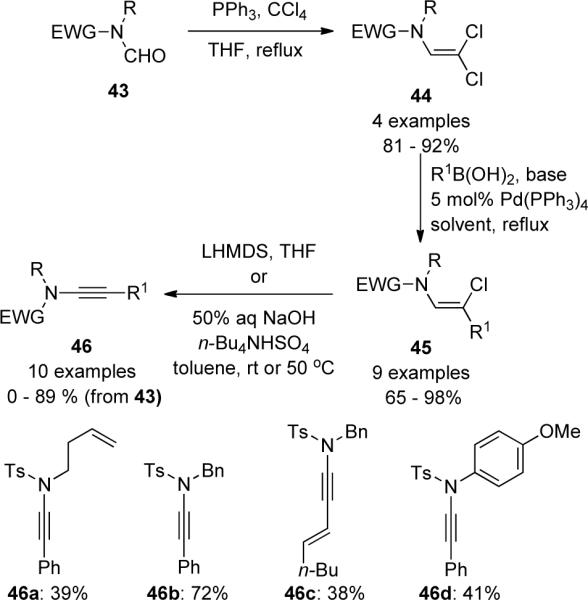
2.3. Alkynyliodonium Salts
The method involving the reaction of alkynyliodonium salts 48 with lithiated amine 47 was first described in Stang's pioneering work42 for the synthesis of ynamines 49 (Scheme 13). It is presumed that a vinylcarbene is first generated via nucleophilic addition β to the iodine followed by a 1,2-shift forming the acetylide. This transformation is discussed in more detail vide infra and has proven to be an excellent protocol for synthesizing sulfonyl-substituted ynamides.
Scheme 13.
In the study of the inter- and intramolecular addition/cyclization of sulfonamide anions with alkynyliodonium triflates, Feldman43 found that when more electrophilic iodonium triflates 51 and 54 were used, chiral ynamides 52 and 55 were isolated in moderated yields instead of the originally desired 1,5-C-H insertion products, representing the first successful synthesis of chiral ynamides (Scheme 14).
Scheme 14.
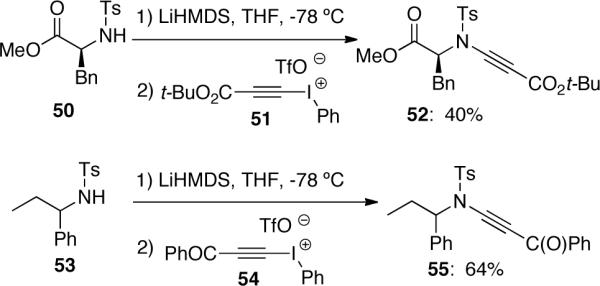
Witulski44 reported a more detailed discussion on the 1,2-migration after in situ formation of carbenes 58. They also discussed the effect of steric hindrance of amides 57 in this methodology for the preparation of ynamides (Scheme 15). A variety of ynamides 59 were obtained in up to 89% yield with lower yields observed in the cases when α-branched amides were used, reflecting an increase of steric hindrance in the nucleophilic addition of 57 to 56. Upon the treatment of ynamides 59 with TBAF, terminal ynamides 60 could be obtained in good to excellent yields. In their later study, it was found that terminal ynamides could also be synthesized directly by using ethynyliodonium triflate 56b.
Scheme 15.

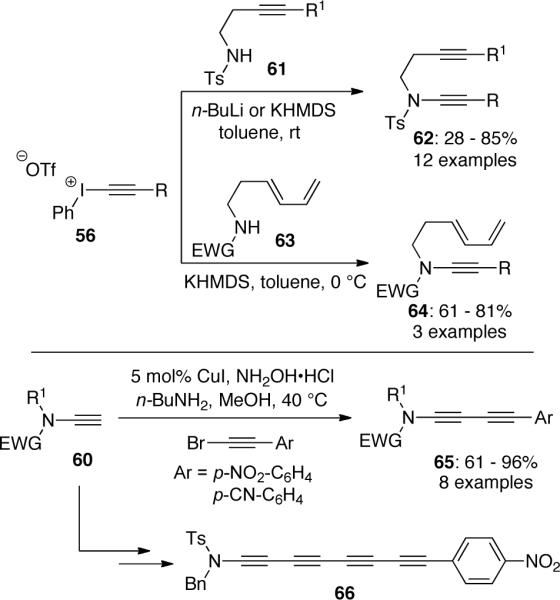
Some highly functionalized ynamides45 such as 62 and 64 were also generated via this route in variable yields (Scheme 16). These ynamides were poised for metal-catalyzed intramolecular cycloaddition reactions. More recently, it was reported that the conjugated polyynes 65 or 66 could be synthesized via the same route as shown in Scheme 16 followed by a Cadiot-Chodkiewicz cross coupling reaction.
Scheme 16.
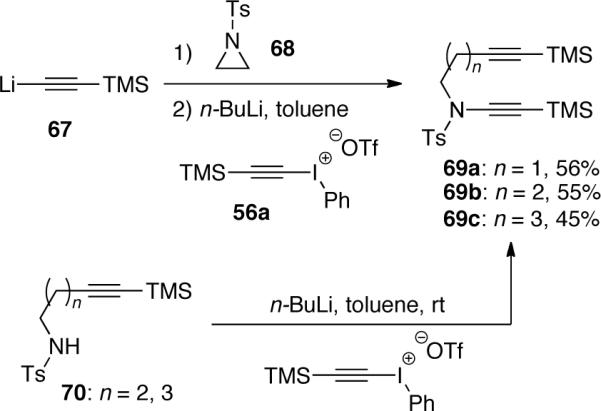
Rainier46 synthesized three yne-ynamides 69 with different tether lengths using this iodonium triflate strategy (Scheme 17). The ring opening of aziridine 68 by lithium trimethylsilyl acetylene 67 provided the nitrogen nucleophile for the synthesis of diyne 69a.
Scheme 17.
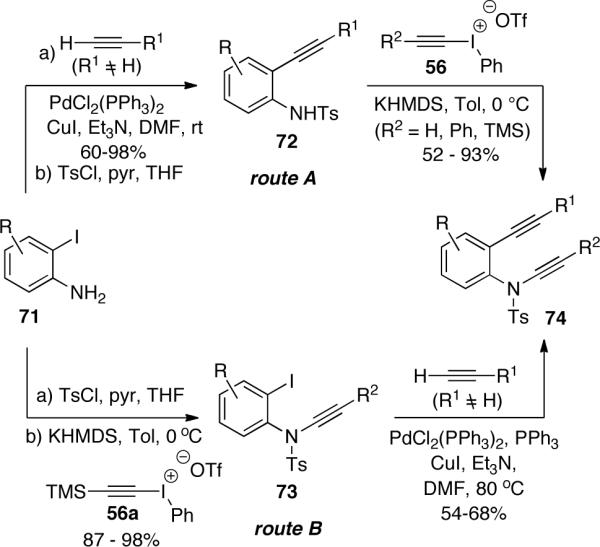
In the study of inter- and intramolecular alkyne cyclotrimerizations, Witulski47 reported the preparation of other functionalized diynes 74 via two sequences from aniline 71 as showed in Scheme 18. If the Sonogashira coupling reaction was introduced after the ynamide formation (route B), higher reaction temperature was required, probably due to the increased steric hindrance in the intermediate 73.
Scheme 18.
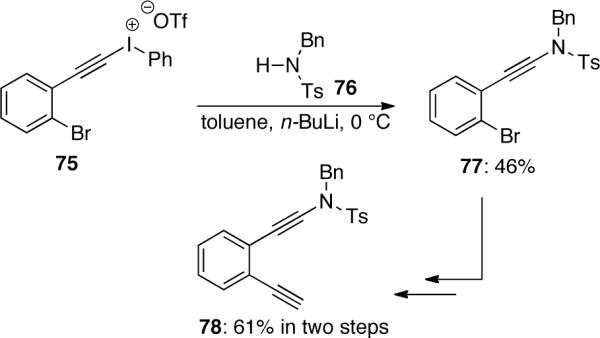
König48 also synthesized ynamide 78 as a target for their study of cyclizations. The reaction of iodonium salt 75 with amide 76 produced ynamide 77, which was followed by Sonogashira coupling and subsequent desilylation to give ynamide 78 in good overall yield (Scheme 19).
Scheme 19.
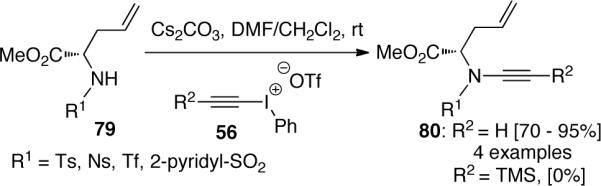
In 1999, Witulski49 published their results for the synthesis of chiral α-branched ynamides 80 using a similar route. The reaction was only effective when ethynyl(phenyl)iodonium triflate 56b (R2 = H) was used (Scheme 20). Lack of reactivity was observed if more sterically hindered iodonium triflate 56a (R2 = TMS) was used, which is consistent with their previous results for low yields of achiral ynamides 61 when the α-position of amides is hindered. The increased steric hindrance inherent in α-branched amino acid derivatives 79 interferes with the nucleophilic addition of the amide nitrogen to the β-carbon of the alkyne.
Scheme 20.
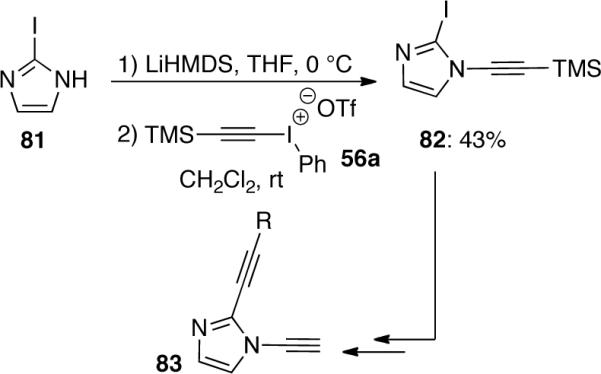
The synthesis of 1,2-dialkynylimidazoles 83 was reported by Kerwin50, in which the ynamide 82 was synthesized as a common intermediate through the reaction of 56a with 2-iodoimidazole 81 (Scheme 21).
Scheme 21.
Diederich51 tried to synthesize azoacetylenes via protected 1,2-bis-ynamides. However, it was found that while the preparation of mono ynamides such as 86a and 88 was successful, the second ynamide formation via similar chemistry failed (Scheme 22). Hydrazodicarboxylate such as 90 also failed to produce desired bis-ynamide 89.
Scheme 22.

2.4. Amidative Cross-Coupling
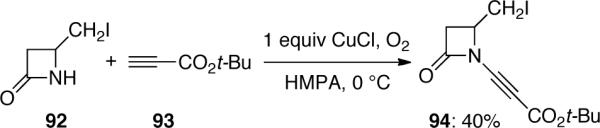
The first synthesis of ynamides using metal-mediated reactions was discovered by Balsamo and Domiano52 in 1985. The authors attempted to react the copper-acetylide of 93 with amide 92 with an intention to displace the iodine of 92, however 94 was found to be the only product and its structure was confirmed by X-ray crystallographic analysis (Scheme 23). It is noteworthy that a stoichiometric amount of CuCl was required in this reaction.
Scheme 23.
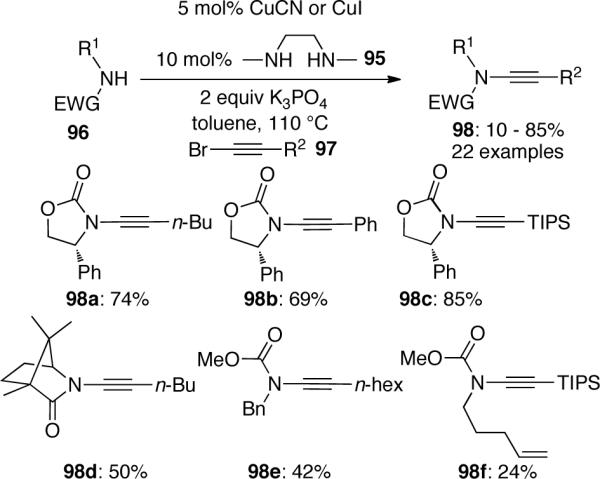
In 2003, Hsung53 published the first copper-catalyzed ynamide formation reaction, which provided the first direct and atom-economical entry to various ynamides (Scheme 24). In this process 5 mol% CuCN or CuI was used as the catalyst and 10 mol% DMEDA (N,N'-dimethylethylenediamine) was used as the ligand. The reaction between amides 96 and alkynyl bromides 97 occurs in refluxing toluene in the presence of K3PO4 as the base. A wide range of substrates including oxazolidinones and lactams were found to be effective substrates and gave corresponding ynamides 98 in good to excellent yields. However, the reactions with imidazolidinones and sulfonamides gave unsatisfactory results. The authors suggested that the course of the catalytic cycle is related to the process proposed by Buchwald54 for the N-arylation of amides.
Scheme 24.
In the same year, Danheiser55a reported a general method for the synthesis of ynamides via copper-mediated coupling of amides with alkynyl bromides. The process involves the conversion of amide substrates 99 to their copper derivatives before the addition of alkynyl bromides 97. Although a stoichiometric amount of CuI was necessary, the reaction proceeded at room temperature and in almost all cases the desired ynamides 100 were isolated in good to excellent yields (Scheme 25). The coupling reaction could be conveniently carried out at gram scale to give ynamide 102 in excellent yield.55b Preparation of more complex substrates such as 100g and 100h can also be accomplished in synthetically useful yields.55c, d
Scheme 25.

Subsequently, Hsung56 disclosed an improved, more practical copper-catalyzed cross-coupling process of amides with alkynyl bromides. The new catalyst system applied inexpensive CuSO4•5H2O and 1,10-phenanthroline as ligand (Scheme 26), and compares quite favorably with the original CuCN catalyst system, as 1) the new system could be used for a large variety of substrates including previously unreactive substrates such as sulfonamides and imidazolidinones, and 2) the reaction temperature could be as low as 60 °C, minimizing ynamide decomposition. It was found that the new process could be carried out conveniently at 100 mmol scale and consistently give excellent yields.56c One limitation for Hsung's system is that the coupling conditions were not effective for the preparation of several simple ynamide substrates such as 98s or 98t.56d
Scheme 26.

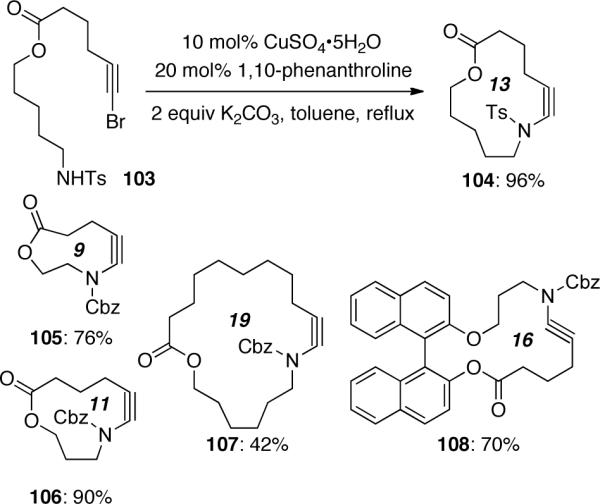
This process was equally effective for intramolecular amidation reactions. As shown in Scheme 27, intramolecular coupling was successfully applied to the synthesis of several unique macrocyclic ynamides 104–108 that contain up to a 19-membered ring system.
Scheme 27.
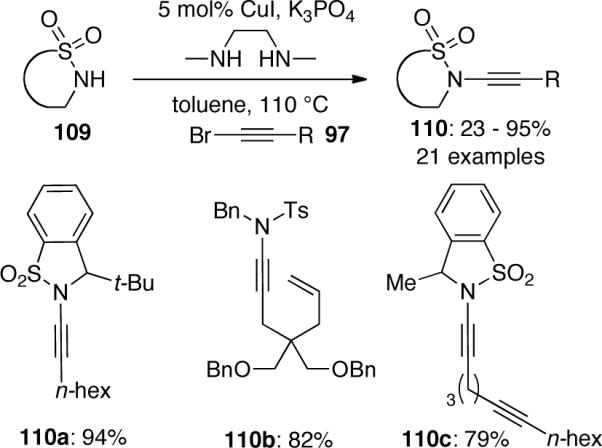
A modified Cu(I)-catalyzed process was described by Urabe57 for the coupling of sulfonamide 109 employing CuI and DMEDA (Scheme 28), Another interesting modification of Hsung's system was described by Skrydstrup,58 who discussed in detail about the water content of potassium phosphate and its effect in this CuSO4 catalyzed coupling reaction.
Scheme 28.
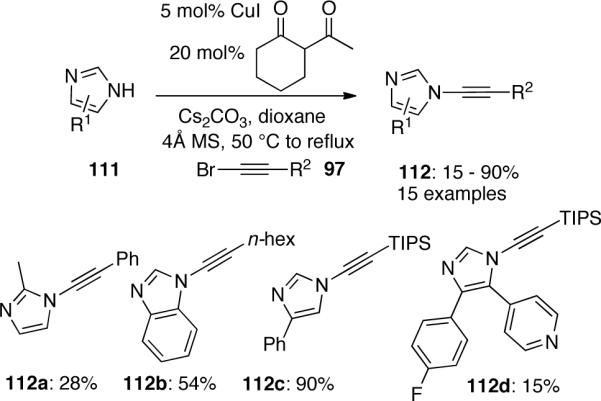
Kerwin59 published a copper-catalyzed coupling of bromoalkynes 97 with imidazoles 111, which provided access to novel N-alkynylimidazoles 112 (Scheme 29). The authors mentioned that diamine ligands such as 1,10-phenanthroline or DMEDA were not effective. In contrast, a catalyst system that utilized CuI and using 2-acetylcyclohexanone as the ligand produced the desire products in variable yields.
Scheme 29.
The first iron-catalyzed coupling of amides and alkynyl bromides was recently published by Zhang.60 It was demonstrated that FeCl3•6H2O as an inexpensive, environmentally benign alternative to copper salt, was an equally efficient and practical catalyst for ynamide synthesis (Scheme 30).
Scheme 30.
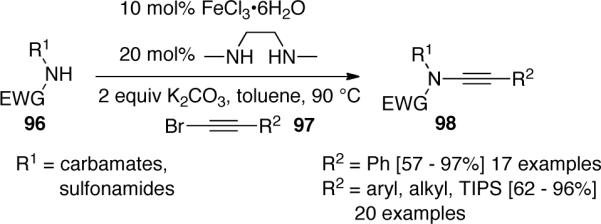
More recently, Evano61 documented the direct synthesis of ynamides 114 from 1,1-dibromo-1-alkenes 113. A combination of CuI and DMEDA was found to be the best catalyst system for the reaction between 96 and 113, and Cs2CO3 was the optimal base (Scheme 31).
Scheme 31.
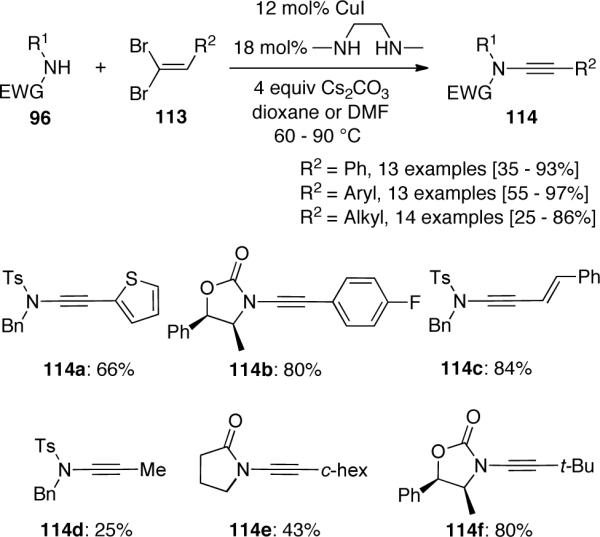
A plausible mechanism is illustrated in Scheme 32. The authors described that 1) a regioselective coupling of the amide and dibromide would produce 115 as a result of the known higher reactivity of the trans C–Br bond toward oxidative addition,62 and 2) Cs2CO3 mediated elimination of 116 would produce 114. This step is supported by the fact that an isolated sample of 116a could be converted to the corresponding ynamide by reaction with only Cs2CO3 via E2 elimination.
Scheme 32.
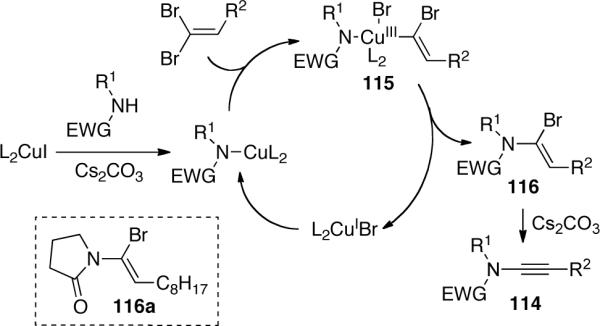
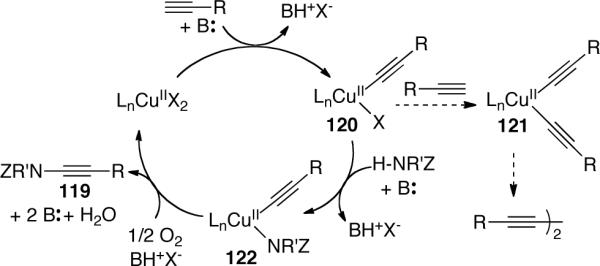
Continuing the with the work started by Balsamo, Domiano, and Peterson vide supra, Stahl63 reported the first copper-catalyzed aerobic oxidative amidation of terminal alkynes. An extensive screening of various Cu sources, Brønsted bases and solvents led to the optimal conditions shown in Scheme 33. It was found that a wide range of nitrogen nucleophiles 117 and alkynes 118 could be used and in most cases the desired ynamides 119 were isolated in good to excellent yields. The only shortcoming of this system lies in the fact that 5 equivalent of amide was necessary to achieve satisfactory yields.
Scheme 33.
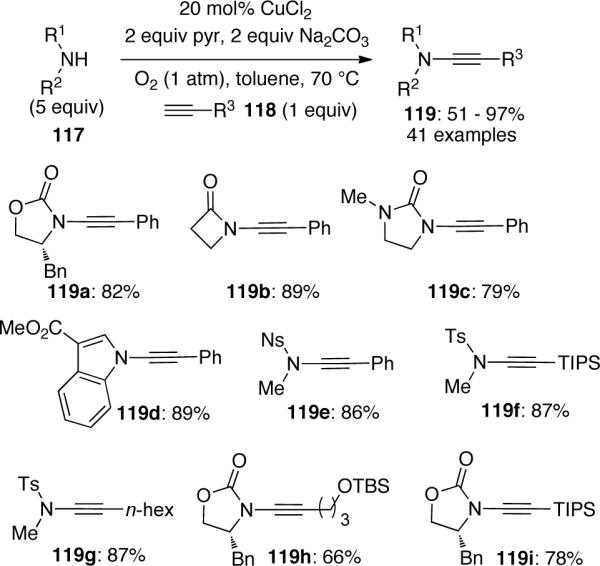
Stahl63 proposed a mechanism that involves sequential activation of the alkyne and amide, followed by reductive elimination and regeneration of Cu(II) catalyst via aerobic oxidation (Scheme 34). The authors rationalized that an intermediate 120 may be involved in two possible pathways. The desired formation of the mixed Cu(II)-(alkynyl)(amidate) species 122 was expected to compete with the undesired formation of bis-alkynyl-Cu(II) species 121. These two pathways thus rationalize the necessity to use excess amount of amide nucleophile.
Scheme 34.
2.5. Other Methods
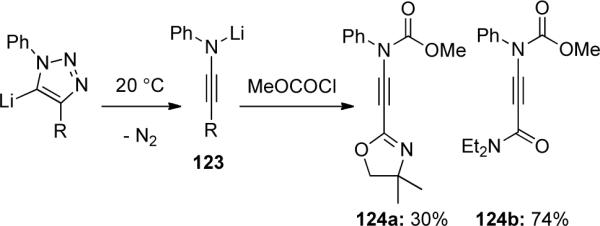
Gilchrist64 demonstrated in 1991 that ynamides 124 could be constructed by the electrophilic trapping of lithiated ynamines 123 generated by decomposition of 1,2,3-triazoles with chloroformate (Scheme 35).
Scheme 35.
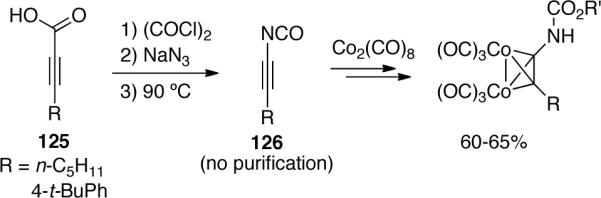
Chen65 synthesized a variation of ynamides known as alkynyl isocyanates. Two isocyanates 126 were prepared from alkynoic acids 125 (Scheme 36) and were directly protected with octacarbonyl dicobalt without purification before further manipulation. The yields were estimated based on the isolation of the protected alkynylcarbamates.
Scheme 36.
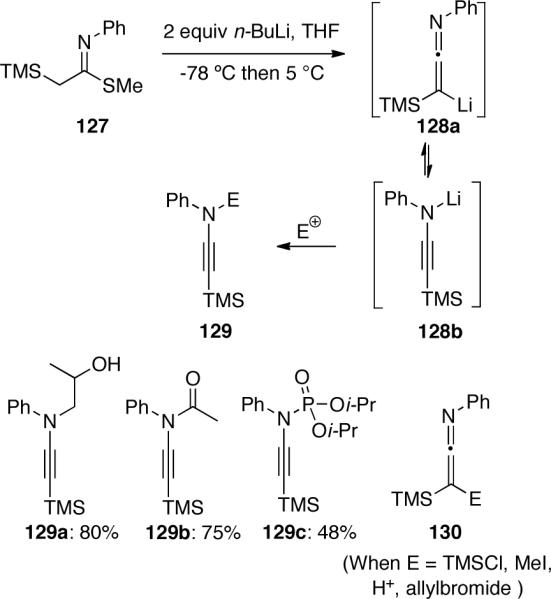
In a manner similar to Gilchrist's previous work64, Masson66 reported that treatment of S-methyl-α-(trimethylsilyl)-ethanimidothiolate 127 with n-BuLi led to the formation of β-lithiated-β-silylated ketenimine 128a (Scheme 37), which is in equilibrium with N-lithiated ynamine 128b. Intermediate 128a could be selectively protonated, methylated, or allylated, yielding ketenimines 130. However, if the lithiated intermediate was treated with more reactive electrophiles such as excess propylene oxide, acetyl chloride, or diisopropyl chlorophosphate, the reaction would favor the nitrogen addition to give ynamines or ynamides 129.
Scheme 37.
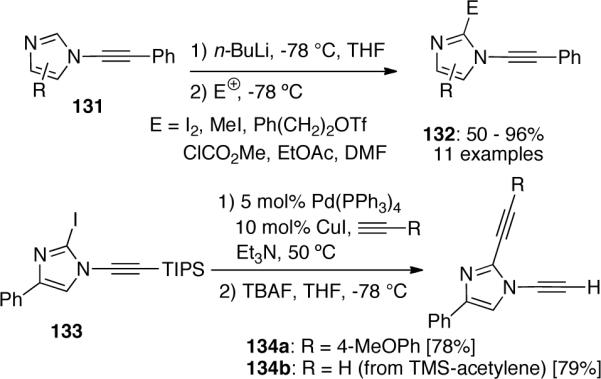
A recent paper by Kerwin67 described their efforts on the functionalization of imidazoles 131 at the 2-position (Scheme 38). Their optimized condition allows a successful deprotonation/lithiation strategy on imidazoles with an adjacent N-alkynyl substituent to give more functionalized ynamides 132. When substrate 133 was subjected to a Sonogashira coupling and desilylation to demonstrate the utility of their strategy, 1,2-dialkynylimidazoles 134 could be obtained in good yields.
Scheme 38.
3. Reactions of Ynamides
3.1. Addition Reactions
The unique nature of ynamides allows for the regioselective addition of electrophiles or nucleophiles onto the ynamide due to the electron-donating ability of the nitrogen.

3.1.1. Addition of Hetero-Nucleophiles
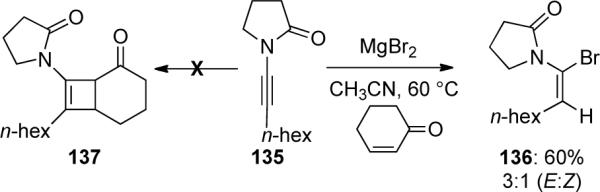
Heteroatom nucleophiles have played a significant role in the addition to ynamides leading to complex heterocycles as well as providing a route to cross-coupling precursors. In 2003, Hsung68 reported a highly stereoselective preparation of chiral α-haloenamides via an unexpected hydrohalogenation of ynamides. While attempting to facilitate a Lewis acid-mediated [2 + 2] cycloaddition of ynamide 135 using magnesium salts, the authors obtained α-haloenamide 136 as a 3:1 mixture of E/Z isomers instead of the desired cycloadduct 137 (Scheme 39). The reaction was highly regioselective as no β-haloenamide was found.
Scheme 39.
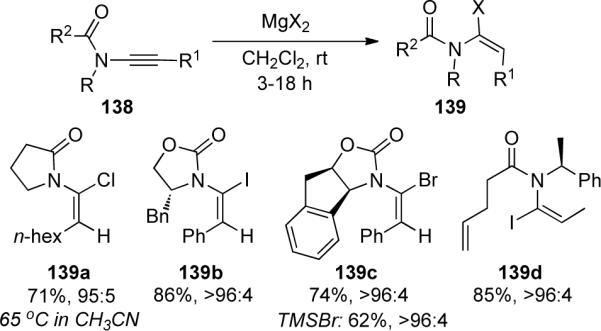
Further optimization found that various magnesium salts in CH2Cl2 gave high yields and excellent diastereoselectivity for the (E)-α-haloenamide at ambient temperature. As shown in Scheme 40, a number of cyclic and acyclic chiral ynamides 138 gave the corresponding α-haloenamide 139 with E/Z ratios of ≥96:4 in almost all cases. It is noteworthy that the use of TMSBr also provided 139c in good yield as a single E-isomer, however attempts to extend this system to include TMSCN and TMSOTf failed. The stereochemistry suggests a syn-addition of HX generated from trace amounts of water with the magnesium salt. This was supported by the observation that freshly prepared MgBr2 in distilled CH2Cl2 gave a much slower conversion rate for the hydrohalogenation compared with the same reaction run in “wet” CH2Cl2 out of a bottle.
Scheme 40.
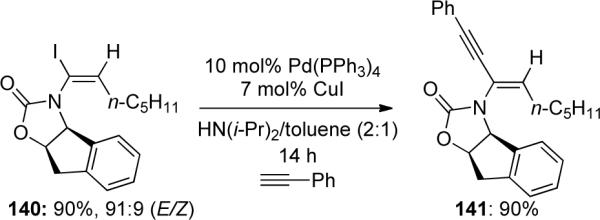
The authors were able to further demonstrate the utility of these α-haloenamides in a Sonogashira coupling reaction (Scheme 41). The α-haloenamide 140 was coupled with phenyl acetylene to give chiral enyne 141, which could be useful in a number of other applications.
Scheme 41.
A gold-catalyzed hydroamination of ynamides was recently reported by Skrydstrup69 for the formation of amidine products and their application toward indole synthesis (Scheme 42). Nucleophilic addition of aniline derivatives onto substituted ynamides under mild conditions using the Gagosz catalyst70 provided amidine products 143 in excellent yields. Cyclic and acyclic ynamides were tolerated with various substituents on the terminal position. Several of the amidines were further used in indole synthesis via Pd(0)-catalyzed ring closure.
Scheme 42.
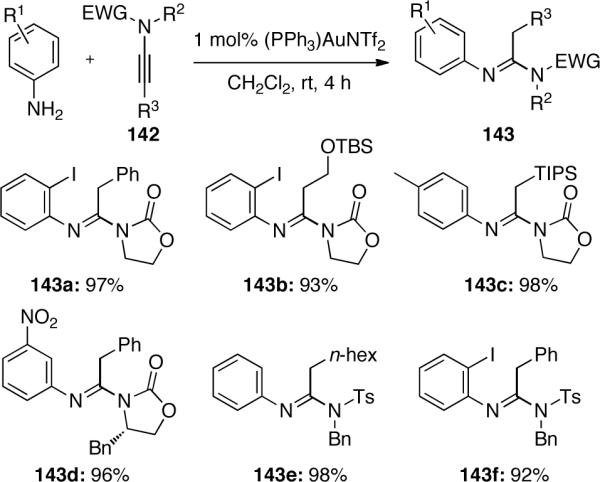
Other metals have also been shown to catalyze the addition of hetero-nucleophiles to ynamides. In Hsung's71 research using rhodium to catalyze a [2 + 2 + 2] cycloaddition of aryl-terminated ynamides vide infra, silver salts were employed as a key additive to enhance the catalytic efficiency. However, the use of AgBF4 led to a surprisingly different reaction pathway in a tandem Rh(I)-catalyzed demethylation-cyclization of o-anisyl ynamides 144, giving benzofuran 146 as the major product instead of the desired chiral biaryl M-145 (Scheme 43).
Scheme 43.
Although the reason for this interesting counteranion effect is unclear, the use of other silver salts also led to benzofuran product in the absence of any diyne. It is reasonable to suggest that the demethylation involves only the cationic rhodium complex 147, which could possess additional coordination sites for a bidentate complexation with the o-methoxy oxygen after being stripped of the ligand present in complex 148.
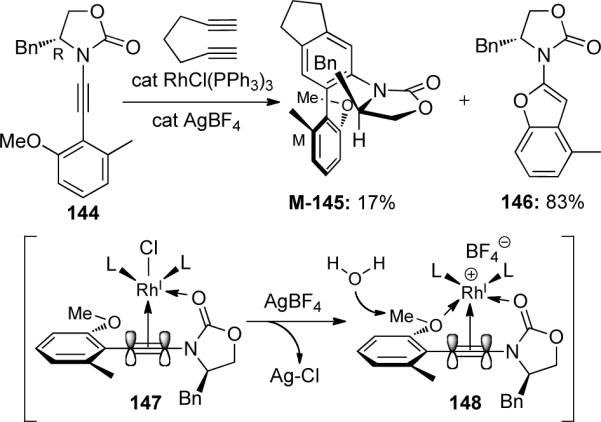
Essentially, this methodology allowed o-anisyl ynamides to serve as protected benzofuran precursors. Using these conditions, the authors were able to construct a variety of functionalized benzofurans, as shown in Scheme 44.
Scheme 44.
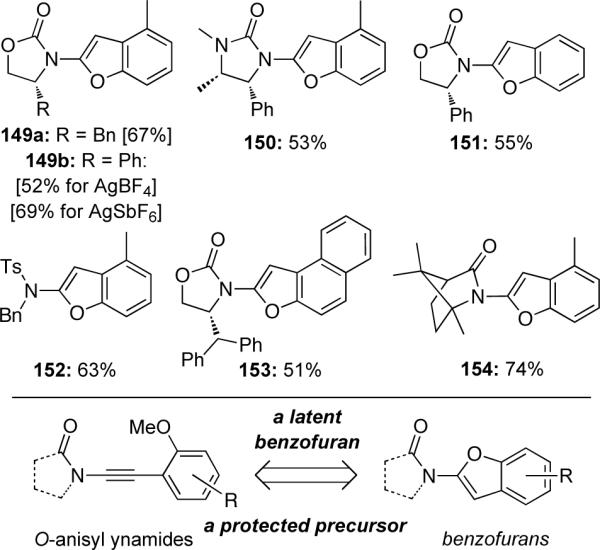
The addition of sulfur nucleophiles to ynamides has been demonstrated by Oshima and Yorimitsu72 leading to interesting N,S-acetal derivatives (Scheme 45). The hydrothiolation of sulfonyl-ynamides 155 with diphenyldithiophosphinic acid 156 led to (E)-alkenyl thioesters 157 as single isomers. Deuterium studies suggest the formation of a reactive keteniminium intermediate 158, whereby addition of the dithiophosphinate anion from the less hindered face leads to the observed (E)-isomer. Furthermore, treatment of ynamide 155a with thiobenzoic acid 159 led to the O-addition product 160 while the use of benzenethiols or alkyl thiols as nucleophiles gave no reaction, suggesting the acidity of nucleophiles is important.
Scheme 45.
3.1.2. Hydroarylation
Zhang73,74 has reported a mild Brønsted acid-catalyzed addition of indoles and other heterocycles as an equivalent process to the hydroarylation of ynamides. As shown in Scheme 46, various sulfonyl-substituted ynamides as well as carbamate- and aza-camphor-derived ynamides could be coupled with indoles, furans, and pyrroles giving hydroarylation products 163 in good yields and with high regio- and stereoselectivities favoring the Z-isomer. Trifluoromethanesulfone imide (Tf2NH) proved to be the optimal catalyst for protonation of ynamide 161 to give the reactive keteniminium ion 162, while platinum and palladium catalysts as well as sulfonic acids were ineffective.
Scheme 46.
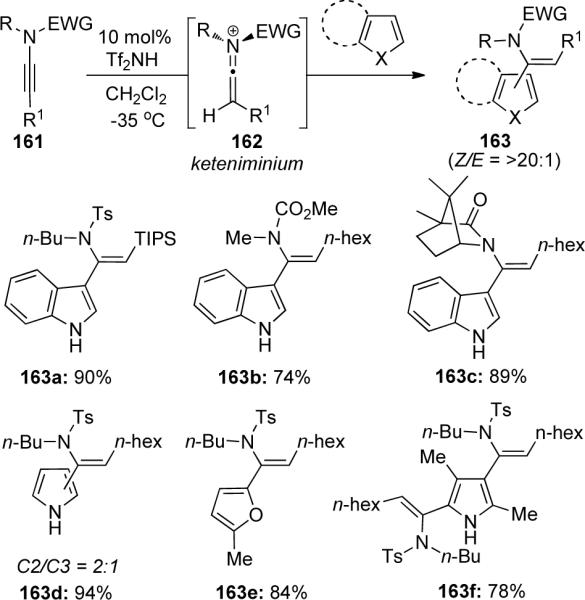
It was noted that no protecting groups were necessary on the indole or pyrrole nitrogen atom and strong electron-withdrawing carbonyl or sulfonyl groups actually led to no desired product formation. However, electron-withdrawing and electron-donating substituents were tolerated on the aryl ring and divinylation products could be obtained when using excess ynamide. When using unsubstituted pyrroles, a 2:1 mixture of C-2 and C-3 vinylation products 163d was isolated. This method provides a synthetically useful route utilizing ynamides to gain access to substituted enamides.
In a similar process, Hsung reported an intramolecular variant in a Pictet-Spengler cyclization of ynamides catalyzed by Brønsted acids. This example is included in Section 3.3.1.
3.1.3. M-H/M-X Additions
The addition of metals to ynamides has allowed for rapid access to a variety of highly functionalized enamides which can be useful in a number of applications and especially in the area of cross-coupling reactions. In 2000, Witulski75 reported the first hydroboration of N-sulfonyl ynamide 164 utilizing catecholborane giving E-vinylborane 166 as the only stereo- and regioisomer (Scheme 47). However, due to the instability and difficulty in isolating the hydroboration product, a one-pot hydroboration/Suzuki-Miyaura coupling procedure was developed with aryl and indolyl halides as coupling partners leading to various β-substituted enamides 167.
Scheme 47.
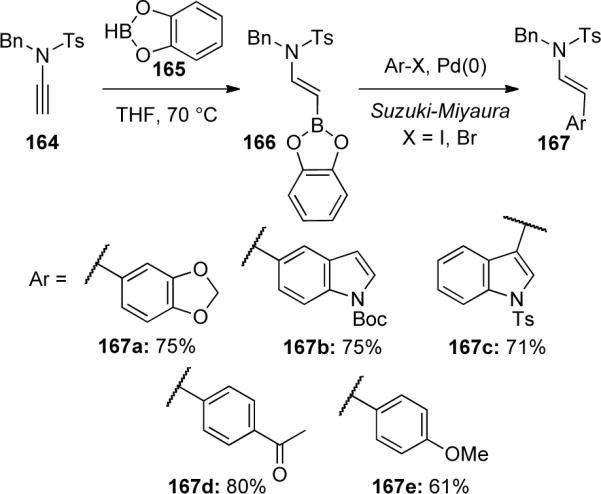
Hoffmann76 was also able to successfully demonstrate a zirconocene-catalyzed hydroboration of ynamide 168 with pinacolborane to generate vinyl boronate 169 in 89% yield (Scheme 48). Further homologation followed by a domino hydroformylation-allylboration-hydroformylation sequence gave the lactol 170 as a 1:1 mixture of anomers. Both hydroboration methods allow for exclusive formation of β-substituted enamides as addition to the ynamide occurs regioselectively.
Scheme 48.
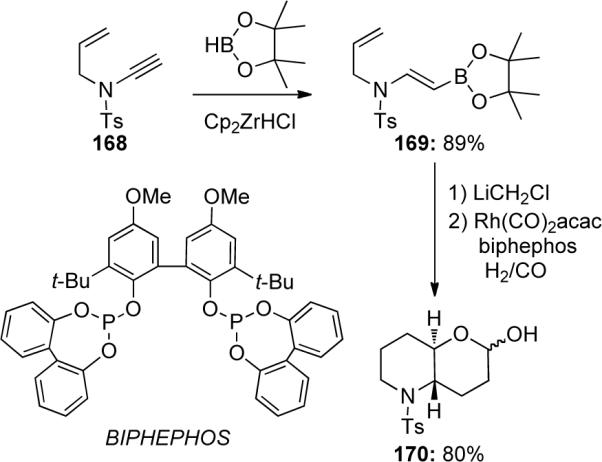
A complementary approach was developed by Cintrat77 through a palladium-catalyzed hydrostannation of ynamides, thereby gaining access to α-substituted enamides. The first attempt at the hydrostannation of silyl-protected ynamide 171 led to a mixture of (E)- and (Z)-enamides 172 (Scheme 49). However, the use of terminally unsubstituted ynamide 173 allowed for a facile synthesis of only the (Z)-silylstannylated product 174. The hydrostannation product 174b was obtained as a 91:9 mixture of regioisomers in favor of the α-stannylated product. The authors also reported significant protodestannylation of the β-isomer when the mixture was subjected to column chromatography.
Scheme 49.
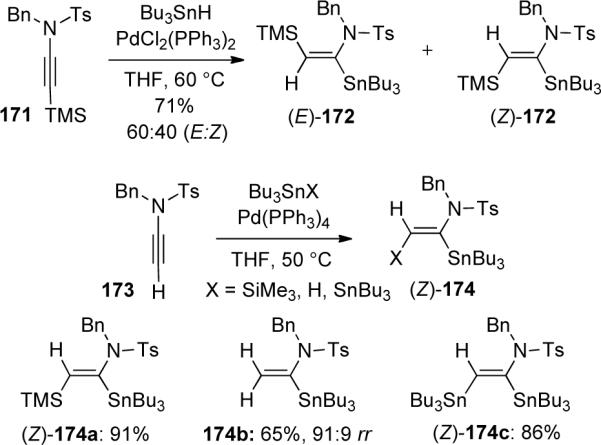
Furthermore, it was shown that the α-stannylated enamides could be selectively functionalized to give a wide array of products78 (Scheme 50). Using a modified Stille coupling procedure, a variety of acyl, allyl, and aryl halides could be coupled with silylstannylated enamide 174a to selectively functionalize the α-position. Iododesilylation at the β-position would then allow for further diversity in the formation of disubstituted enamides 176. It was noted that in the absence of the silicon group, heating was necessary for complete conversion of the Stille coupling.
Scheme 50.
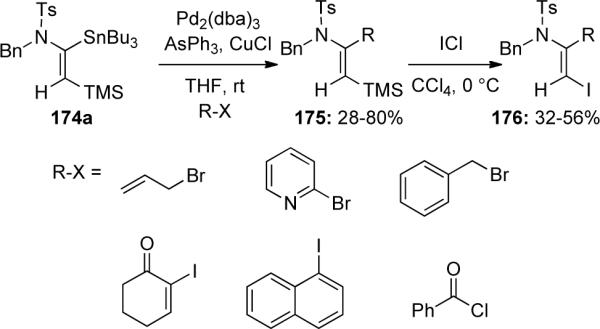
Cintrat79 later reported an α-selective hydrostannation of terminally-substituted ynamides. They found that use of cyclic amide-derived ynamides 177, specifically with oxazolidinone or imidazolidinone moieties, gave the best selectivity for the α-stannylated enamide 179, presumably through an intermolecular chelation of the carbonyl oxygen to tin, as shown in 178 (Scheme 51). The hydrostannation worked with a variety of terminal substituents as well as chiral and achiral auxiliaries, giving high regioisomeric ratios (rr) in favor of the α-stannylated product with little or none of the (Z)-isomer.
Scheme 51.
3.1.4. Umpolung-Type Additions
Much of the earlier chemistry involving ynamides has taken advantage of the electron-donating ability of nitrogen for the regioselective addition of nucleophiles and electrophiles to the α- and β-carbons of ynamides, respectively. More recently, several groups have focused on “umpolung-type” additions of nucleophiles to ynamides via metal-catalyzed processes, providing a facile entry to the opposite regioisomeric addition products.

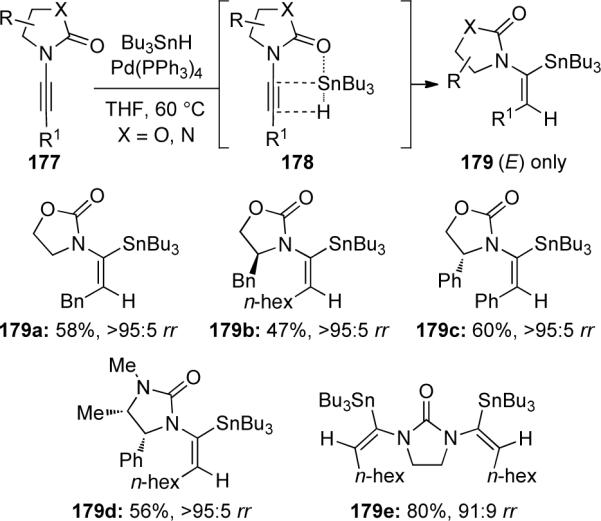
Indeed, the unique nature of ynamides, combining an electron-withdrawing substituent with a chelating moiety, was utilized by Marek80 in a regiochemically controlled carbometallation of ynamides (Scheme 52). Two sets of conditions could be used in a carbocupration (Condition A) or copper-catalyzed carbomagnesiation (Condition B) of ynamide 180. Chelation of the organometal reagent to the carbamoyl moiety controls the regioselective addition of the carbon nucleophile to the β-position. This provides the α-metallated intermediate 181 which can subsequently be trapped with various electrophiles to give substituted enamides 182. In general, both conditions gave similar results, with the carbomagnesiation giving slightly higher yields. The carbometallation could also be carried out on N-sulfonyl ynamides, although both conditions were slower and required higher temperatures.
Scheme 52.
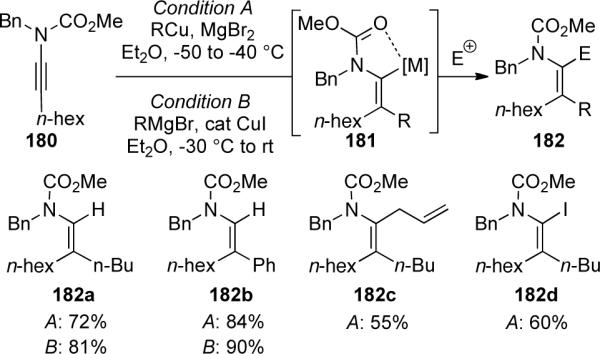
Marek81 further demonstrated the utility of ynamides in an efficient one-pot carbocupration/Znhomologation/allylation sequence to obtain product 186 with the formation of all-carbon quaternary stereocenter (Scheme 53). A possible mechanism suggests that after initial carbocupration with the diethylcuprate, transmetallation of vinyl cuprate intermediate 184 followed by homologation with the Simmons-Smith-Furukawa82 zinc carbenoid (generated in situ) would give allylzinc species 185. Subsequent trapping with benzaldehyde then provides the aldol product 186. Several other aldehydes and organocuprates were also used giving similar yields and high diastereoselectivities. The absolute stereochemistry of the major isomer of 186 can be rationalized by the Zimmerman-Traxler transition state, in which the benzyl group of the auxiliary shields one face in the chelated six-membered chair conformation.
Scheme 53.

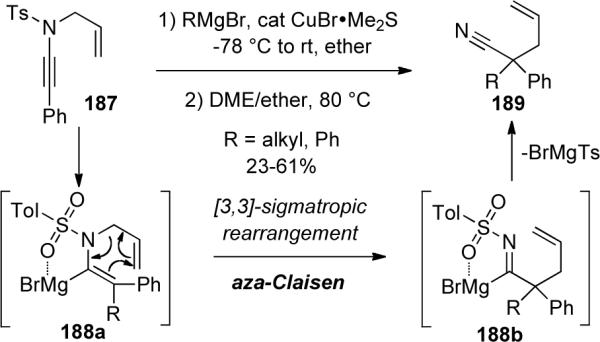
Oshima and Yorimitsu83 reported a similar silylcupration and copper-mediated carbomagnesiation of N-sulfonyl ynamides followed by an aza-Claisen rearrangement (Scheme 54). After the initial carbomagnesiation of ynamide 187, instead of trapping the α-metallated enamide 188a with an electrophile, they found that heating the reaction mixture in 1,2-dimethoxyethane (DME) led to 4-pentenyl nitriles 189 via a two-step [3,3] sigmatropic rearrangement/elimination sequence. While it is unclear whether the elimination occurs before or after the rearrangement, the reaction did not occur with a protonated enamides, suggesting the presence of the magnesium metal is essential for the reaction to proceed.
Scheme 54.
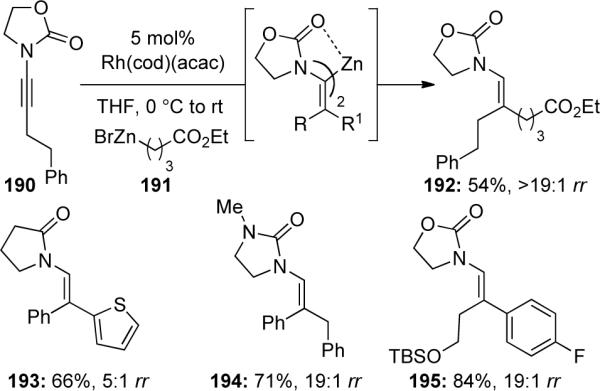
One limitation to these types of carbometallation reactions is the use of Grignard reagents, which limits certain functional groups that can be tolerated on the ynamide. In an effort to improve upon this, Lam84 recently communicated their efforts on a rhodium-catalyzed carbozincation of ynamides. A representative example is shown in Scheme 55 in which ynamide 190 reacts with alkylzinc bromide 191 containing an ester moiety yielding the substituted enamide 192. Using the previous carbometallation conditions with Grignard reagents, the ester functionality would not be tolerated. They were also able to functionalize the vinylzinc intermediate through simple acylations or Negishi couplings.
Scheme 55.
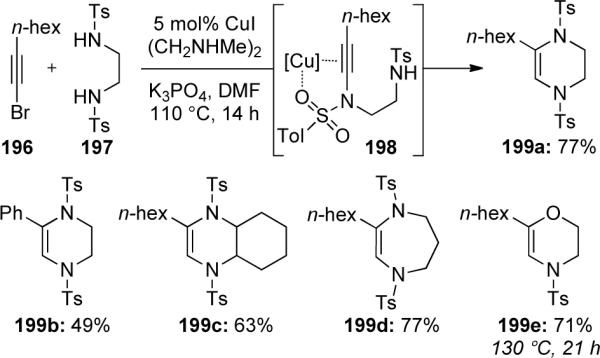
In addition, nucleophiles other than carbon have been shown to add to ynamides in an umpolung-type manner. In 2008, Urabe57b discovered an unexpected copper-catalyzed double amination of 1-bromoalkynes leading to tetrahydropyrazines while attempting an N,N'-dialkynylation of diamines (Scheme 56). The reaction of bromo-acetylene 196 with diamine 197 gave tetrahydropyrazine 199a using standard coupling conditions at elevated temperatures. Presumably, after the first alkynylation of the sulfonamide, the second amination occurs intramolecularly through a 6-endo-dig pathway on the intermediate 198. The sulfonyl group is thought to play an important role in mediating the endo-type pathway through coordination to the copper salt. This method is useful with aliphatic and aromatic acetylenes. Branched 1,2-diamines, 1,3-propanediamines would lead to seven-membered heterocycles, while oxygen nucleophiles require slightly higher temperatures.
Scheme 56.
3.1.5. Radical Processes
Although only a few radical processes involving ynamides have been reported, they provide another complementary approach toward constructing complex heterocycles and substituted enamides.
Malacria85 reported a radical cascade reaction of ynamides to gain access to nitrogen heterocycles (Scheme 57). Two types of ynamides were used based on the position of the carbonyl group. The radical cascade occurs through a 5-exo-dig cyclization followed by a 6-endo-trig trapping to provide the heterocyclic products. Acyclic or cyclic alkenes could be used with Type I (Malacria's classification) ynamides for the 6-endo-trig radical trapping as well as an aromatic acceptor with varying yields depending on the rate of hydride addition and the halide used. While simple alkenes also worked for Type II ynamides as in 201a, the cascade reaction was halted after the 5-exo-dig cyclization when using an aromatic acceptor giving a 1:1 mixture of the Z- and E-isoindolinones 201b. Photolysis of hexa-n-butylditin was necessary to obtain the cascade product 201c, suggesting the carbonyl may have a steric and an electronic effect on the radical trapping.
Scheme 57.

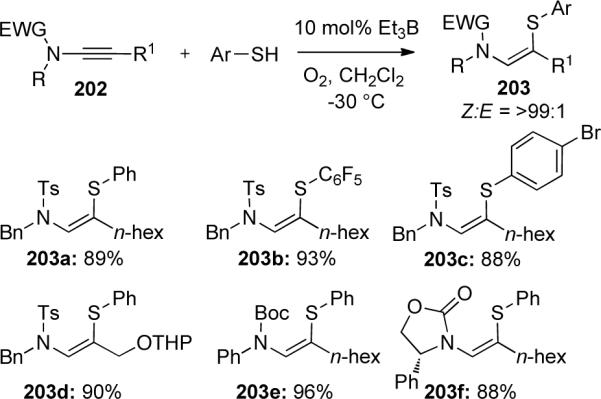
Yorimitsu and Oshima86 reported a radical hydrothiolation of ynamides giving (Z)-1-amino-2-thio-alkenes 203 (Scheme 58). Cyclic and acyclic ynamides were reacted with various aryl thiols using triethylborane and O2 as the radical initiator. The reaction is believed to proceed by regioselective addition of the electron-deficient thiyl radical to the more electron-rich 2-position of ynamides 202. The resulting stereoselectivity for the Z-isomer arises from sterics between the terminal substituent and the amide, as smaller terminal substituents led to a mixture of stereoisomers. Furthermore, electron-deficient arenethiols were well tolerated (203b and 203c) while electron-rich arenethiols gave much lower yields, suggesting electron-donation lowers the reactivity of the electrophilic thiyl radical.
Scheme 58.
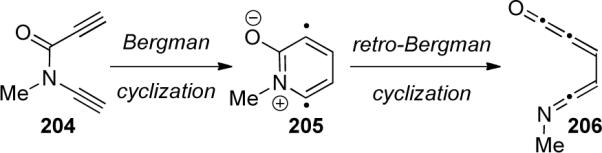
The use of enediynes as potential anticancer drugs is an important area of research because of their ability to cleave DNA through a presumed Bergman cyclization.87 Kraka and Cremer88 reported a theoretical investigation of heteroatom-substituted enediynes and the effect on possible Bergman cyclization (Scheme 59). Calculations on ynamide 204 showed very little enediyne character and the energy barrier for the retro-Bergman cyclization was much lower than needed for 204 to be useful as a drug candidate.
Scheme 59.
3.2. Cycloadditions
3.2.1. [2 + 1] Cycloadditions
Hsung89 recently published a Rh(II)-catalyzed cyclopropenation of ynamides providing a facile access to highly substituted 2-amido-furans (Scheme 60). Both diazo dimethyl malonates 208 and phenyl iodonium ylides 209 could be successfully used as the cyclopropenating agent, however the diazo compounds gave better yields. Unsymmetrical cyclopropenating agents (R = Me) resulted in highly regioselective furan formation. It is noteworthy that this methodology could be applied to terminally substituted ynamides, resulting in tetrasubstituted furans 210.
Scheme 60.
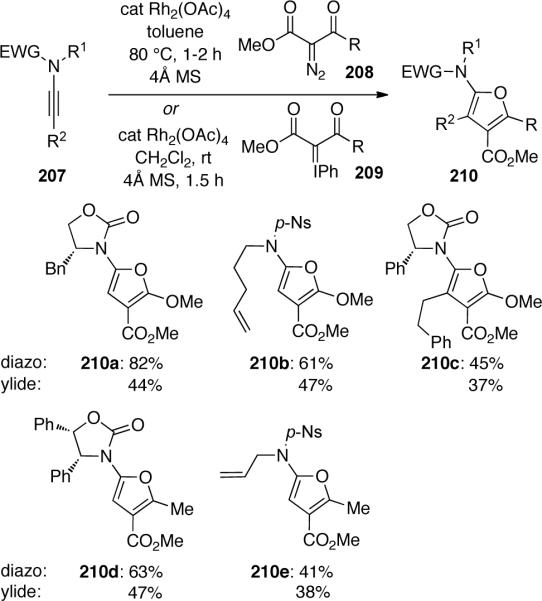
The authors suggested that the mechanism for this transformation may proceed through amido-cyclopropene 211 formed by a Rh-carbene-mediated [2 + 1] cycloaddition (Scheme 61, path a). Ring-opening of the cyclopropene followed by subsequent ring-closer would yield the observed furan products 213. Alternatively, 211 may not be involved (path b). Metallo-keteniminium 214 could tautomerize to 215 followed by cyclization and reductive elimination to give 213. Formally, this transformation represents a [3 + 2] cycloaddition.
Scheme 61.
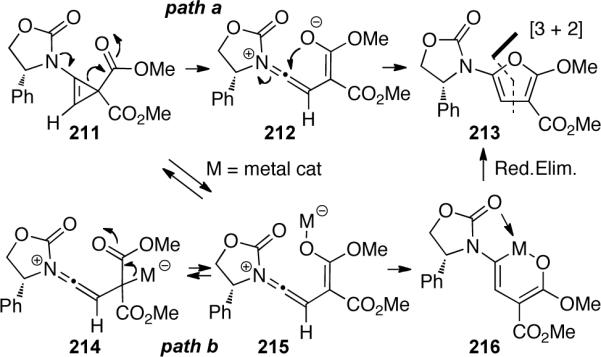
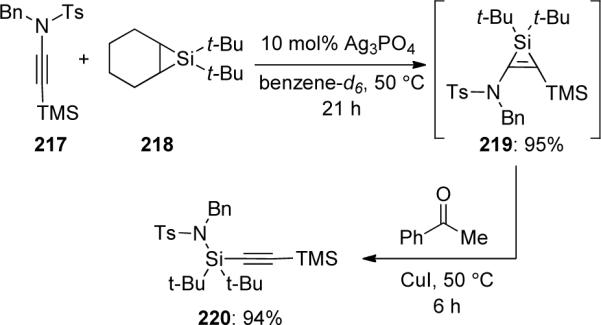
Woerpel90 found that amido-silacyclopropene 219 could be formed through silver-catalyzed silylene transfer from silacyclopropane 218 to ynamide 217 (Scheme 62). Alternatively, in the presence of CuI and acetophenone, the in situ generated silacyclopropene 219 rearranges to alkynylsilane 220 in 94% yield. This silylene transfer may proceed through Cu-mediated ring opening of 221 to give 222, which may undergo intramolecular silyl transfer and loss of CuI to generate alkynylsilane 224. An O-to-N silyl transfer gives the energetically preferred silyl acetylene 225 (Scheme 63).
Scheme 62.
Scheme 63.
Cossy and Meyer91 reported a novel example of a chemoselective DMDO [methyl(trifluoromethyl)-dioxirane] 227 epoxidation of 1,6-ene-ynamide 226 to generate aza-[3.1.0]bicycle 231 through an α-oxocarbene intermediate 229 (Scheme 64).
Scheme 64.
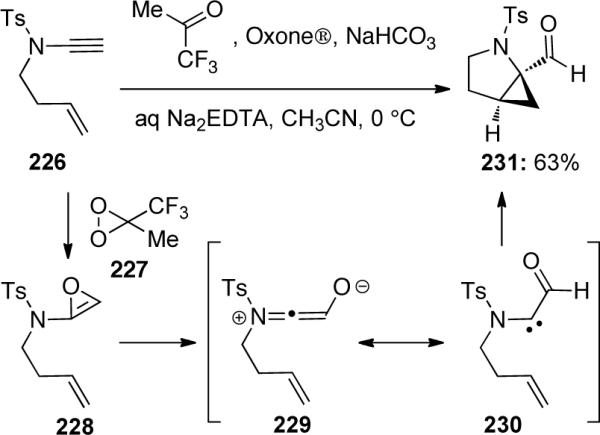
The above method, however, did not extend well to terminally-substituted ynamides (Scheme 65). The authors discovered that the use of t-BuOOH with VO(acac)2 could successfully catalyze the epoxidation and subsequent cyclization of ynamides 232, 234 and 236, tethering through either the N- or C-terminus of the ynamide. Unfortunately, there was no diastereomeric induction with this catalyst, the reaction of 232 give 233a and 233b as 1:1 diastereomeric mixtures. The epoxidation and cyclization of 236 resulted in a 70:30 diastereomeric mixture of 237a : 237b.
Scheme 65.
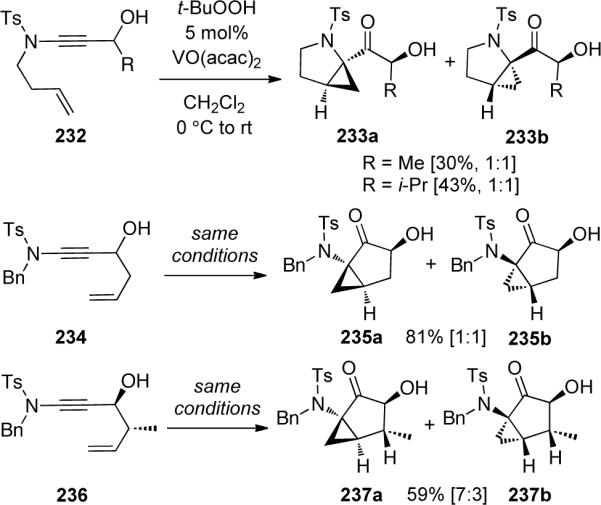
In the same year, Hsung and Al-Rashid92 described an efficient preparation of α-keto-imides 239 through both RuO2−NaIO4 and DMDO mediated oxidation of ynamides 238 (Scheme 66). The RuO2−NaIO4 protocol provided novel syntheses of vicinal tricarbonyl containing 239e and 239f, while the DMDO oxidation allowed for the chemoselective oxidation of olefin-tethered ynamides 240 to 241 (Scheme 67).
Scheme 66.
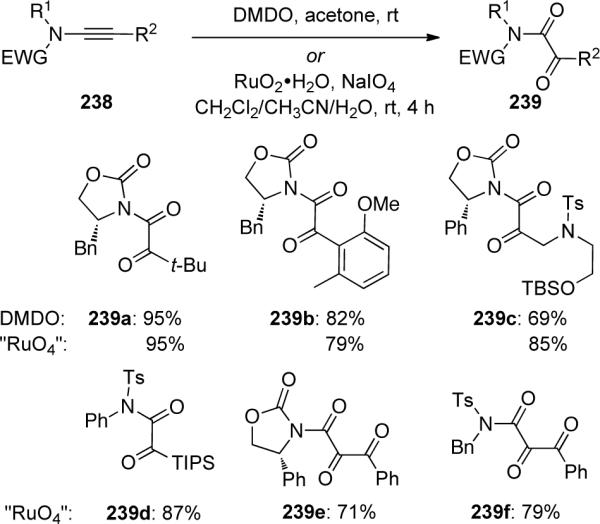
Scheme 67.
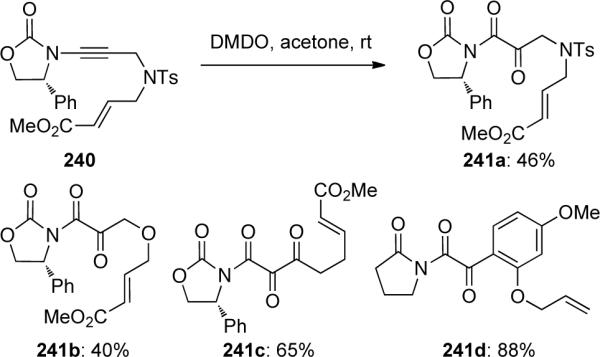
Hsung and Al-Rashid93 also demonstrated the intramolecular DMDO-mediated cyclopropanation of chiral ene-ynamide 242 through the generation of push-pull carbene 244 (Scheme 68). Amido-cyclopropane 243 was obtained in good yield and 3:1 dr with an α-keto-imide being isolated as a side product.
Scheme 68.

The authors were able to probe the formation of amido carbenes 248 by varying the electronics of the tethered olefin. Ynamides containing electron-deficient olefins gave only α-keto-imides 247c and 247d, while with electron-rich olefins the amido-cyclopropanes 246a and 246b could be isolated with no traces of bicyclo[3.1.0]hexane 249, which should result from cyclopropanation via oxocarbene 248.
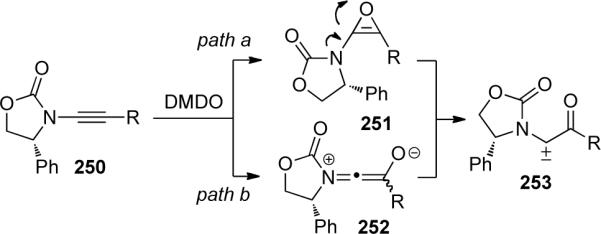
The formation of push-pull carbene 253 may proceed through two reaction pathways (path a or b, Scheme 69). In path a, ynamide epoxidation and ring opening of the intermediate oxirene 251 would give 253. Alternatively, zwitterionic oxyketeniminium 252 may be involved in the formation of 253 via path b.
Scheme 69.
3.2.2. [2 + 2] Cycloadditions
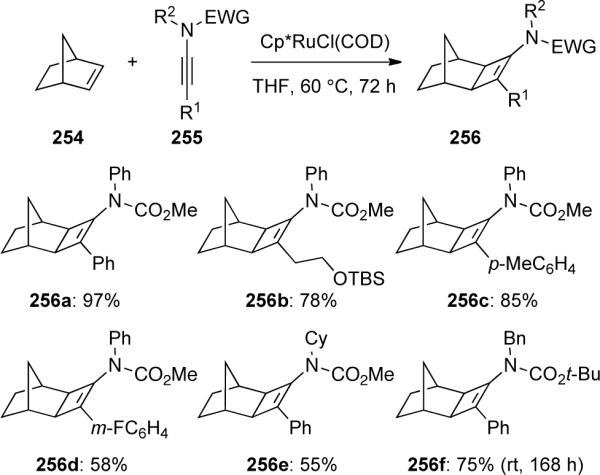
Tam94 reported the first examples of a ruthenium-catalyzed [2 + 2] cycloaddition between norbornene 254 and ynamides 255 to give amido-cyclobutenes 256 (Scheme 70).
Scheme 70.
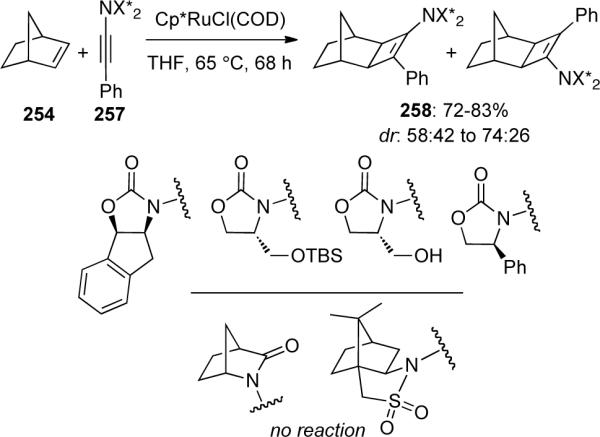
When the authors tried to extend this methodology to include chiral ynamides 257, the yields of products 258 were good, however the diastereoselectivity was only moderate, and no reaction was observed using bicyclic amide or sultam derived ynamides, presumably due to the increased steric hindrance (Scheme 71).
Scheme 71.
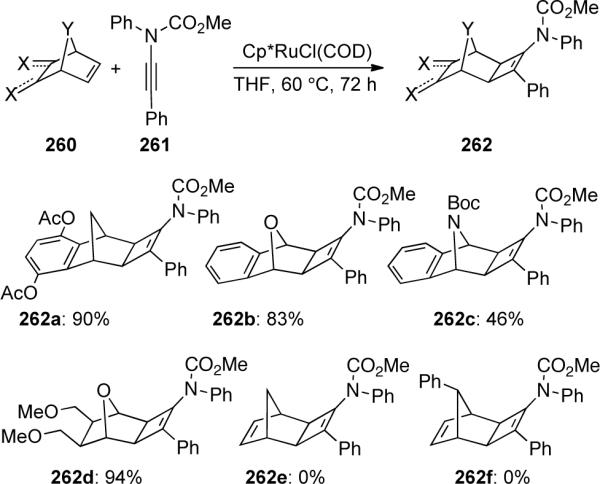
Tam95 later disclosed that a diverse array of bicyclic alkenes 260 could partake in these Ru-catalyzed [2 + 2] cycloadditions providing functionalized cycloadducts 262 in moderate yields while norbornadienes failed to produce 262e and 262f (Scheme 72). The authors believed this could be attributed to chelation of ruthenium to both alkenes of 260e and 260f in a bidentate fashion, thereby disfavoring coordination of the ynamide 261.
Scheme 72.
Danheiser96 has also reported some beautiful [2 + 2] cycloadditions of ynamide 263 with several classes of ketenes, providing substituted 3-amidocyclobutenones 264–267 in good yields (Scheme 73).
Scheme 73.

Hsung97 explored the reactivity of ynamide 268 with various aldehydes in BF3•Et2O-promoted hetero-[2 + 2] cycloaddition reactions (Scheme 74). The resulting amides 269 were obtained in good to excellent yields with high E-selectivity. This transformation presumably proceeds through a step-wise [2 + 2] cycloaddition, followed by electrocyclic ring opening.
Scheme 74.
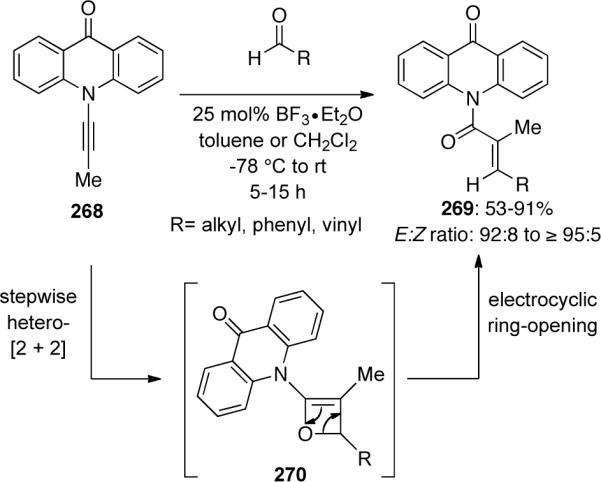
Ynamides 272 bearing a tethered carbonyl could also be utilized in this Lewis acid-catalyzed intramolecular hetero-[2 + 2] cycloaddition, ring-opening sequence to synthesize cyclic α,β-unsaturated amides 273 (Scheme 75). Notably, imide-containing ynamide 274 also led to bicyclic lactam 275 in good yield.
Scheme 75.
A tandem cross-coupling, cycloaddition, ring opening sequence98 was also explored by Hsung (Scheme 76). In situ ynamide formation from the cross-coupling of amide with bromide 276 gave chromene 277 in 60% yield. In another case, oxidation of the primary alcohol in 278 could trigger the formation of quinolizidine 280 in 53% yield. The process was also successfully applied to the facile synthesis of pyrrolizidine motif. The optically enriched bromide 281 was coupled with amide to give ynamide 282, deprotection of TBS group followed by a one-pot tandem oxidation-cycloaddtion-ring opening to produce 284 in good overall yield.
Scheme 76.

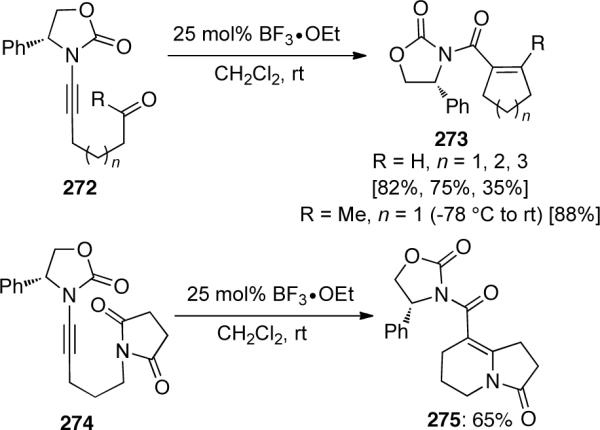
In 2007, Hsung99 reported a two-carbon homologation of aldehydes and ketones employing ynamides. Ynamides 285 could react with aldehydes under BF3•OEt2-promoted conditions to give 286 exclusively as the E-isomer (Scheme 77).
Scheme 77.
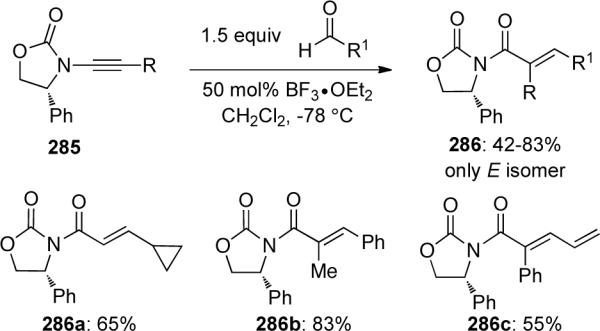
The reaction of terminal ynamide 287 with a variety of ketones also led to unsaturated amides 288 in moderate yields and with low E/Z selectivity in the examples using unsymmetrical ketones. When phenyl-substituted ynamide 289 was employed in this transformation, deconjugated products such as 290 could be obtained with high diastereoselectivity (Scheme 78).
Scheme 78.
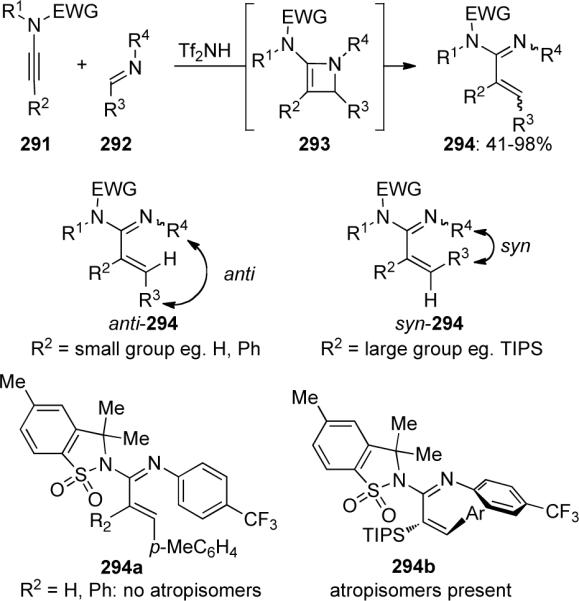
Takemoto and Takasu100 described a new method for the synthesis of α,β-unsaturated amidines through a cascade reaction between ynamides 291 and imines 292 consisting of a stereoselective [2 + 2] cycloaddition and a thermal cycloreversion or ring-opening (Scheme 79). When R2 was small, anti-294 was isolated, whereas, when R2 was large, the product was syn-294, which existed as a mixture of atropisomers.
Scheme 79.
3.2.3. [3 + 2] Cycloadditions
The first examples of “click” employing ynamides was described by Cintrat101 in 2006, in which N-tosyl ynamide 295 was reacted with alkyl azides to give 4-amino-1,2,3-triazoles 296 with a variety of functionalities at the 1-position (Scheme 80). Cu(OAc)2-Na ascorbate was found to be the best catalytic system for these transformations, while CuI could also be employed.
Scheme 80.
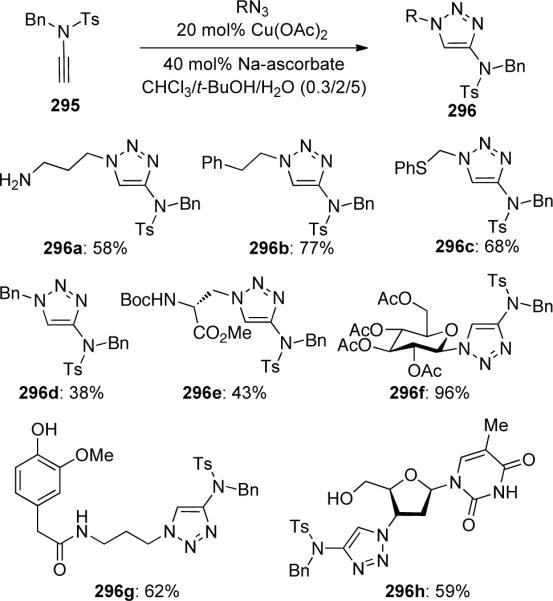
The authors found that although both cyclic carbamate and urea-derived ynamides could not be converted to the expected triazole products, N-benzoyl ynamide 297 afforded triazole 299 in 55% yield (Scheme 81)
Scheme 81.
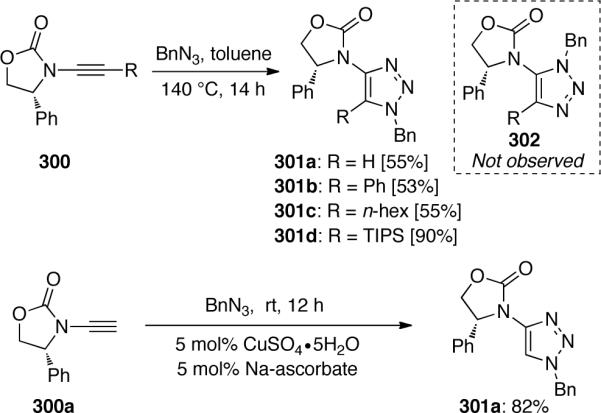
In the same year, Hsung102 reported a variation of Huisgen's [3 + 2] cycloaddition involving chiral ynamides (Scheme 82). The carbamate-derived ynamide 300 reacted with benzyl azide to provide the 1,4-adduct 301 under thermal conditions without observation of the 1,5-adduct 302. The same 1,4-regioselectivity was also observed in the thermal cycloaddition of internal ynamides. The yield of 301a was increased to 82% under the Fokin-Sharpless Cu(I) catalytic conditions.
Scheme 82.
The authors also found an unexpected tandem hydroazidination-Huisgen [3 + 2] cycloaddition of terminally-unsubstituted ynamide 300a that provided vinyl triazole 305 as the major product (Scheme 83). The expected triazole 304 could be preferentially formed by syringe pump addition of the ynamide. By excluding both PhI and L-proline from the reaction mixture, 305 could be isolated in 92% yield, presumably through transition state 303.
Scheme 83.
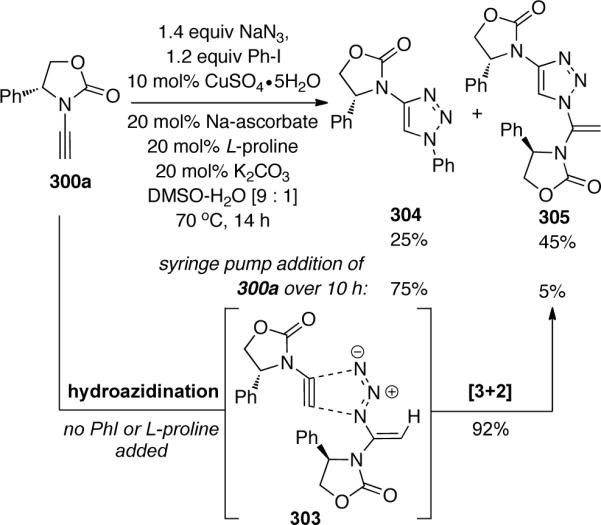
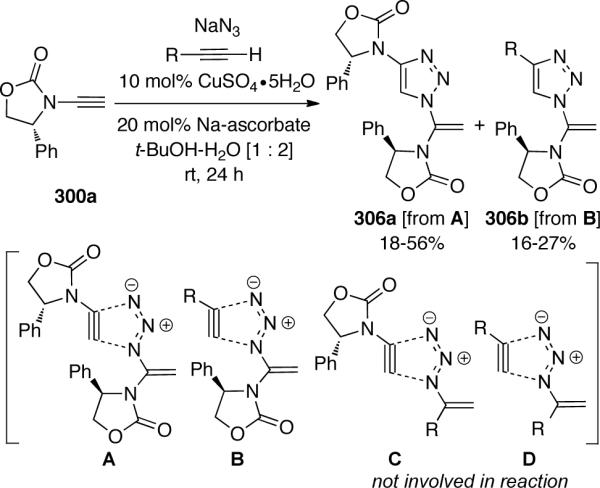
By introducing a second terminal alkyne to the reaction mixture, the authors were able to investigate the electronic sensitivity of this tandem sequence (Scheme 84). In the presence of several alkyl and aryl acetylenes, only 306a and 306b were formed resulting from transition states A and B, respectively. Under these conditions, there was no formation of products resulting from C or D, implying that the hydroazidination was chemoselective in favor of the more electron-rich ynamides.
Scheme 84.
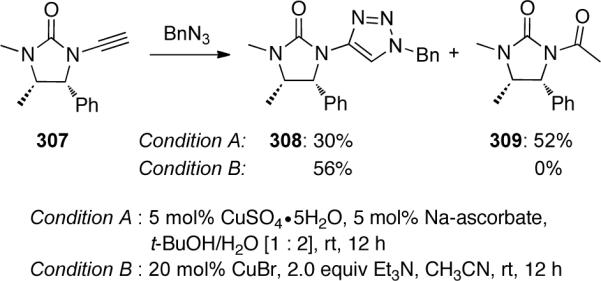
Later, it was found that ynamide hydrolysis could be a competing reaction in this [3 + 2] cycloaddition,103 especially with less stable ynamides such as 307 (Scheme 85). When 307 was reacted with benzyl azide under CuSO4•5H2O catalyzed conditions (Condition A), only 30% of the expected triazole 308 was isolated, with the major product being 309 resulting from ynamide hydrolysis. The authors were able to circumvent the hydrolysis issue by using CuBr as the catalytic copper species providing 308 in 56% yield.
Scheme 85.
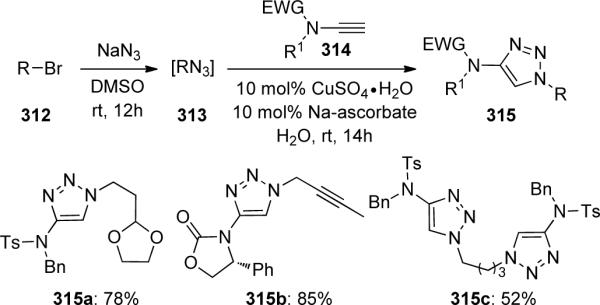
This reaction sequence could also be carried out in one pot by in situ formation of the alkyl azides 313 from the corresponding alkyl bromides 312 (Scheme 86). As anticipated from their previous work, the only observed product was the 1,4-disubstituted isomer. Both primary and secondary alkyl bromides could be used while the latter afforded triazoles 315 in a lower yield. This one-pot protocol could also be applied to the synthesis of the novel bis-triazole 315c.
Scheme 86.
In a similar manner, bis-triazoles 317 and 318 could be prepared in synthetically useful yields from the corresponding bis-ynamides 316 (Scheme 87). As another example of the utility of this transformation, macrocyclic ynamides 319 could be functionalized to the 1,4-disubstituted macrocyclic fused triazoles 320 in good yields.
Scheme 87.
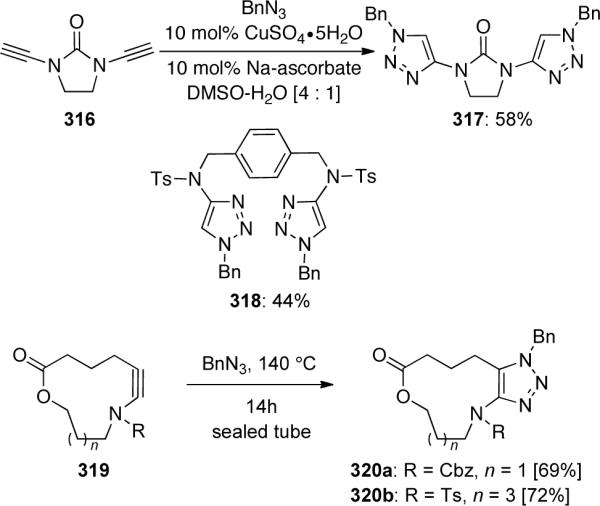
With the anhydrous CuBr-catalyzed conditions overcoming the problem of ynamide hydrolysis, another intriguing observation was soon discovered104 (Scheme 88). When ynamide 321 was treated with benzyl azide, a mixture of triazole 322 and alkyne 323 was isolated. N-sulfonyl-substituted ynamides afforded even higher yields of ene-ynamides 324, which were presumably derived from reductive elimination of intermediate 326, implying the presence of vinyl copper intermediate 325.
Scheme 88.
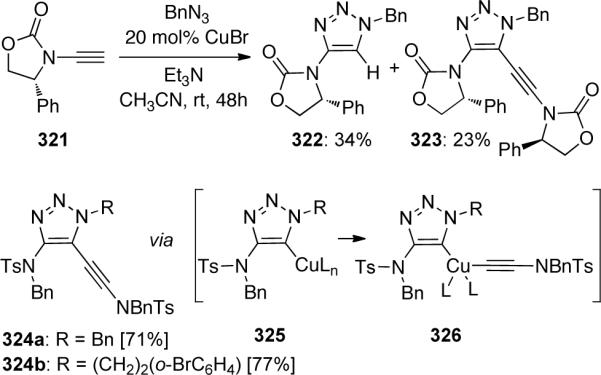
The presence of this vinyl copper species afforded an excellent opportunity to prepare unique trisubstituted-amido-triazoles (Scheme 89). By trapping the vinyl copper intermediate prepared from ynamide 327 with allyl iodide, triazoles 328 could be formed. These diallyl-triazoles could be directly subjected to Grubbs' II catalyzed ring-closing metathesis to construct a variety of bicyclic triazoles 329.
Scheme 89.
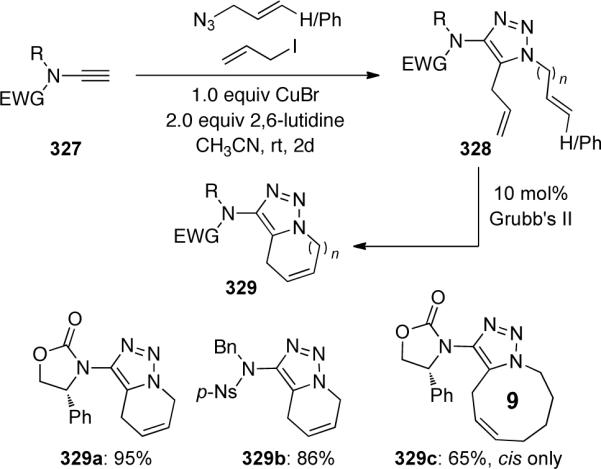
In a variation of this transformation, the authors found that N-allyl ynamides 330 could be functionalized to triazoles 331 allowing subsequent ring-closing metathesis to afford bicyclic triazoles 332 with the amide functionality incorporated into the ring (Scheme 90).
Scheme 90.
Chang105 described a copper-catalyzed three-component coupling reaction of ynamides 333, tosyl azide and various amines to prepare α-amido amidines 334 (Scheme 91). In this reaction, the 1,2,3-triazol-5-yl copper species 335 was first formed by [3 + 2] cycloaddition, followed by the generation of intermediate 336 or 337, which would lead to formation of highly reactive amido-ketenimine 338. The ketenimine could then be trapped by amines to provide α-amido amidines 334 or by methanol to give α-amido imidate 340.
Scheme 91.

Cintrat106 reported a highly regioselective ruthenium-catalyzed [3 + 2] cycloaddition to provide 5-amido 1,2,3-triazoles 343 (Scheme 92). This methodology compliments the copper-catalyzed cycloaddition discussed previously that yields 1,4-substituted triazoles. Both sulfonyl and carbamate-derived ynamides 341 worked well in this system affording the corresponding triazoles in moderate to excellent yields.
Scheme 92.
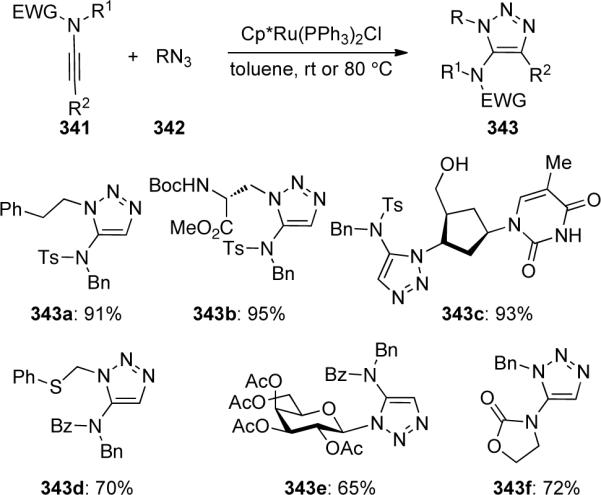
Lin, Jia and Fokin107 also reported a similar ruthenium-catalyzed [3 + 2] cycloaddition of ynamide 344 with azides 345 and 347 to efficiently provide 5-amido 1,2,3-triazoles 346 and 348, respectively (Scheme 93).
Scheme 93.
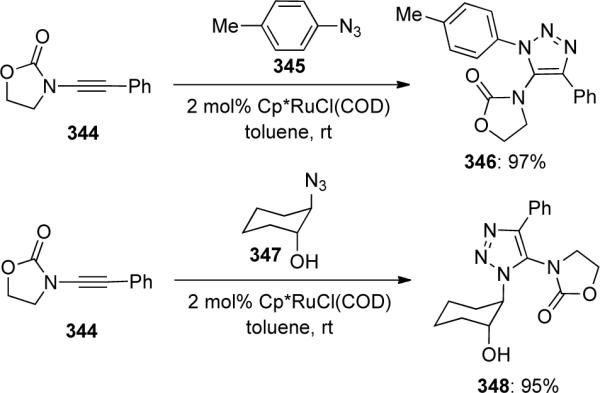
In 2007, Hsung108 disclosed a highly regioselective [3 + 2] cycloaddition between terminally-unsubstituted ynamides and nitrile oxides generated in situ from α-chloro oximes 350 to synthesize 5-amido-isoxazoles 351 (Scheme 94). Significantly, no 4-amido-isoxazoles were observed in these reactions, denoting the excellent regioselectivity present in these transformations.
Scheme 94.
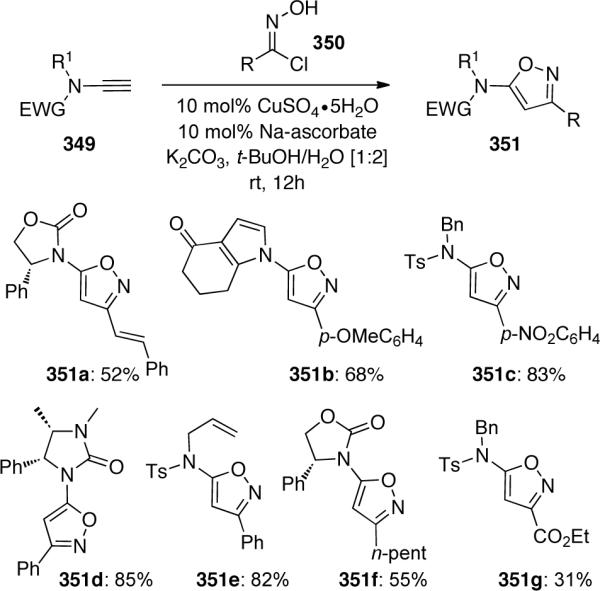
Also discussed was the preparation of 3-amido-pyrazole 353 from the [3 + 2] cycloaddition of ynamide 352 and ethyl diazoacetate to give intermediate 354 (Scheme 95). An ensuing 1,5-H shift results in preferential formation of 353.
Scheme 95.
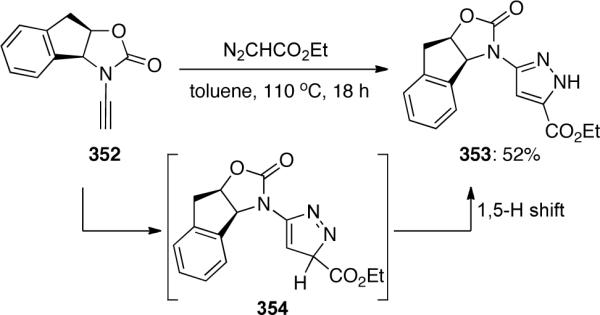
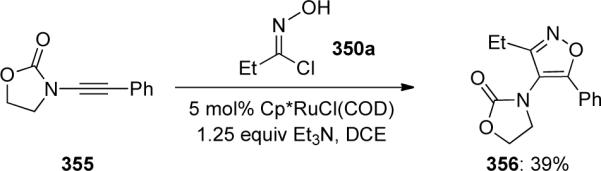
Fokin109 reported a complementary ruthenium-catalyzed [3 + 2] cycloaddition of ynamide 355 with oxime 350a providing isoxazole 356 as a single regioisomer in 39% yield. Notably, this methodology provided the opposite regioisomer to the copper-catalyzed cycloadditions discovered by Hsung (Scheme 96).
Scheme 96.
Hsung110 later demonstrated a highly diastereoselective Kinugasa nitrone-[3+2] cycloaddition involving ynamides to synthesize chiral α-amino-β-lactams 361 (Scheme 97). The mechanism involves an initial [3 + 2] cycloaddition of nitrone 358 with chiral ynamide 357 to give metallated-isoxazole 359, which may afford 361 through intramolecular trapping of ketene 360.
Scheme 97.

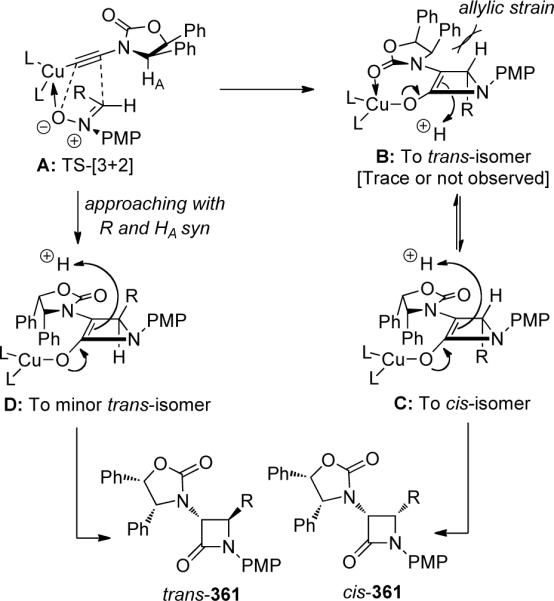
The authors attributed the excellent diastereoselectivity in this Cu(I)-promoted nitrone-[3 + 2] cycloaddition to a divergence in reactivity of intermediate A, which could follow two reaction pathways determining the β-carbon stereochemistry (Scheme 98). The preferred pathway would involve the approaching nitrone with its vinyl hydrogen being syn to HA on the chiral auxiliary and the larger R group anti to HA to minimize steric interactions. This pathway would lead to intermediate B and while B could undergo protonation at the more open bottom face away from the phenyl rings, it would lead to trans-361 that was not observed. Therefore, the authors reasoned that a facially selective protonation took place instead via intermediate C on the top face to give cis-361 because C is more stable than B in light of the allylic strain. On the other hand, the less favorable cycloaddition pathway would involve the larger R group approaching syn to HA, and should lead to the minor trans-361 isomer via related intermediate D.
Scheme 98.
3.2.4. [4 + 2] Cycloadditions
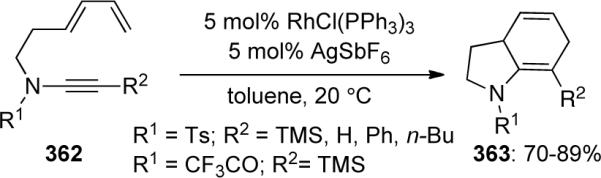
The first intramolecular [4 + 2] cycloaddition employing diene-ynamides 362 was reported by Witulski45b in 2003 (Scheme 99). Their protocol provided an efficient and versatile access to functionalized dihydroindolines 363 employing cationic rhodium(I) prepared in situ from RhCl(PPh3)3 and AgSbF6. It is noteworthy that the authors did not observe the typical Diels-Alder side products usually resulting from subsequent isomerization or aromatization under these mild conditions.
Scheme 99.
Hsung56b later expanded on this work with an intermolecular Diels-Alder cycloaddition between chiral ynamide 364 and several symmetrical dienes (Scheme 100). Anilide 365 (5:1 atropisomeric ratio) and bicyclic chiral enamides 366 were obtained in moderate yields. The intramolecular cycloaddition of ynamide 367 proved to be highly diastereoselective, affording dihydroindoline 368 as a single isomer. Hsung97 also showcased a hetero-Diels Alder cycloaddition of ynamide 369 with methyl vinyl ketone to give 370.
Scheme 100.

Danheiser55d described the first intramolecular thermal [4 + 2] cycloaddition giving highly substituted indolines 372 and 374 employing enyne-tethered ynamides 371 and yne-tethered ene-ynamides 373 (Scheme 101, BHT = butylated hydroxytoluene). Likewise, interesting acetylene tethered indolines 372e, 372f, and 374b could be prepared from the respective diyne-ynamides. The indoline product could be conveniently oxidized to the corresponding indole 375 employing o-chlorocil.
Scheme 101.

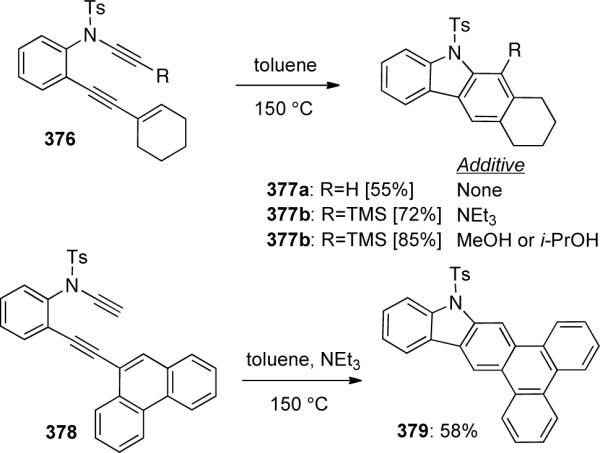
An intramolecular dehydro-Diels-Alder reaction of ynamides 376 and 378 was soon after reported by Saá111 for the synthesis of several carbazole skeletons (Scheme 102). Depending on the substrate, various additives including NEt3, MeOH, and i-PrOH were required for optimal yields.
Scheme 102.
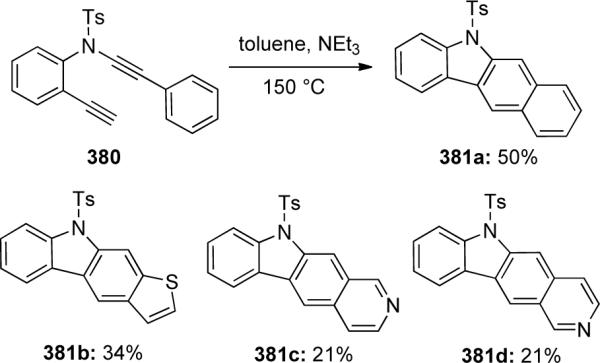
In 2008, it was discovered that by introducing various aromatic functionalities to the C-terminus of the yne-ynamides 380 instead of to the N-terminus, heteroarylcarbazoles 381 could be isolated, albeit in low yield using NEt3 as an additive (Scheme 103).
Scheme 103.
Movassaghi112 reported an acid-catalyzed aza-[4 + 2] cycloaddition of ynamide 382 with enamides 383 to provide a direct synthesis of pyridines 384 (Scheme 104). This methodology proved compatible with a variety of N-vinyl and N-aryl amides and π–nucleophiles. The authors proposed that the mechanism involves acidic activation of 383 to give bis-iminium 385, which is subsequently attacked by the ynamide to give keteniminium 386. The ensuing intramolecular aza-[4 + 2] cycloaddition would afford the observed pyridine derivatives.
Scheme 104.

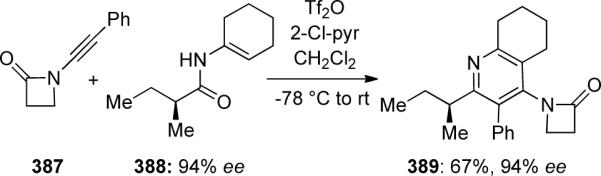
It is noteworthy that carrying out this reaction with enantiomerically enriched enamide 388 resulted in formation of the cycloadduct 389 with no loss of optical activity (Scheme 105).
Scheme 105.
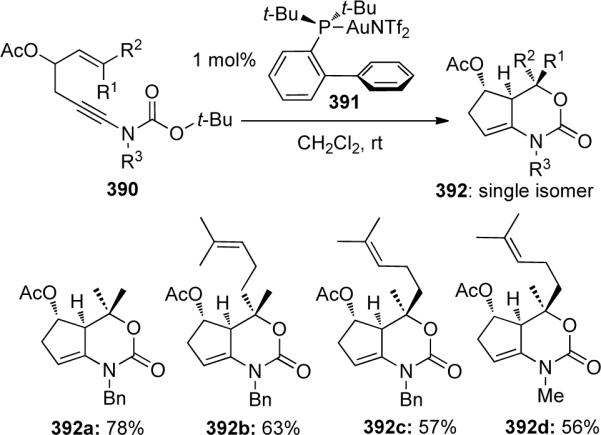
Gagosz113 reported a formal Au-catalyzed hetero-[4 + 2] cycloaddition of Boc-protected ynamides 390 bearing a tethered tri-substituted alkene to give enamides 392 all as single isomers (Scheme 106).
Scheme 106.
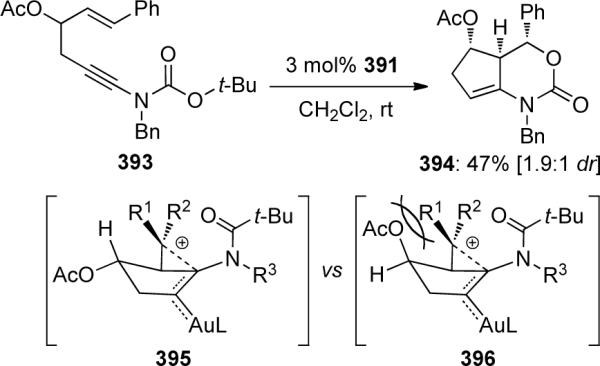
In the case of ynamide 393 containing a trans-disubstituted alkene, the diastereoselectivity was only moderate. The authors attributed this selectivity to a strong steric interaction between the acetoxy group and R1 in the intermediate 396 compared to the intermediate 395. The use of chiral N-auxiliaries was also explored, however no asymmetric induction was observed (Scheme 107).
Scheme 107.
3.2.5. [2 + 2 + 1] Cycloadditions
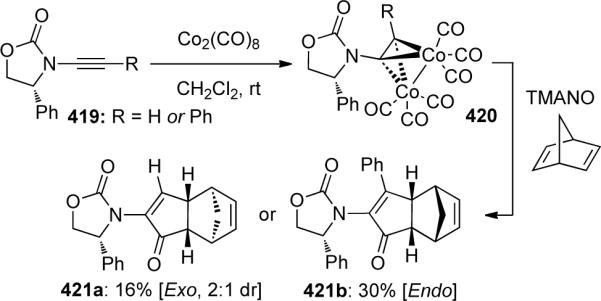
In 1998, Witulski44a,114 elegantly demonstrated that ynamides could serve as electron-rich alkynes in highly regioselective Pauson-Khand [2 + 2 + 1] cycloadditions with a variety of olefins (Scheme 108). Initially formed is an isolable dicobalt complex 398, which when treated with trimethylamine N-oxide (TMANO) in the presence of norbornadiene or methylenecyclopropane yielded α-amidocyclopentenones 399 as the single exo diastereomer or a 5:1 mixture of 400a to 400b, respectively. Several other olefins (not shown) also underwent desired cycloaddition to give cycloadducts 401–403 in moderate yields.
Scheme 108.
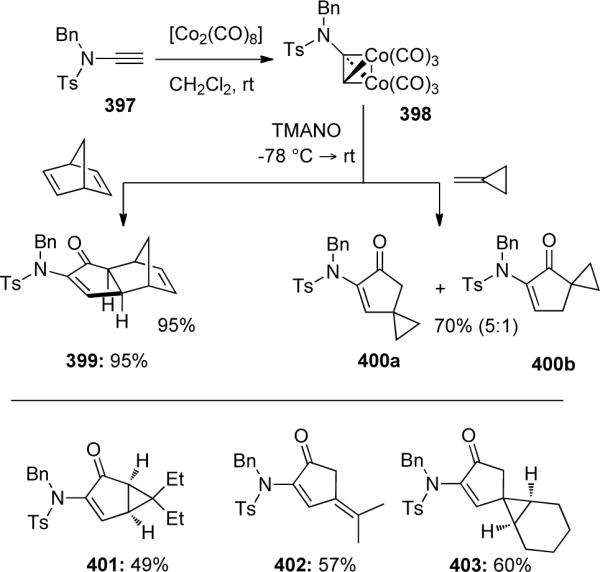
By incorporating an N-tethered olefin into ynamides 404, the authors were able to construct bicyclic vinylogous amides 405 in 45–60% yield (Scheme 109). It is noteworthy that both α and β-branched ynamides afforded the products as single diastereomers. The authors rationalized this exceptional diastereoselectivity by comparing reaction intermediates 406a and 406b, where 406b is less favored due to a pseudo-diaxial interaction between the R-group and a carbonyl fragment.
Scheme 109.
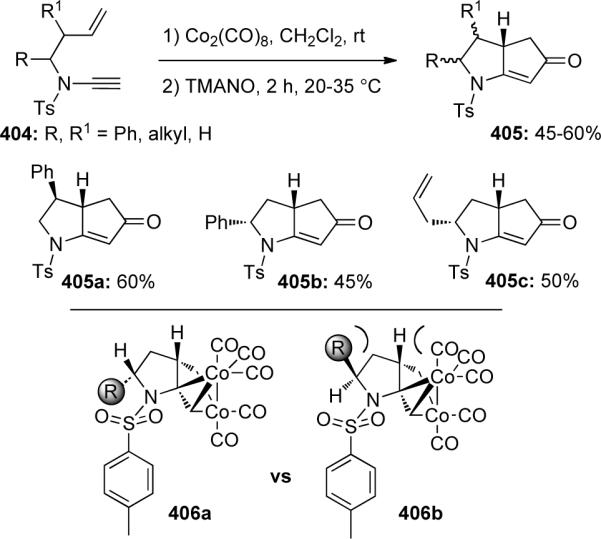
Shortly thereafter, Witulski49 demonstrated that the N-protecting group of ynamides 407 could be altered, still resulting in high levels of diastereoselectivity in 408 in the Pauson-Khand reaction (Scheme 110).
Scheme 110.
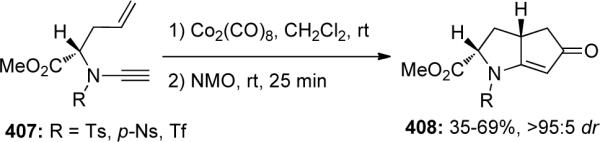
Rainer reported46a that when yne-ynamides 409 was subjected to Volhardt's conditions (CpCo(CO)2 in the presence of light), they undergo an intramolecular Pauson-Khand reaction giving cobalt bound cyclopentadienones 410 in 57% yield (Scheme 111). Oxidative cleavage of the cobalt led to in situ generation of cyclopentadienone 411 that was then trapped by either heterodienes or dienophiles via intermolecular Diels-Alder reactions yielding products such as 412 or 413 through loss of CO.
Scheme 111.
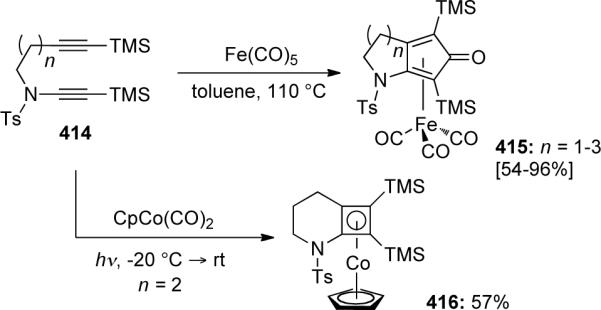
Rainer46b later discovered in 2000 that the CpCo(CO)2 catalyzed reaction of yne-ynamide 414 resulted in formation of cyclobutadiene complex 416. In contrast, Fe(CO)5 could be used to catalyze the Pauson-Khand cycloaddition of 414 to give 415 in moderate to excellent yields (Scheme 112). The use of Fe(CO)5 represented a novel catalytic system for these transformations.
Scheme 112.
In an attempt to carry out a Pauson-Khand cycloaddition with ynamide 417, Pérez-Castells115 was able to isolate the desired annulated indole 418 in low yield (Scheme 113).
Scheme 113.

Hsung116 expanded on this body of work by developing intermolecular [2 + 2 + 1] cycloadditions with chiral ynamides 419 (Scheme 114). Interestingly, the endo vs. exo selectivity in product 421 was affected by the alkyne substitution, with terminally substituted ynamides favoring unusual endo addition of norbornadiene. The mechanistic rationale for this unusual endo selectivity remains unclear.
Scheme 114.
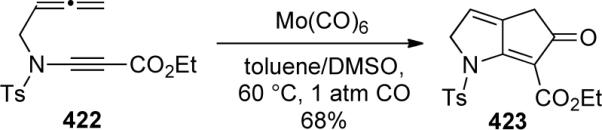
In 2005, Oh117 reported the use of Mo(CO)6 to catalyze the transformation of allene-ynamide 422 to 423 in 68% yield, as shown in Scheme 115.
Scheme 115.
3.2.6. [2 + 2 + 2] Cycloadditions
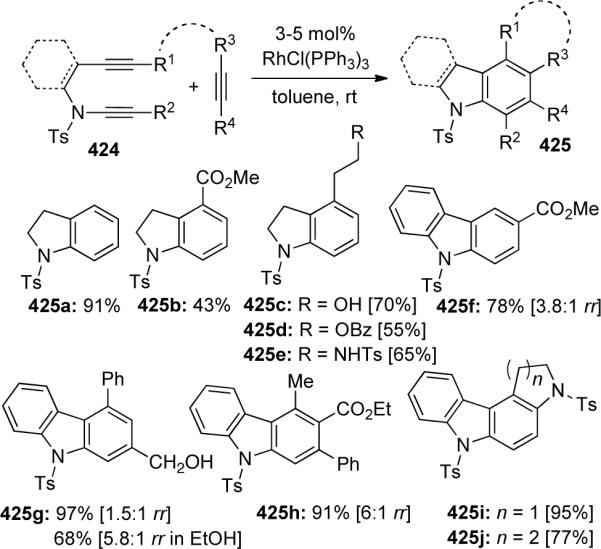
Witulski47, 118 was the first to demonstrate the power of the [2 + 2 + 2] cycloaddition with ynamides. The authors were able to rapidly construct indolines and carbazoles 425 through a Wilkinson's catalyst promoted cyclotrimerization of yne-ynamides 424 with a variety of alkynes (Scheme 116). The regioselectivity of the addition was highly dependent on both steric factors and solvent effects, as demonstrated by the increase in regioselectivity in some cases using ethanol instead of toluene (see 425g). Diyne-ynamides could similarly be used to construct annulated carbazoles in good to excellent yields.
Scheme 116.
It was later reported by Witulski119 that the regioselectivity of the cycloaddition could be completely reversed by using Grubbs' second-generation catalyst instead of Wilkinson's catalyst (Scheme 117). It was proposed that Grubbs' catalyst induces a cascade of metathesis steps beginning by addition of the electrophilic Ru-benzylidene to the electron rich ynamide motif, resulting in the change in regioselectivity.
Scheme 117.
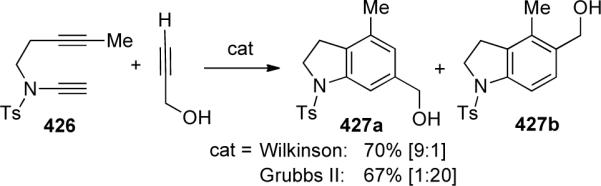
In 2006, Tanaka120a reported an asymmetric Rh-catalyzed [2 + 2 + 2] cycloaddition of diynes 428 with ynamide 429. As shown in Scheme 118, a variety of chiral ligands were screened, the best being (R)-xyl-BINAP which provided 430 with up to 98% ee. They were then able to construct a variety of highly functionalized axially-chiral anilides in good yields with good to excellent enantiomeric induction. The yields were heavily dependent on the ynamide's substitution, as terminal ynamides resulted in no reaction.
Scheme 118.

The authors proposed model TS-431 to rationalize the observed enantioselectivity, where the ynamide preferentially coordinates to the rhodium center so as to minimize the steric interaction between the benzyl group of 429 and the PAr2 of the (S)-xyl-BINAP.
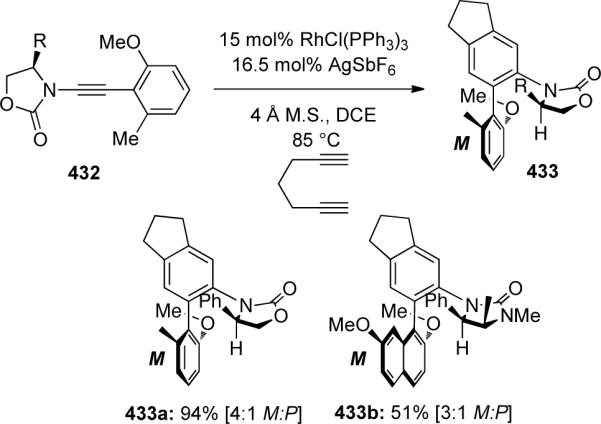
By introduction of a chiral moiety, Hsung121 was able to use ynamides 432 in a diastereoselective rhodium-catalyzed [2 + 2 + 2] cycloaddition for the synthesis of axially-chiral biaryls 433 (Scheme 119). Initial screening revealed a significant counterion effect, as AgSbF6 was essential for complete conversion, as was the addition of molecular sieves to prevent demethylation Vida supra. The best diastereoselectivity was observed using phenyl-substituted Evans' auxiliary, resulting in 433a with a moderate dr of 4:1 in favor of the M atropisomer. Biaryl 433b could be synthesized in a 3:1 dr using Close's auxiliary. It is noteworthy that there is no interconversion of the M and P atropisomers under the reaction conditions as the energy barrier to thermal equilibration was calculated to be 29.4 kcal mol−1 and no equilibration was observed even at 120 °C.
Scheme 119.
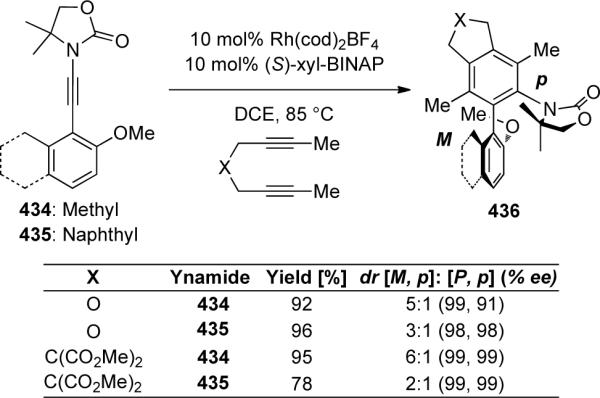
Using the rhodium-catalyzed protocol developed by Tanaka with terminally substituted ynamides 434 and 435 in the presence of (S)-xyl-BINAP, Hsung122 was able to construct axially-chiral N,O-biaryls 436 in up to a 6:1 diastereomeric ratio of M:p to P:p with both diastereomers having up to 99% ee (Scheme 120). The procedure was general for a variety of aryl-substituted ynamides as well as with diynes containing either a carbon tether or one containing a heteroatom.
Scheme 120.
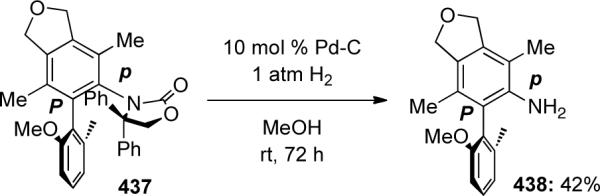
Removal of the achiral diphenyl-2-oxazolidinone auxiliary such as in 437 under mild hydrogenation conditions allowed access to interesting chiral N,O-biaryl compounds (Scheme 121).
Scheme 121.
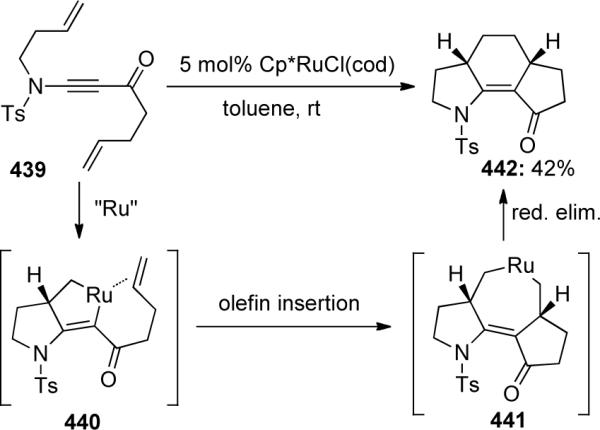
Sato and Mori123 were able to construct tricyclic vinylogous amide 442 via a [2 + 2 + 2] cyclization of diene-ynamide 439 under ruthenium catalyzed conditions (Scheme 122). Initial formation of ruthenacyclopentene 440 is followed by olefin insertion to give 441, which then undergoes reductive elimination yielding 442. The authors discovered in the case of non-ynamide substrates that the carbonyl moiety was not essential for reactivity, however, the carbon-tethering length was. This work provided rapid access to heterocyclic tricycles from linear substrates.
Scheme 122.
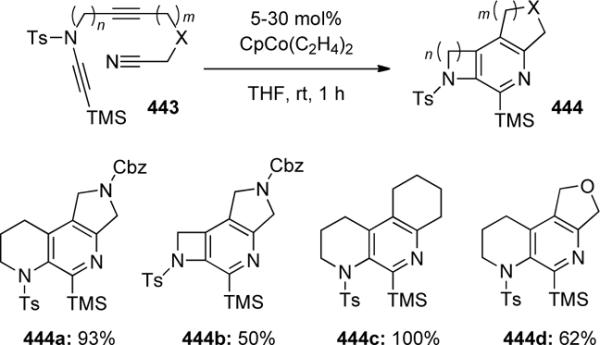
In work recently reported by Aubert and Malacria,124 yne-ynamide nitriles 443 were used for the construction of functionalized pyridines 444 by a cobalt-catalyzed intramolecular [2 + 2 + 2] cycloaddition. As shown in Scheme 123, a variety of tethering lengths with or without heteroatom substitution gave the desired product, often in high yield. Furthermore, the mandatory trimethylsilyl group provided a handle for functionalization via Hiyama cross-coupling, allowing access to substituted biaryl compounds.
Scheme 123.
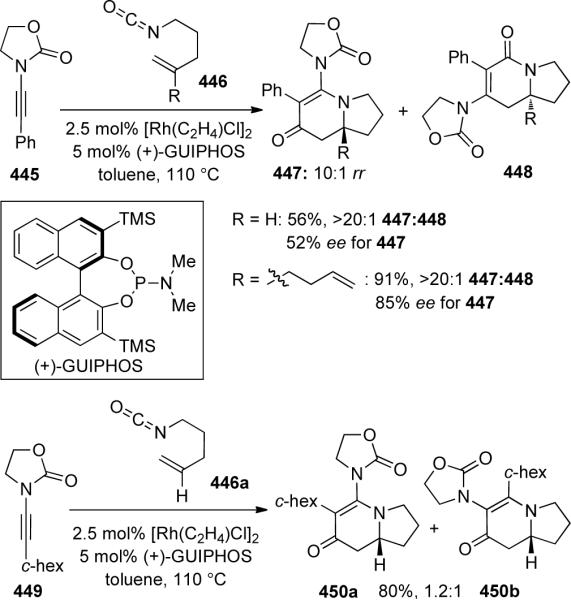
Rovis125 demonstrated a rhodium-catalyzed [2 + 2 + 2] of achiral ynamide with alkenyl isocyanates 446 to afford vinylogous amides 447 with moderate to good enantioselectivity using a chiral GUIPHOS ligand (Scheme 124). With phenyl-substituted ynamide 445, the ratio of 447 to 448, resulting from distinct reaction pathways (see Scheme 125), was better than 20: 1 with 427 being isolated as an inseparable 10:1 mixture of enamide regioisomers. However, with cyclohexenyl-substituted ynamide 449, a 1.2:1 mixture of regioisomers 450a:450b was obtained.
Scheme 124.
Scheme 125.

The authors proposed that the vinylogous amides and lactams observed originated from different reaction pathways as shown in Scheme 125. Product selectivity was determined in the initial oxidative cycloaddition which may lead to 452 or 456. Electronic effects were shown to be significant, as demonstrated by the difference in selectivity using phenyl or cyclohexenyl-terminated ynamides.
3.3. Cycloisomerization Reactions
3.3.1. Metal and Acid-Catalyzed Cyclizations
Over the past three decades, there have been significant advances in the area of transition metal catalyzed cycloisomerization reactions of ene-ynamides.
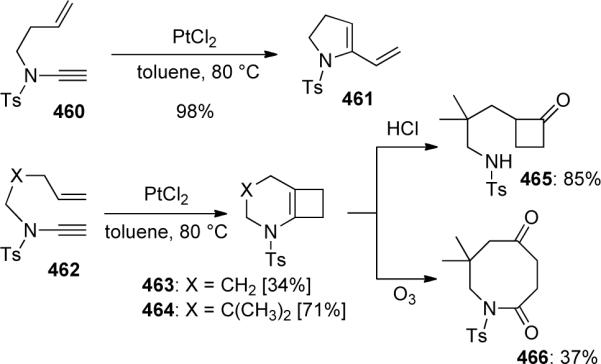
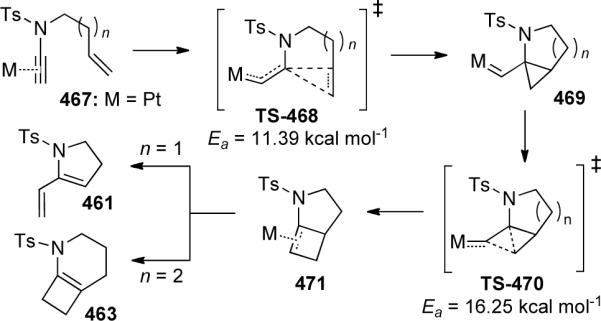
Ene-ynamides were introduced into this field in 2004 when Malacria and Fensterbank126 developed the first platinum-catalyzed ene-ynamide cycloisomerization leading to either aza-1,3-dienes 461 or aza-bicyclo[4.2.0] compounds 463 and 464, depending on the number tethering carbons (Scheme 126). Mori had previously observed 461 obtained through Ru-catalyzed RCM of ynamide 460 vide infra. As the bicyclic products were prone to decomposition, they were directly functionalized via a one-pot hydrolysis or ozonolysis to give 465 or 466.
Scheme 126.
Soriano and Marco-Contelles127 explored the mechanism of this transformation through DFT theoretical calculation (Scheme 127). They proposed initial complexation of the electrophilic metal to the nucleophilic ynamide followed by exo-attack of the tethered alkene to give cyclopropyl metallacarbene 469. A 1,2-alkyl shift into the carbene would form the key intermediate metallocyclobutene 471. Depending on the tethering length and ultimately the ring stain, either formal metathesis product 461 or bicyclo[4.2.0]octene 463 is preferentially formed.
Scheme 127.
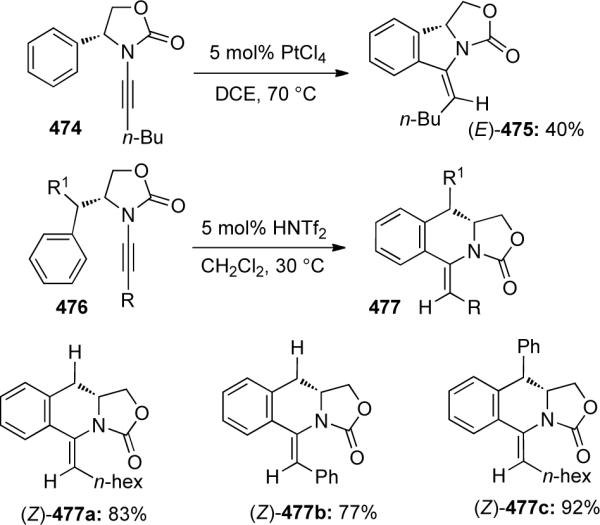
Hsung128 further demonstrated the power of ynamides as functional groups through the synthesis of chiral isoindole and tetrahydroisoquinoline derivatives via acid-catalyzed keteniminium Pictet-Spengler cyclization (Scheme 128). Interesting, the use of an (R)-phenyl oxazolidinone as a nucleophile only afforded isoindole 475 with an E-exocyclic olefin in moderate yield when using PtCl4. By adding a methylene unit such as in 476, HNTf2 could catalyze the reaction to give Z-olefin containing isoquinolines 477 in good yields with a variety of different functionalities.
Scheme 128.
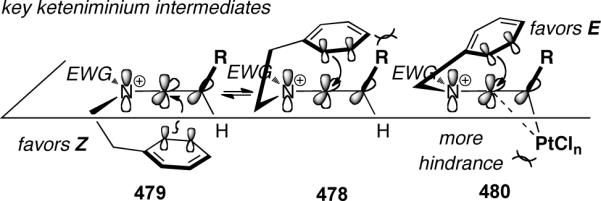
The divergence in selectivity could be rationalized by analyzing the conformations of the keteniminium intermediate in both the Brønsted-acid and Lewis-acid catalyzed cyclizations (Scheme 129). In the Brønsted-acid case, the Z-conformation 479 is preferred to minimize steric interaction between the aromatic nucleophile and R-group. Alternatively, the use of bulky PtCl4 favors the E-keteniminium 480, leading to the observed selectivity.
Scheme 129.
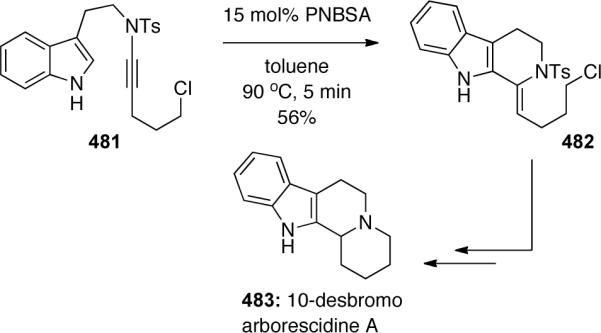
This methodology was then applied to the total syntheses of two arborescidine alkaloids. Shown in Scheme 130 is a facile total synthesis of 10-desbromo arborescidine A, beginning with the Brønsted-acid catalyzed keteniminium Pictet-Spengler cyclization of ynamide 481 to enamide 482. This and another related alkaloid synthesis is further discussed in the Applications to Natural Products section of this review.
Scheme 130.
A recent study by Popik129 showed that cyclic ynamide 484 containing enediyne moiety could undergo cycloaromatization more than two orders of magnitude faster than the analogous carbocyclic enediyne (Scheme 131). This finding supports the theoretical calculations that π-donors and σ-acceptors directly attached to the acetylenic termini of enediynes enhance the rate of Bergman cyclization. Surprisingly however, 489 was never isolated, implying that in this system, Bergman cyclization did not occur.
Scheme 131.
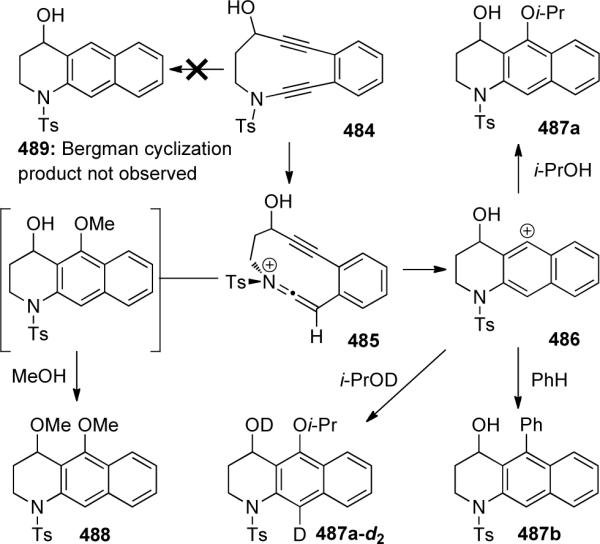
The rate of the cyclization was heavily dependent on solvent polarity, with the half-life of 484 being 105 min in hexane, 65 min in chloroform, and 20 min in 2-propanol. In addition, by running the reaction in benzene, the single major product was 487b via analogous Friedel-Crafts trapping of the intermediate carbocation. The cycloaromatization was two times slower in 2-propanol-d1 to give 487a-d2, supporting a kinetic isotope effect (kH/kD) in the normal direction, consistent with the proton transfer being at least partially rate limiting.
Collectively, these results indicate a polar pathway rather than diradical. The initially formed keteniminium 485 is trapped by the nearby alkyne and later by an external nucleophile such as an alcoholic solvent. When methanol was used, 488 was isolated presumably through displacement of the protonated hydroxyl group.
Gagosz130 attempted to cyclize ynamide 490 featuring a tethered carbonate under gold-catalyzed conditions (Scheme 132). They initially reported 494 as the cyclized product, proceeding through nucleophilic attack of the carbonate onto the metal-activated alkyne, followed by olefin migration, cyclization, and loss of isobutene. Upon further investigation,130b they recently rescinded their original structure in favor of α,β-unsaturated amide 497, resulting from 1,3-migration of the carbonate and fragmentation of the resulting allenamide 496.
Scheme 132.
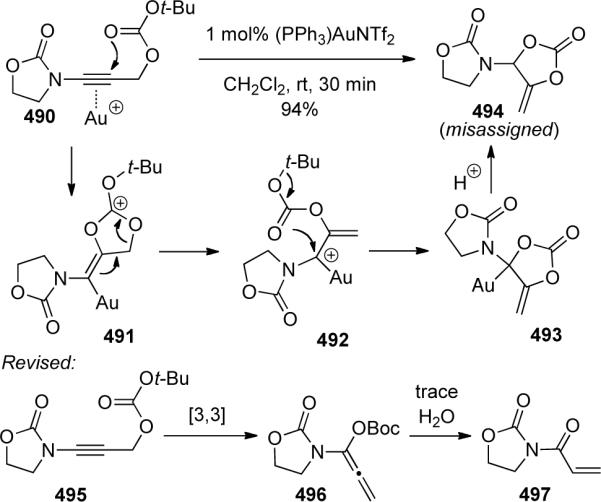
Starting from carbamate-protected ynamides 498, Hashmi131 was able to accomplish a similar gold-catalyzed transformation yielding a variety of oxazolidinones 501 (Scheme 133). Mechanistically, this involved 5-endo-dig cyclization of the Boc-protecting group onto the gold-activated alkyne, followed by loss of isobutene and protodemetallation. The only reported examples were with terminal or silylated ynamides, limiting the substrate scope.
Scheme 133.

Hashmi132 later expanded on this concept employing Echavarren's cationic [Ph3P-Au-(NCCH3)]+SbF6−catalyst (Scheme 133), improving the generality of the transformation and allowing the authors to isolate oxazolidinones 501e–i in good to excellent yields. It is noteworthy that the use of AgNTf2 could also catalyst this transformation in come cases, however the results were not general.
In a similar manner, Liu133 recently reported a silver-catalyzed cycloisomerization of epoxide-alkyne functionalities to quickly generate complex carbocyclic skeletons (Scheme 134). In one such example, treatment of ynamide 502 with AgBF4 triggered a 1,2-alkyl shift ring expansion through intermediate 503. Consequently, cyclization of 504 and elimination of silver from 505 gave 506, which could be subsequently reduced to access the core of gibberic acid.
Scheme 134.
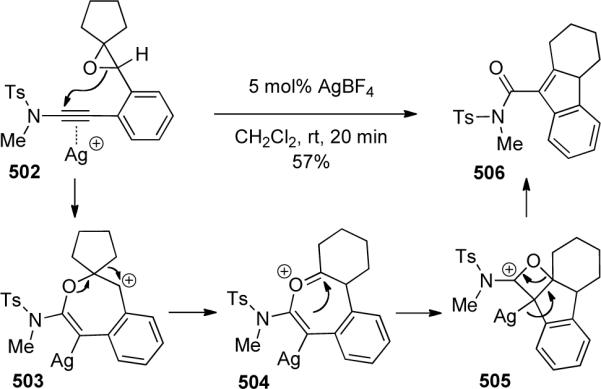
Hashmi134 found that AuCl3 could be used to catalyze the transformation of ynamido-furans 507 to functionalized indoline or tetrahydroquinoline derivatives 511, depending on the tether length (Scheme 135). The authors proposed the intermediacy of gold-stabilized carbocation 508, which may undergo ring open to give 509 primed for insertion into the nearby carbonyl to give epoxide 510. Subsequent tautomerization would result in the formation of 511.
Scheme 135.
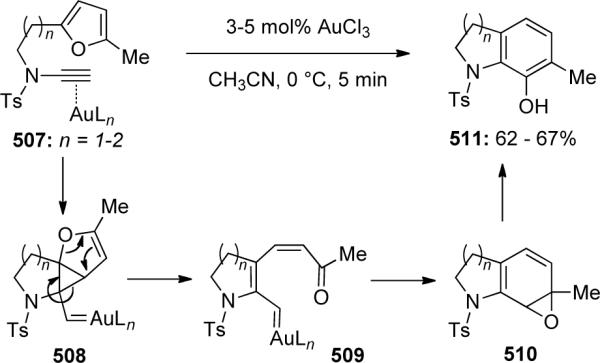
In 2008, Hsung and Zhao135 reported a sequential Pd-catalyzed C–N bond formation for the synthesis of 2-amido-indoles 515 (Scheme 136). The tandem sequence involves the amination of aryl halides to give intermediate 513 followed by metal-promoted 5-endo-dig cyclization. The generality of this reaction was demonstrated using a variety of primary amines, N-functionalized ynamides, and aryl halides.
Scheme 136.

Recently, Skrydstrup136 developed a very nice variation of this indole synthesis involving the Pd-catalyzed coupling of terminal ynamides 516 with various o-iodoaniline and phenol derivatives (Scheme 137). The authors suggested this reaction proceeds through Sonogashira coupling of the terminal ynamide with the aryl iodide, followed by a Pd-mediated hydroamination to yield product 517.
Scheme 137.
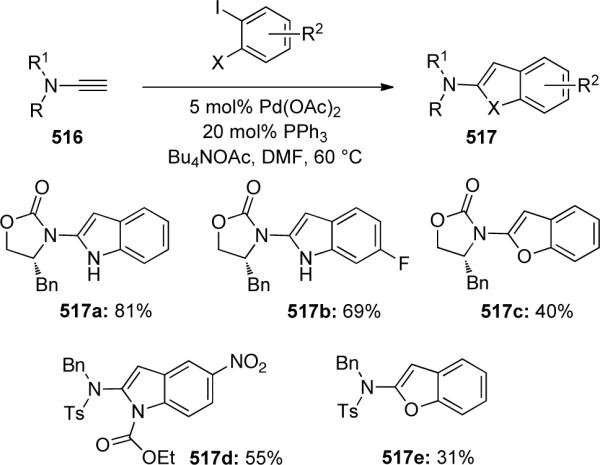
Cossy137 elegantly demonstrated the feasibility of constructing substituted cyclobutanones 519, amido-lactones 520 and 522, and aza-bicycles 524 from ene-ynamides via highly diastereoselective gold-catalyzed cycloisomerizations (Scheme 138). Due to the instability of the resultant amino-cyclobutanones 519, they were directly functionalized via Baeyer-Villiger oxidation to lactones 520 or 522. Ynamides containing both α and β-substituents were tolerated to give products in good yields and with high diastereoselectivity, as shown from 518 and 521. Interestingly, a tethered hydroxide functionality in 523 altered the reaction pathway leading to aza-bicycles 524 presumably through an oxygen-mediated hydride shift.
Scheme 138.
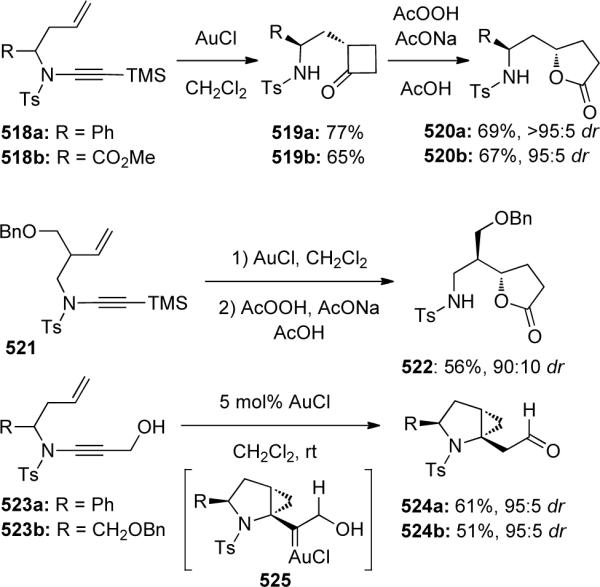
The authors were intrigued by the mechanism for this transformation and furthermore by the high levels of diastereoselectivity, which must originate during the formation of gold-stabilized carbocations 527 and 530, as shown in Scheme 139.
Scheme 139.
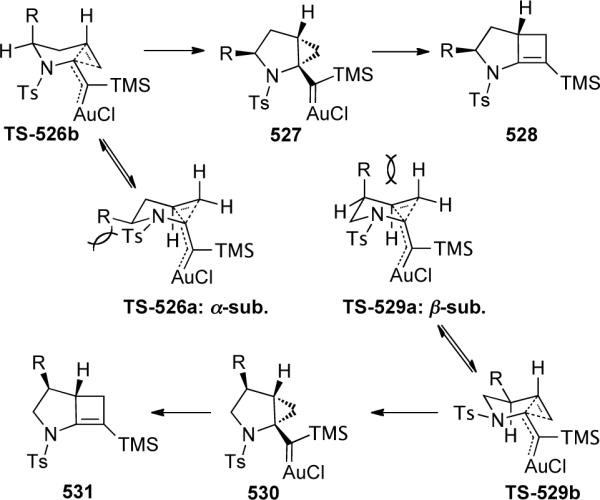
Conformational drawings of the two possible chair-like transition states leading to the formation of 527 from α-substituted ynamides reveal a distinct steric preference for TS-526b, avoiding the gauche interaction between the sulfonyl and α-substituent. This preference is further enhanced by nitrogen stabilization of the developing carbocation becoming similar to A1,2 strain. In a similar manner, TS-529b is favored for β-substituted ynamides avoiding 1,3-diaxial interactions. A subsequent 1,2-alkyl shift in both cases would form the respective cyclobutene, maintaining the high level of diastereoselectivity.
Further analysis137b via deuterium labeling supported the initial formation of silylated cyclobutene 533, followed by desilylation and hydrolysis to afford the isolated di-deuterated amino cyclobutanone 536 (Scheme 140).
Scheme 140.
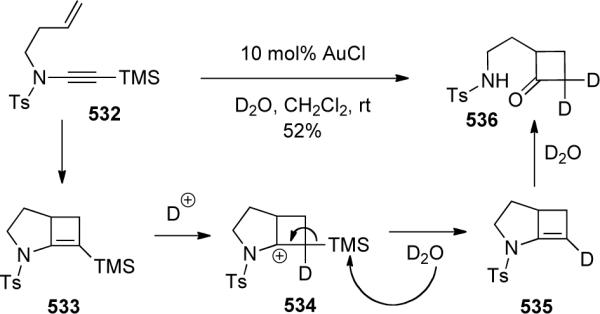
Cossy also reported the use of E and Z-substituted ene-ynamides 537 and 539 to give cis or trans-disubstituted lactones (Scheme 141). As expected from the diastereoselectivity analysis provided Vida supra, the relationship across the olefin was conserved in the formation of product.
Scheme 141.
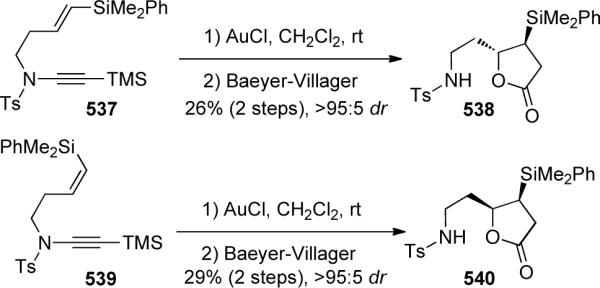
3.3.2. Pericyclic Rearrangements
In 2002, Hsung138 showcased the first Ficini-Claisen rearrangement with chiral ynamides, proceeding with high levels of diastereoselectivity and in good to excellent yields (Scheme 142). The use of p-nitrobenzenesulfonic acid (PNBSA) minimized hydrolysis of the ynamide, allowing for the optimal yields of 542 to be obtained. The highest level of diastereomeric control was achieved using a benzyl-substituted oxazolidinone auxiliary [up to 96:4 dr]. It is noteworthy that a variety of allyl alcohols could be used with good yields and dr favoring the syn isomers, many of which cannot be accessed through classical Evans' asymmetric alkylation.
Scheme 142.
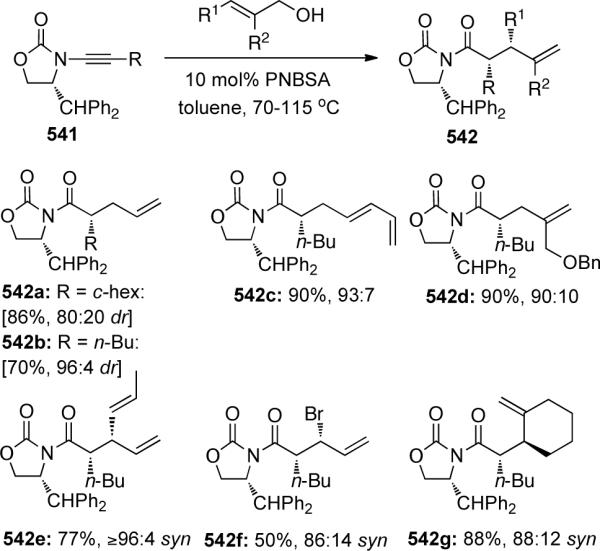
A plausible mechanistic model to explain the stereochemical outcome of this transformation is shown in Scheme 143. Protonation of 541 gives keteniminium 543, which would be attacked by the allyl alcohol over H rather than the R group. By comparison to Evan's model for asymmetric aldol chemistry, minimization of the dipole moments in ketene aminal 544 and positioning of the R1 group equatorial would favor Claisen-rearrangement from the less-hindered back face to give the observed syn diastereomer.
Scheme 143.
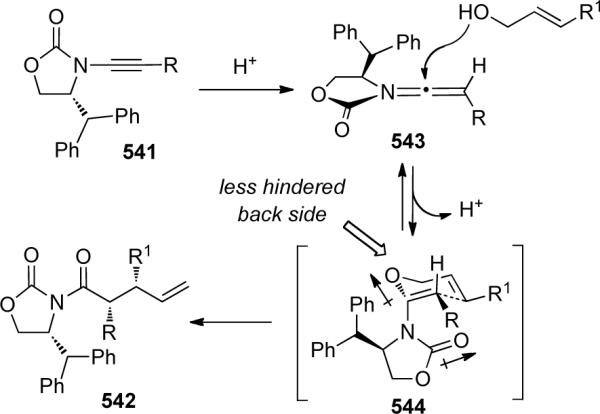
Cleavage of the chiral auxiliary and iodolactonization under standard conditions proceeded smoothly to give chiral lactones 546 in 3:1 dr favoring the R-conformation (Scheme 144).
Scheme 144.
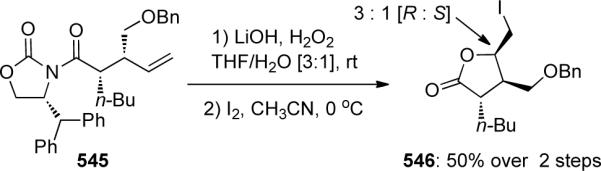
This research was later expanded upon with the first example of a Saucy-Marbet139 rearrangement of chiral ynamides with chiral propargyl alcohols140 (Scheme 145). After addition of the alcohol to give either 548 (matched) or 550a (mismatched), the resulting yne-enamides undergo a concerted [3,3]-sigmatropic rearrangement. In the case where the two chiral entities are sterically matched, the allenyl imides 549 are produced in good yields with excellent diastereoselectivities. For the mis-matched examples, the equilibrium between 550b and 550c governs the diastereomeric outcome, with a bulky substituent on the chiral alcohol favoring 550b by minimizing steric interactions between R1 and the chiral auxiliary.
Scheme 145.
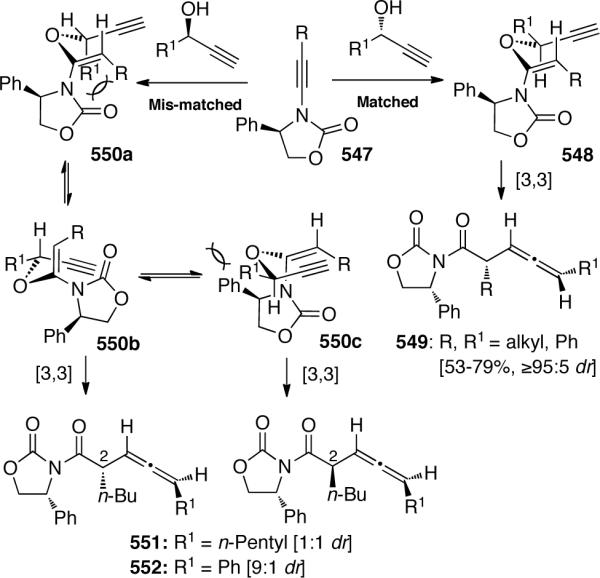
In order to determine the origin of the lower diastereoselectivity in the mis-matched examples (i.e. from either C2 or the axial chirality of the allene), the allene was hydrogenated in both 551 and 552, destroying any axial chirality141. As the resultant imides maintained the original diastereomeric ratio, it became clear that the lack of stereochemical control was centered at C2 and the chirality transfer from the chiral alcohol to the allene was in fact high (Scheme 146).
Scheme 146.
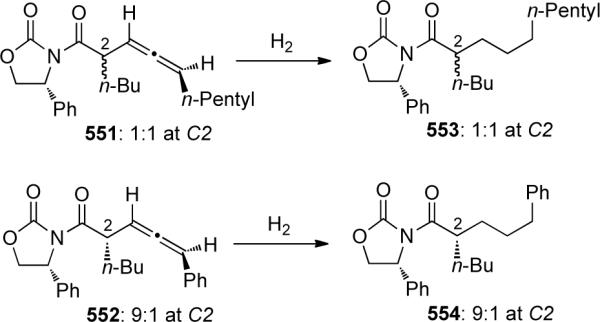
Hsung142 recently disclosed a thermal aza-Claisen rearrangement of silylated ynamides 555 to isolable ketenimines 556, which can be trapped in situ with nucleophilic amines (Scheme 147). In refluxing toluene, the reaction proceeded efficiently to give allylated amidines 557 in high yields as a complementary method to the palladium-catalyzed reaction (Section 3.7).
Scheme 147.
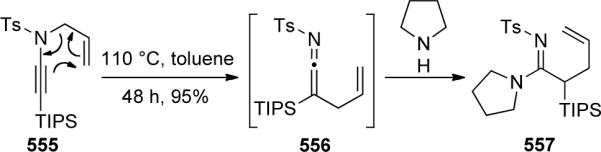
Another pericyclic rearrangement process was described by Oshima,83 it involves a metal-mediated aza-Claisen rearrangement of ynamide following the addition of alkyl or aryl Grignard reagents (Scheme 54 in Section 3.1.4).
3.3.3. Metal-Catalyzed Ring-Closing Metathesis
In 2002 Mori143 reported the first ring-closing metathesis (RCM) of ene-ynamides 562 using Grubbs' second-generation catalyst (Scheme 148). When n = 1, the reaction proceeded best in toluene at 80°C under an atmosphere of ethylene, however with a longer tether (n = 2), refluxing dichloromethane led to the highest yields. Intriguingly, the reaction proceeded in similar yield under an argon atmosphere for both substrates.
Scheme 148.
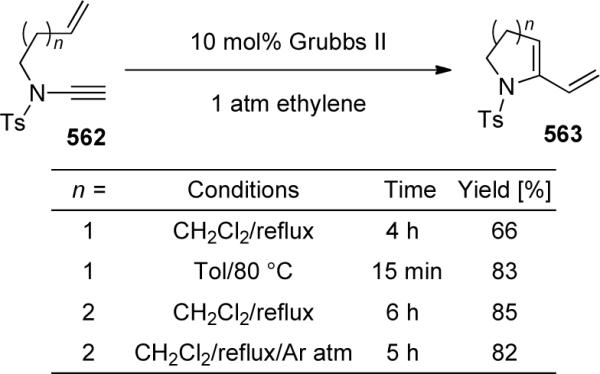
Mori144 later expanded on the scope of the reaction to include a variety of terminally-substituted ynamides 564. Shown in Scheme 149 are two representative examples. Both electron rich and electron poor substituents could be tolerated producing the respective aza-1,3-dienes 565 in good yields. An interesting reaction dichotomy was observed for (Z)-ene-ynamide 566. Under one atmosphere of argon, the expected metathesis product 567 was isolated in 41% yield as a 5.9:1 mixture of Z:E isomers. Alternatively, one atmosphere of ethylene gas provided aza-diene 568 in 66% yield as a result of ethylene exchange with the ruthenium alkylidene.
Scheme 149.
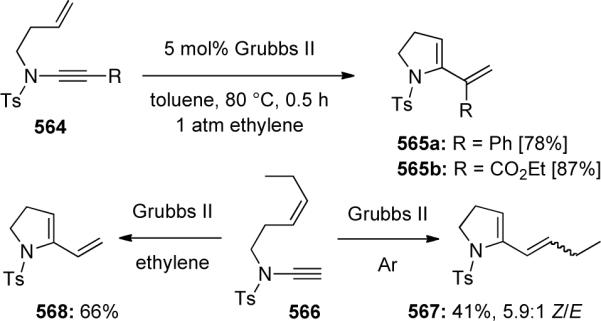
Mechanistically, the transformation occurs through a series of [2 + 2] and retro-[2 + 2] cycloadditions initiated at the alkyne to give carbene-complex 572 (Scheme 150). Intramolecular cycloaddition and cycloreversion yields the product 574 and regenerates the reactive ruthenium alkylidene to continue the catalytic cycle.
Scheme 150.
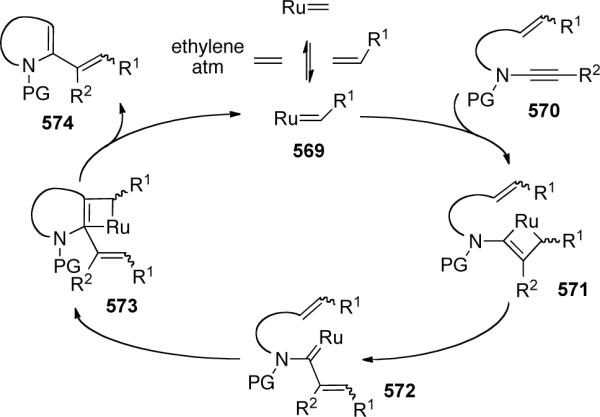
Soon after Mori reported their initial discovery of the ene-ynamide RCM, Hsung145 developed a modification by incorporating the tethered alkene into the electron-withdrawing N-substituent, allowing the use of a chiral auxiliary as demonstrated by the transformation of 575 to 576 (Scheme 151). This route provided an entry to chiral 2-amido dienes containing either a fused 6- or 7-membered lactam in good yields, which could be further functionalized through Diels-Alder chemistry to give 577 with moderate diastereoselectivity.
Scheme 151.

Expanding on this concept, Hsung was able to develop the first tandem RCM of diene-ynamides such as 578 and 581. The sequence progressed smoothly for 578 yielding a 6:1 mixture of 579:580, depending on which alkene was involved in the first RCM. By extending the tethering length for one of the alkenes, a mixture of mono-RCM product 582 and double-RCM products 583 and 584 was isolated. However, re-subjecting 582 to the reaction conditions provided the double-RCM products in good yield.
Pérez-Castells146 attempted to use the RCM of ene-ynamide 585 for the synthesis of natural product scaffolds, however they were only able to detect the desired vinyl indole 586 in the crude 1H NMR (Scheme 152). Attempts at isolation were unsuccessful.
Scheme 152.

While initially probing the reactivity of enynes in RCM, Mori found that enyne 587 yielded a mixture of the formal metathesis product and unprecedented diene 588 (Scheme 153). Six years later147, they revisited this issue and by exploring the use of other ruthenium catalysts, they were able to optimize the yields of 1-azadienes such as 593 from ene-ynamide 589. They proposed that the mechanism for these transformations occurs through ruthenacyclopentene 590 into which ethylene can undergo olefin insertion to give ruthenacycloheptene 591. β-Hydride elimination followed by reductive elimination would yield the observed 1-amidodienes 593.
Scheme 153.
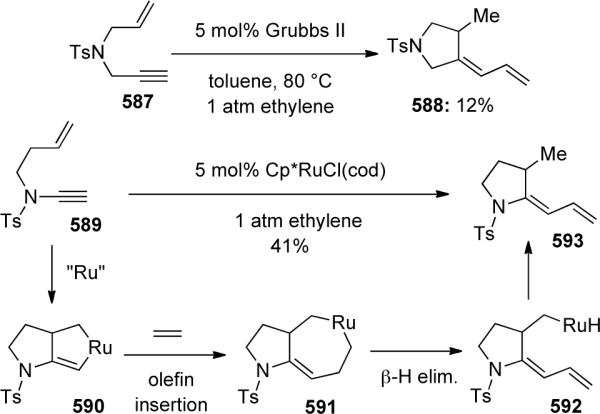
3.4. Metal-Catalyzed Coupling Reactions
One of the most concise methods for synthesizing complex ynamides and enamides has proven to be via metal-catalyzed coupling reactions. Yamamoto148 demonstrated the synthesis of polysubstituted anilines via Pd-catalyzed cross-benzannulations. The conjugated enyne 604 was prepared in moderate yield by dimerization of ynamide 603 (Scheme 154). Ene-ynamide 605 could similarly be subjected to the cross benzannulation reaction with conjugated diyne 606 to prepare the highly functionalized aniline 607 in 59% yield with high regioselectivity under relatively mild reaction conditions.
Scheme 154.
The homocoupling of ynamide 608 was first reported by Saá149, 150 in 2004. The dimerization could be catalyzed by CuI/TMEDA to afford diynes 609 in high yield (Scheme 155).
Scheme 155.
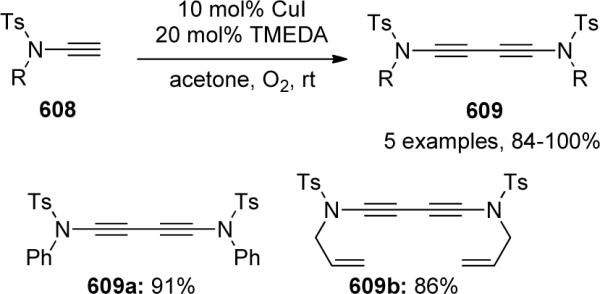
Later, Hsung151 succeeded in the first Sonogashira cross-coupling of ynamides with aryl and vinyl iodides under Pd-catalyzed conditions (Scheme 156). This methodology could tolerate a variety of heteroatoms including N, S, and O to give terminally-substituted ynamides 610. Notably, competing dimerization pathways (as shown in Scheme 155) were not observed. Hsung also applied this protocol towards the construction of extended conjugated phenylacetylenic systems. The first coupling reaction between ynamides 603 and aryl iodide 611 afforded TMS-capped acetylenes 612 in 44% yield. Subsequent basic desilylation of 612 and a second Sonogashira coupling of the resulting ynamide 613 with another aryl iodide 614 gave the conjugated yne-ynamide 615 in high yield.
Scheme 156.
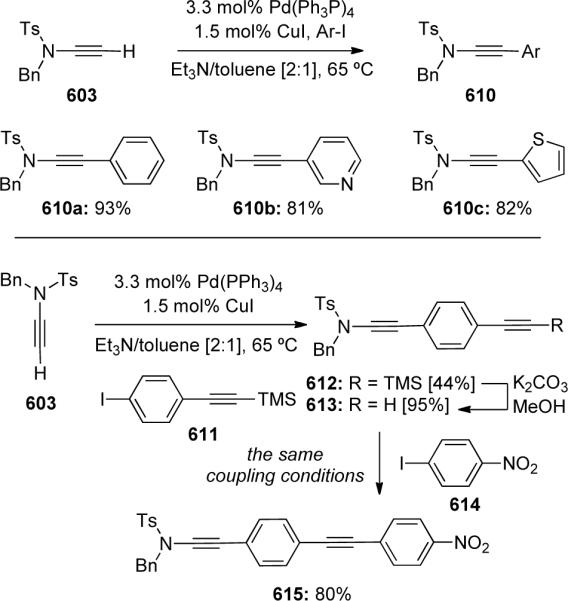
In 2004, Saá39 demonstrated a ZnBr-mediated Negishi coupling of ynamides to give similar arylated ynamides, which was discussed in Scheme 10.
Skrydstrup152 accomplished a very interesting intermolecular Mozoroki-Heck-type enyne coupling reaction between electron deficient olefin 617 and ynamide 616 to access synthetically rare push-pull 2-amido-diene 618 in moderate yield with high regioselectivity and E/Z control (Scheme 157). Mechanistically, the catalytic Pd(II) hydride species 619 was generated from iso-butyryl chloride likely by oxidative addition of Pd0 into the C–Cl bond, followed by decarbonylation and subsequent β-hydride elimination. The Pd-H complex underwent hydropalladation with alkynes providing alkenyl Pd(II) intermediates 620. Association of the alkene followed by olefin insertion and β-hydride elimination would afford 618 and regenerate the reactive Pd-H species.
Scheme 157.
A diversity-tolerant 2-amidoindole synthesis via Pd-catalyzed heteroannulation was reported by Witulski153. Their catalytic system allowed for the preparation of a wide variety of 2-amidoindoles (Scheme 158). The proposed mechanism involves in situ formation of Pd0, followed by oxidative addition into the C–X bond to give 626. Addition of amines to the activated ynamide 626 would generate the vinyl-Pd intermediate 627. Reductive elimination would afford product 625 and regenerate the active Pd0 catalyst.
Scheme 158.

Cossy154 reported a concise access to isoindolinones via a Pd-catalyzed Heck-Suzuki-Miyaura domino reaction of ynamides (Scheme 159). After ynamide 628 underwent cyclization, the resulting vinyl-Pd intermediate 631 could be transmetallated with various aryl boronic acids 629 to afford 3-(arylmethylene)isoindolinones 630 in good yield with high E-selectivity. In the natural product synthesis section of this review, the application of this transformation in total synthesis will be discussed.
Scheme 159.
In addition to a number of classic homo- and cross-coupling protocols shown in this section, Urabe and Sato155 discovered a unique coupling protocol via formation of acetylene- or ynamido-titanium complexes (Scheme 160). Treatment of alkyne 632 with Ti(Oi-Pr)4/2i-PrMgCl generated the Ti-cyclopropene intermediate 633 that further reacted with ynamide 634 providing the titanacyclopentadiene 635. Acid hydrolysis of 635 afforded dienamides 636 in excellent yields with high functional group tolerance. In several cases involving unsymmetrical alkynes, a mixture of regioisomeric dienes (shown with 636c) was observed.
Scheme 160.
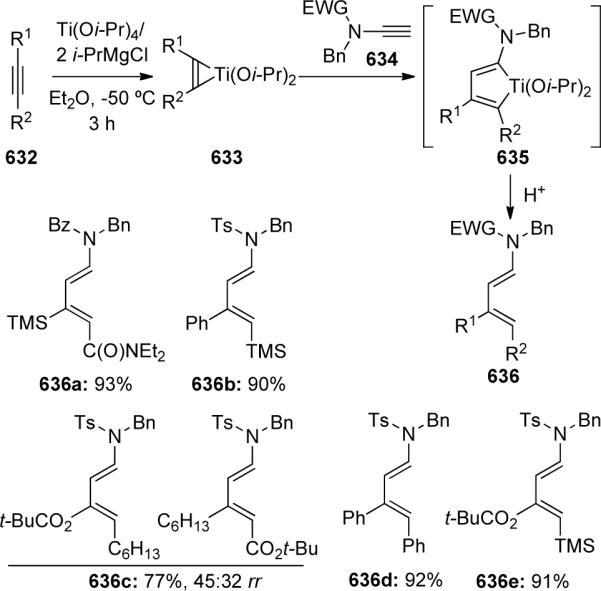
In a similar manner, N-substituted Ti-cyclopropene intermediates 638 could be formed from ynamides 63757b,156. This allowed the formation of oxatitanacycles 639 via coupling reactions with a variety of aldehydes. The remaining vinyl-titanium bond in 639 was confirmed by deuteriolysis (97% D in 640a). Hydrolysis of 639 gave a wide range of amido-substituted allylic alcohols 640 with good diastereoselectivity via notable 1,5-remote asymmetric induction (Scheme 161).
Scheme 161.
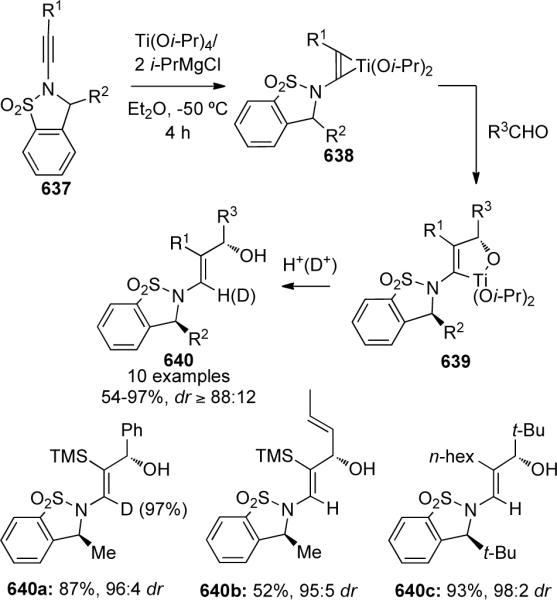
Titanacyclopentadienes 643 were also capable of reacting with nitriles 644 to generate titanacycles 645 or 646157 (Scheme 162). Depending on the type of N-protecting group, two reaction pathways were observed. From tosyl-protected ynamides, formation of 646 was followed by rearrangement to metallated pyridine 647, which was confirmed by deuteriolysis (path a).
Scheme 162.
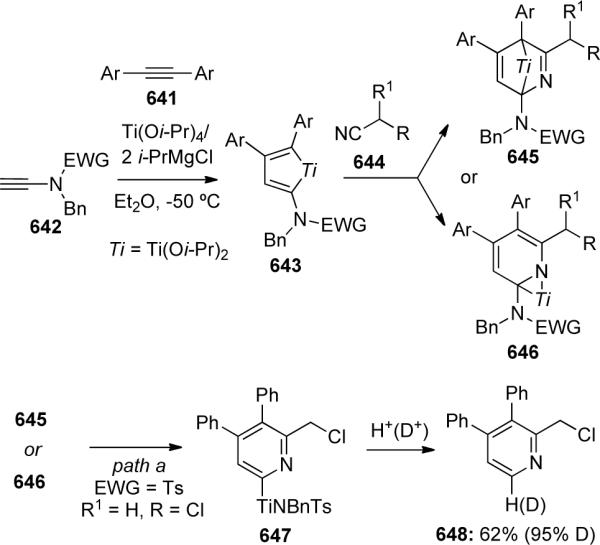
Using mesyl-protected ynamides, formation of 646 was followed by elimination of the mesyl group to provide 1-amino-pyridines 647 in high yields (path b) (Scheme 163).
Scheme 163.
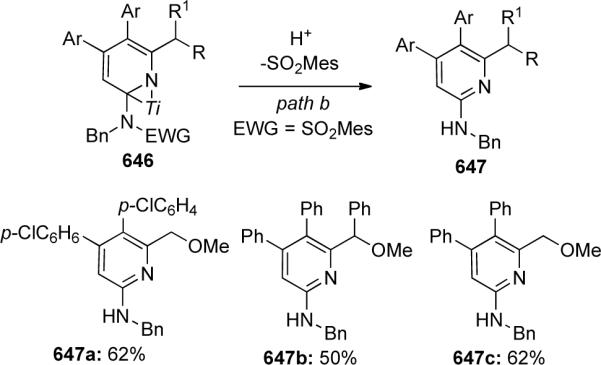
Recently, Sato158 reported a Ni-NHC-catalyzed multi-component coupling of ynamides, aldehydes and silanes to access functionalized enamides (Scheme 164). Reactions between ynamides 648 and various aldehydes resulted in regioselective formation of oxanickelacycle intermediate 650, and then hydride reduction of the metallacycle by Et3SiH gave the desired enamides 651 in high yield with a high degree of E/Z control.
Scheme 164.
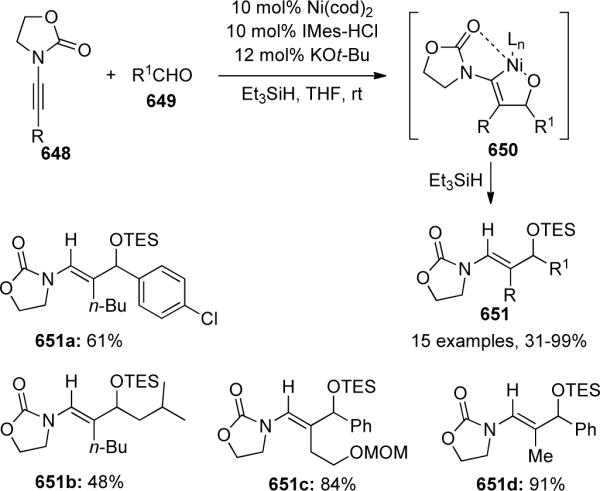
3.5. Reductions of Ynamides
Hsung56b accomplished the first stereoselective reduction of ynamides. A variety of Z-enamides 660 could be prepared by Lindlar hydrogenation of ynamides 659 in high yields or E-enamide 662 could be accessed by DIBAL-H reduction at −78 °C in moderate yield (Scheme 165).
Scheme 165.
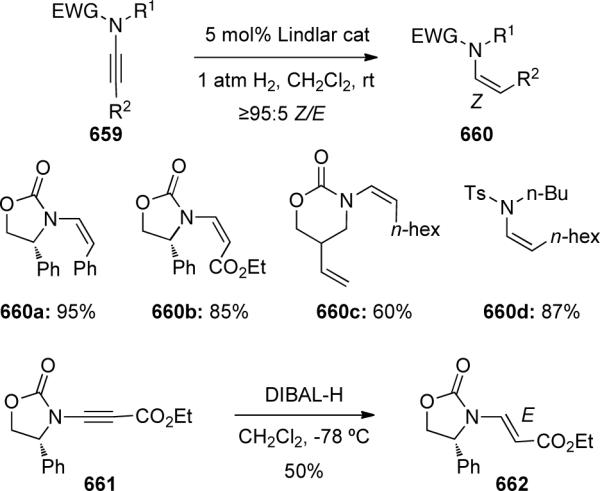
Alternatively, Cossy and Meyer159,160 prepared Z-enamides Z-664 via Ti(II)-mediated reduction of ynamides 663 or E-enamides E-664 via Red-Al-mediated reduction (Scheme 166).
Scheme 166.
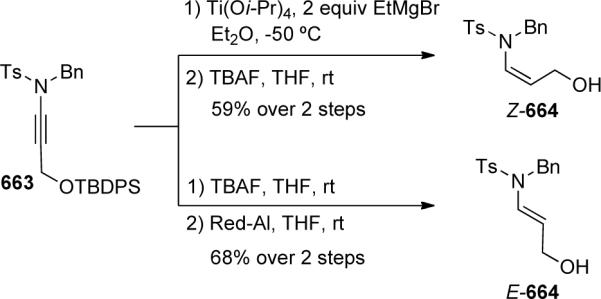
3.6. Reactions Involving Ynamido-Metal Intermediates
While there have been many recent and notable improvements in ynamide chemistry, perhaps one of the newest and most exciting areas in this field involves the generation and reactivity of ynamido-metal intermediates.
Hsung and Zhang142 recently reported a possible metallo-ynamido complex in their work on the synthesis of amidines 668 from N-allyl ynamides 665 (Scheme 167). Pd(0) was found to catalyze an N-to-C allyl transfer presumably through oxidative addition into the C—N bond to form an ynamido-Pd-π-allyl complex in equilibrium with the ketenimine-Pd complex 666. Nucleophilic addition of various amines into the reactive ketenimine complex 666 followed by reductive elimination of 667 provides the allyl-transferred amidine products 668. Within their work, they found that use of the Xantphos ligand was necessary to accelerate reductive elimination preventing de-allylation of the Pd-complex, especially when using more nucleophilic secondary amines.
Scheme 167.

It was found that a variety of amidines could be prepared using primary or secondary amines and various terminally-substituted N-sulfonyl ynamides.
Other metals have also been reported to form ynamido-metal intermediates as described in previous sections. In particular, ynamido-copper intermediates generated by a formal [3 + 2] cycloaddition have been recognized as a very useful synthon in multi-component reactions.

Chang161 reported the synthesis of various N-sulfonyl amides and amidines via copper(I)-catalyzed “click” chemistry, both of which could be obtained through a common ynamido-copper intermediate 670 (Scheme 168). The reaction of various terminal alkynes with alkyl- or arylsulfonyl azides in the presence of Cu(I) salts led to amide products 672 when using water as the nucleophile or amidines 673 with the use of amines. After initial formation of triazole 669, elimination of N2 would give the ynamido-copper complex 670, which could be protonated to give a reactive ketenimine 671. Addition of the nucleophile to the ketenimine would then lead to the desired product. Various amine nucleophiles could be tolerated as well as a variety of other functional groups such as alkynes and azides.
Scheme 168.

Fokin162,163 later reported a similar procedure for the formation of amide products, in which the use of tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine was found to accelerate the reaction and sodium ascorbate was used to prevent the formation of oxidation byproducts.
Both Fokin164 and Xu165 also cleverly applied the use of these ynamido-copper intermediates in cycloaddition chemistry (Scheme 169). Fokin showed that ketenimine intermediates 675 underwent [2 + 2] cycloaddition reactions with imines 677 resulting in azetidinimine products 678. In most cases, the trans stereochemistry was observed in the product. However, the cis-product could be obtained predominately with electron-deficient imines. Similarly, this key intermediate 675 was shown by Xu165 to undergo [2 + 2] cycloadditions with carbodiimides 676 to synthesize a variety of 2,4-diiminoazetidines 679.
Scheme 169.

Additionally, Chang166 reported am efficient three-component reaction of terminal alkynes, sulfonylazides, and alcohols for a facile access to N-sulfonylimidates 680. This reaction proceeded through a similar ynamido-copper intermediate generated through formal [3 + 2] cycloaddition (Scheme 170).
Scheme 170.
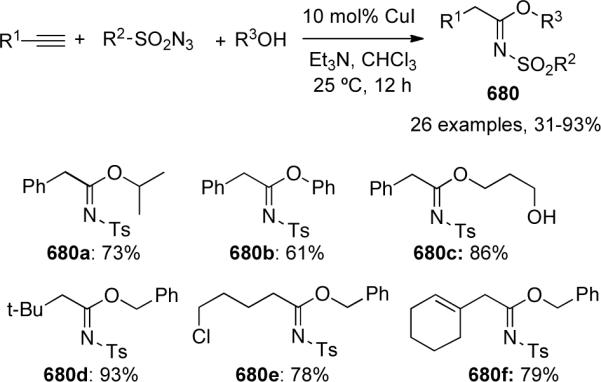
Wang167 has beautifully demonstrated a variety of multi-component annulation reactions via equilibration of ynamido-copper intermediate 681 and ketenimine intermediate 682 which were again generated through a formal [3 + 2] cycloaddition (Scheme 171). In the presence of an amine base, addition of salicylaldehydes 683 provided the intermediate 684. Subsequent cyclization followed by elimination of water efficiently afforded a wide range of coumarins 685. Notably, this protocol can be applied to ketones to access even further functionalized coumarins. Similarly, aziridines 686 could undergo addition reactions with 682 to give 687, which could then cyclize and dehydrate to afford 1-aza-bicyclo[3.1.0]-hexenes 688. The subsequent ring-opening of 688 would afford pyrrolines 689. Finally, a formal [2 + 2] cycloaddition of iminophosphoranes 690 and ketenimines 682 was shown to form amidine ylides 692 through ring-opening of the intermediate 1,2-phosphazetidine 691.
Scheme 171.

Also, an intramolecular variant was reported by Fu168 for the formation of medium- and large-sized heterocycles 698 using “click” chemistry. They proposed the intramolecular addition of nitrogen or oxygen nucleophiles to the ketenimine-copper intermediate 697 (Scheme 172). After elimination of nitrogen gas from triazole 695, it is possible for the ynamido-copper complex 696 to exist in equilibrium with a ketenimine-copper complex 697 rather than being protonated. Trapping of the ketenimine-copper complex by the nucleophile followed by elimination of copper would then give the heterocyclic product. Various-sized macrocycles 698 could be obtained in good yields from the ortho-diamides 694.
Scheme 172.
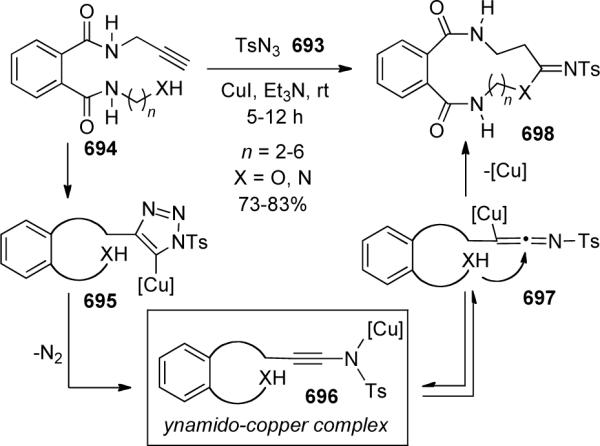
3.7. Other Reactions
Chen65 discovered in 1998 that isocyanate containing ynamides 699 could be protected as dicobalt complexes 700 (Scheme 173), allowing further functionalization of the isocyanate moiety. Introduction of an alcoholic nucleophile proceeded in moderate to good yields to give carbamate complexes 701. The dicobalt protecting group could then be oxidatively cleaved providing access to functionalized malaeic anhydride derivatives 702.
Scheme 173.
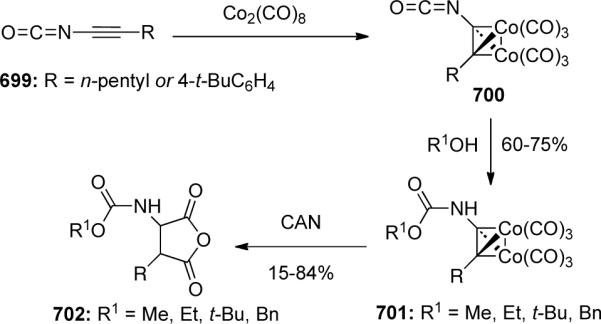
In 2005, Wudl169 published an interesting example of a double-barreled migration of both the tosyl and p-methoxybenzyl groups of ynamide 703 to nitrile 705, as shown in Scheme 174. The authors proposed that the tosyl transfer should be more facile, giving ketenimine 704 as a reactive intermediate.
Scheme 174.
Another example of an ynamide rearrangement was reported by Akai170, involving the transformation from ynamido-alcohol 706 to α,β-unsaturated amide 707 using a Mo, Au, and Ag tri-catalytic system (Scheme 175).
Scheme 175.
4. Ynamides in Total Synthesis of Natural Products
There have been few reports of natural product syntheses employing ynamides during a key step. Hsung128 was the first to utilize ynamides in the total syntheses of 10-desbromoarborescidine A and 11-desbromoarborescidine C (Scheme 176). Indole-tethered ynamides 708 were subjected to Brønsted acid-catalyzed Pictet-Spengler cyclization via the keteniminium intermediate 709 to construct the tricyclic core of the desbromoarborescidines 710 and 711. From 710, reduction of the enamide followed by intramolecular N-alkylation provided 10-desbromoarborescidine A and 11-Desbromoarborescidine C was reached in 7 more steps from 711.
Scheme 176.
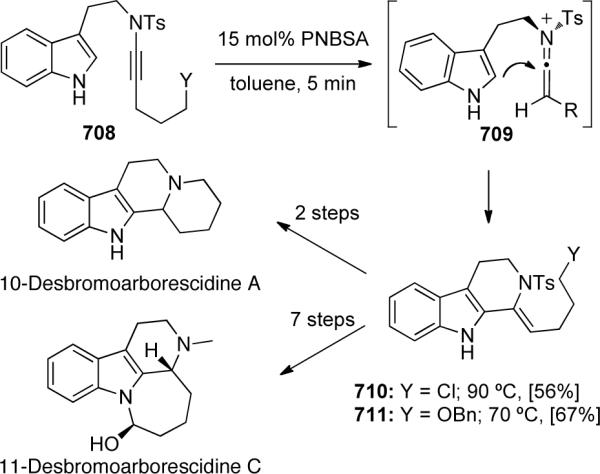
Cossy and Meyer171 accomplished a total synthesis of lennoxamine (Scheme 177) via tandem intramolecular Heck-type cyclizations of ynamides 712 to generate the vinyl Pd(II) intermediate 714, which underwent transmetallation with aryl boronic acid 713 affording the desired product 715 in high yield with moderate E/Z control. Hydrogenation of the resulting enamide 715 followed by construction of the seven-membered ring via a ring-closing Friedel-Crafts reaction and a second enamide reduction completed the total synthesis of lennoxamine.
Scheme 177.
Most recently, Witulski172 completed an elegant total synthesis of antiostatin A1 (Scheme 178). As a key step, a chemo- and regioselective Rh-catalyzed alkyne cyclotrimerization reaction employing ynamide 717 afforded 719 in high yield through trapping of the rhodiacyclopentadiene intermediate 718 with an ynol ether. From the cyclotrimerization product 719, the total synthesis of antiostatin A1 720 was accomplished in four steps.
Scheme 178.
5. Most recent developments
After the manuscript of this review was submitted, Hsung disclosed an extension of his Pd-catalyzed N-allyl ynamide rearrangement for the synthesis of amidines (Scheme 179). They discovered that under Pd-catalyzed conditions without an amine nucleophile present, the intermediate ketenimine 722a could be isolated in near quantitative yield even after silica gel chromatography. Upon treatment with cyclohexylamine, amidine 724 was isolated, supporting 722a as a reactive intermediate. Furthermore, when alkyl and aryl-terminated ynamides were employed in a thermal aza-Claisen variation of this rearrangement, an unusual 1,3-sulfonyl transfer occurred to give nitriles 723 bearing an adjacent quaternary center. In the case of 723a, the nitrile was formed presumably after hydrolysis of the silicon group in 722a.
Scheme 179.
With the isolation of the ketenimine intermediate, it became possible to probe the mechanism of the Pd-catalyzed allyl transfer, namely whether the transfer occurred intra- or intermolecularly (Scheme 180). A cross-over experiment employing a 1:1 mixture of N-crotyl and N-allyl ynamides 725 and 726 resulted in a 10:1 product distribution disfavoring any cross-over. This result supports an intramolecular transfer through a tightly-coordinated ynamido-Pd-π-allyl intermediate rather than via a dissociative Tsuji-Trost type mechanism.
Scheme 180.
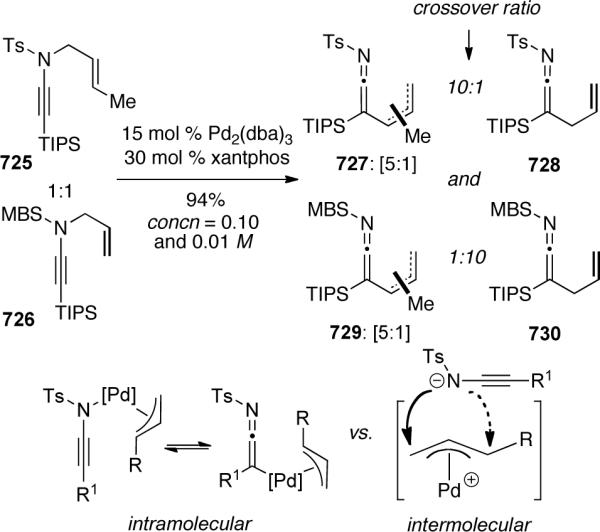
The authors also demonstrated an aza-Rautenstrauch-type cyclization of Pd-ketenimine 731 to give silylated cyclopentenimine 733, presumably through Pd-carbenoid 732 (Scheme 181). Interestingly, subjection of 722a to either Pd-catalyzed or thermal conditions resulted in no formation of 733, implying that the cyclization must occur before reductive elimination and that an imino-Nazarov type cyclization is not in operation.
Scheme 181.
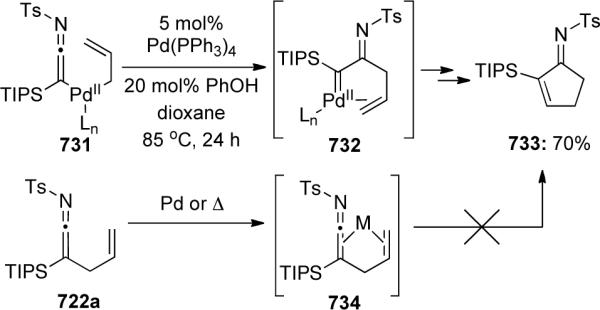
A nickel-catalyzed [3+2+2] cycloaddition of ethyl cyclopropylideneaceate 735 and ynamides 736 was described by Saito (Scheme 182). The reaction procedure involves slow addition of 735 and 736 over several hours to the mixture of catalyst and ligand. It was found that the cocyclization product 737 could be isolated in 36–50% yield, however significant amount of the trisubsituted benzene 738 were also observed. The authors also found that ynamines bearing bulky N-substituents were more efficient substrates.
Scheme 182.
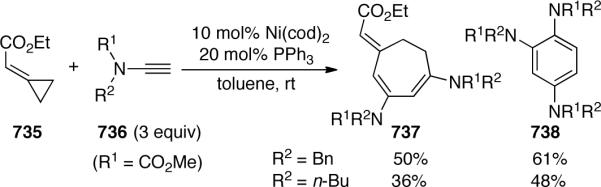
One of the most recent contributions to ynamide chemistry was disclosed by Burley and Davies, who reported a facile and efficient Cu(I)-catalyzed N-alkynylation of imidazoles, benzimidazoles, indazoles and pyrazoles using poly (ethylene glycol))400 (PEG400) as solvent medium (Scheme 183). It was suggested that PEG can act as a efficient phase transfer solvent by providing a bridge between substrates and inorganic base. Meanwhile, PEG may also act as ligand for copper catalyst as it was found that common Cu(I)-stablizing ligands were not necessary for the success of reaction. Another improvement of this process involves the use of microwave irradiation, which significantly reduced the reaction time from 24 hours to 30 minutes. The major side-product isolated from crude reaction mixture were bormoalkenes 740 and 744, and their formation increase as the steric bulk of the heteroarene increases.
Scheme 183.
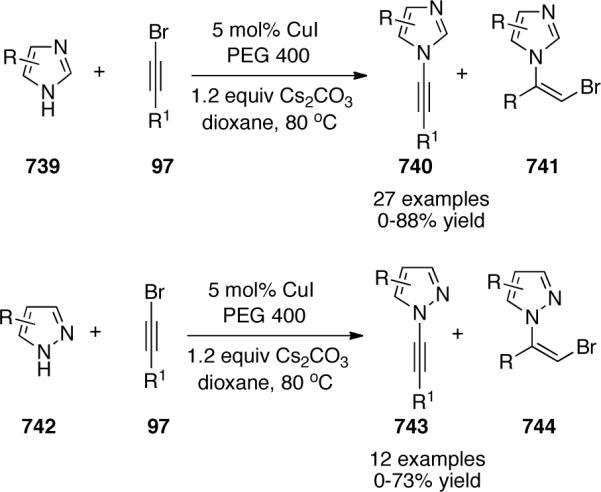
6. Conclusions
The field of ynamide chemistry has experienced rapid expansion in the last decade, fueled by the development of efficient means of preparation. Beautiful work has been accomplished showcasing the unique reactivity of ynamides in a plethora of reactions that have delivered a diverse array of novel carbo- and heterocyclic structures representing prevalent and important pharmacophores in addition to being useful platforms for further transformations. To date, ynamides have yet to be extensively employed in total synthesis, however with all of the recent methodological advancements, we are very interested to see the work that is surely on the horizon.
Perhaps the most exciting area of current ynamide chemistry is the discovery and exploitation of ynamido-metal intermediates. These methods provide unique modes of reactivity and are sure to play a vital role in establishing ynamides as a powerful synthon for the new millennium.
Biographies

Kyle A. DeKorver graduated from Grand Valley State University in Allendale, MI with his B.S. in Chemistry in 2007. While there, he carried out undergraduate research with Professor Felix Ngassa investigating Pd-catalyzed C–N bond formation in nucleosides. In 2007, he moved to the University of Wisconsin at Madison where he has been pursuing a Ph.D. degree in Organic Chemistry under the supervision of Professor Richard Hsung while exploring metal-catalyzed and thermal pericyclic rearrangements of ynamides.

Hongyan Li received her B.S. in Applied Chemistry from the University of Science and Technology of China in 2004. Since 2005, she has been pursuing a Ph.D. degree in Organic Chemistry under the supervision of Professor Richard Hsung at University of Wisconsin at Madison while working on cycloaddition reactions employing ynamides.

Andrew G. Lohse received his B.S. in Chemistry from Calvin College in Grand Rapids, MI, in 2007, and worked with Professors Ron Blankespoor and Eric Arnoys. Since 2007, he has been pursuing a Ph.D. degree in Organic Chemistry under the supervision of Professor Richard Hsung at University of Wisconsin at Madison while working on [4 + 3] cycloadditions employing allenamides.

Ryuji Hayashi graduated from Indiana State University in Terre Haute, IN, with his B.S. in Chemistry and Biochemistry in 2003. He then moved to North Dakota State University in Fargo, ND, and obtained his Ph.D. degree in organic chemistry under the supervision of Professor Gregory Cook in 2008. He was a recipient of the NDSU Graduate School Doctoral Dissertation Award 2007–2008. Since 2008, he has been researching as a postdoctoral research associate in Professor Richard Hsung's group at the University of Wisconsin at Madison. He is currently exploring a variety of tandem reactions via allenamide isomerizations.

Zhenjie Lu obtained her B.S degree in chemistry in 1996 from the East China University of Science and Technology in Shanghai, China. She started as a graduate student at Michigan State University under Professor Bill Wulff's direction in 2002 as a graduate student and fished her Ph.D. degree in 2008. Her research involved studies on the optimization and synthetic application of catalytic asymmetric aziridination reactions.

Yu Zhang carried out his undergraduate research at the East China University of Science and Technology in Shanghai, China. He received his B.S degree in chemistry in 1996 and then in 2000 he joined Professor Bill Wulff's group as a graduate student at Michigan State University. He obtained his Ph.D. degree in 2006 after working in the area of mechanism and methodology development of catalytic asymmetric aziridination. After spending one year at University of Maryland working for Professor Michael P. Doyle, from 2007 to 2009 he was a postdoctoral scholar at the University of Wisconsin at Madison, working with Professor Richard Hsung in the area of ynamide chemistry and natural product synthesis. He is currently a senior chemist in Dow AgroSciences LLC.

Richard P. Hsung obtained his B.S. in Chemistry and Mathematics from Calvin College in Grand Rapid, MI. He then attended The University of Chicago and received his M.S. and Ph.D. degrees in Organic Chemistry in 1990 and 1994, respectively, under supervisions of Professors Jeff Winkler and Bill Wulff. After pursuing a post-doctoral stay with Professor Larry Sita in Chicago and NIH-post-doctoral work with Professor Gilbert Stork at Columbia University, he moved to the University of Minnesota as an Assistant Professor in 1997 and was promoted to Associate Professor in 2002. He was promoted to Professor and moved to the University of Wisconsin in 2006. He was a recipient of Camille Dreyfus Teacher-Scholar Award and National Science Foundation Career Award. He has co-authored 200 publications, delivered over 180 invited lectures, and supervised 120 students and post-doctoral fellows with research interests in developing cycloaddition and annulation approaches to natural product syntheses and stereoselective methods using allenamides, ynamides, enamides, and acetals.
7. References
- 1.Bode J. Ann. 1892;267:268–286. [Google Scholar]
- 2.For a correction of this claim see: Klages F, Drerup E. Ann. 1941;547:65..
- 3.Zaugg HE, Swett LR, Stone GR. J. Org. Chem. 1958;23:1389. [Google Scholar]
- 4.Wolf V, Kowitz F. Ann. 1960;638:33. [Google Scholar]
- 5.Viehe HG. Angew Chem. Int. Ed. Engl. 1963;2:477. [Google Scholar]
- 6.Viehe HG. Angew Chem. Int. Ed. Engl. 1967;6:767. [Google Scholar]
- 7.Viehe HG. Chemistry of Acetylenes. Marcel Dekker; New York: 1969. Chapter 12; pp. 861–912. [Google Scholar]
- 8.Ficini J. Tetrahedron. 1976;32:448. [Google Scholar]
- 9.Pitacco G, Valentin E. Chapter 15. Chemistry of Functional Groups. 1979:623–714. [Google Scholar]
- 10.Collard-Motte J, Janousek Z. Topics in Current Chem. 1986;130:89. [Google Scholar]
- 11.Himbert G. In: Methoden Der Organischen Chemie (Houben-Weyl) Kropf H, Schaumann E, editors. Georg Thieme Verlag; Stuttgart: 1993. p. 3267. [Google Scholar]
- 12.Zificsak CA, Mulder JA, Rameshkumar C, Wei L-L, Hsung RP. Tetrahedron. 2001;57:7575. [Google Scholar]
- 13.Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379. [Google Scholar]
- 14.Katritzky AR, Jiang R, Singh SK. Heterocycles. 2004;63:1455. [Google Scholar]
- 15.The unfortunate and premature loss of Professor Jacqueline Ficini contributes another critical reason for lack of advancement in ynamine chemistry.
- 16.Genet JP, Kahn P, Ficini J. Tetrahedron Lett. 1980:1521. [Google Scholar]
- 17.van Elburg PA, Honig GWN, Reinhoudt DN. Tetrahedron Lett. 1987:6397. [Google Scholar]
- 18.Fischer H, Podschadly O, Roth G, Herminghaus S, Klewitz S, Heck J, Houbrechts S, Meyer T. J. Organomet. Chem. 1997;541:321. [Google Scholar]
- 19.Balsells J, Vázquez J, Moyano A, Pericàs MA, Riera A. J. Org. Chem. 2000;65:7291. doi: 10.1021/jo000278+. [DOI] [PubMed] [Google Scholar]
- 20.Katritzky AR, Singh SK, Jiang R. Tetrahedron. 2006;63:3794. [Google Scholar]
- 21.Nadipuram AK, Kerwin SM. Tetrahedron. 2006;63:3798. [Google Scholar]
- 22.Ishihara T, Mantani T, Konno T, Yamanaka H. Tetrahedron. 2006;63:3783. [Google Scholar]
- 23.For a leading reference, see: Murch P, Williamson BL, Stang PJ. Synthesis. 1994:1255..
- 24.Janousek Z, Collard J, Viehe HG. Angew. Chem. Int. Ed. Engl. 1972;11:917. [Google Scholar]; Also see: Goffin E, Legrand Y, Viehe HG. J. Chem. Res. (S) 1977;105.
- 25.Feldman KS, Bruendl MM, Schildknegt K, Bohnstedt AC. J. Org. Chem. 1996;61:5440. [Google Scholar]
- 26.Hsung RP. Tetrahedron. 2006;62:3781. [Google Scholar]
- 27.Ficini J, Guingant A, d'Angelo J. J. Am. Chem. Soc. 1979;101:1318. [Google Scholar]
- 28.Zhdankin VV, Stang PJ. Tetrahedron. 1998;54:10927. [Google Scholar]; Also see: Kitamura T, Tashi N, Tsuda K, Fujiwara Y. Tetrahedron Lett. 1998;39:3787. Kitamura T, Tashi N, Tsuda K, Chen H, Fujiwara Y. Heterocycles. 2000;52:303..
- 29.Balsamo A, Macchia B, Macchia F, Rossello A, Domiano P. Tetrahedron Lett. 1985;26:4141. [Google Scholar]
- 30.Petersen LI. Tetrahedron Lett. 1968;9:5357. [Google Scholar]
- 31.Galy GP, Elguero J, Vincent EJ, Galy AM, Barbe J. Synthesis. 1979:944. [Google Scholar]
- 32.Mahamoud A, Galy JP, Vincent EJ, Barbe J. Synthesis. 1981:917. [Google Scholar]
- 33.Katritzky Alan R., Ramer Wolfgang H. J. Org. Chem. 1985;50:852. [Google Scholar]
- 34.Majumdar KC, Ghosh SK. Synth. Commun. 1994;24:217. [Google Scholar]
- 35.(a) Wei L-L, Mulder JA, Xiong H, Zificsak CA, Douglas CJ, Hsung RP. Tetrahedron. 2001;57:459. [Google Scholar]; (b) Huang J, Xiong H, Hsung RP, Rameshkumar C, Mulder JA, Grebe TP. Org. Lett. 2002;4:2417. doi: 10.1021/ol020097p. [DOI] [PubMed] [Google Scholar]
- 36.Janousek Z, Collard J, Viehe HG. Angew. Chem., Int. Ed. 1972;11:917. [Google Scholar]
- 37.Joshi RV, Xu Z-Q, Ksebati MB, Kessel D, Corbett TH, Drach JC, Zemlicka J. J Chem Soc, Perkin Trans 1. 1994:1089. [Google Scholar]
- 38.(a) Brückner D. Synlett. 2000:1402. [Google Scholar]; (b) Brückner D. Tetrahedron. 2006;62:3809. [Google Scholar]
- 39.Rodríguez D, Castedo L, Saá C. Synlett. 2004:783. [Google Scholar]
- 40.Rodriguez D, Martinez-Esperon MF, Castedo L, Saá C. Synlett. 2007:1963. [Google Scholar]
- 41.Couty S, Barbazanges M, Meyer C, Cossy J. Synlett. 2005:905. [Google Scholar]
- 42.(a) Murch P, Williamson BL, Stang PJ. Synthesis. 1994:1255. [Google Scholar]; (b) Zhdankin VV, Stang PJ. Tetrahedron. 1998;54:10927. [Google Scholar]
- 43.Feldman KS, Bruendl MM, Schildknegt K, Bohnstedt AC. J. Org. Chem. 1996;61:5440. [Google Scholar]
- 44.(a) Witulski B, Stengel T. Angew. Chem., Int. Ed. 1998;37:489. doi: 10.1002/(SICI)1521-3773(19980302)37:4<489::AID-ANIE489>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]; (b) Witulski B, Gossmann M. Synlett. 2000:1793. [Google Scholar]
- 45.(a) Witulski B, Stengel B. Angew. Chem., Int. Ed. 1999;38:2426. [PubMed] [Google Scholar]; (b) Witulski B, Lumtscher J, Bersträber U. Synlett. 2003:708. [Google Scholar]; (c) Witulski B, Schweikert T, Schollmeyer D, Nemkovich NA. Chem. Comm. 2010 doi: 10.1039/b919275a. DOI: 10.1039/b919275a. [DOI] [PubMed] [Google Scholar]
- 46.(a) Rainier JD, Imbriglio JE. Org. Lett. 1999;1:2037. [Google Scholar]; (b) Rainier JD, Imbriglio JE. J. Org. Chem. 2000;65:7272. doi: 10.1021/jo001044t. [DOI] [PubMed] [Google Scholar]
- 47.Witulski B, Alayrac C. Angew. Chem. Int. Ed. 2002;41:3281. doi: 10.1002/1521-3773(20020902)41:17<3281::AID-ANIE3281>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 48.Klein M, König B. Tetrahedron. 2004;60:1087. [Google Scholar]
- 49.Witulski B, Gössmann M. Chem. Commun. 1999:1879. [Google Scholar]
- 50.Kerwin SM, Nadipuram A. Synlett. 2004:1404. [Google Scholar]
- 51.Denonne F, Seiler P, Diederich F. Helv. Chim. Acta. 2003;86:3096. [Google Scholar]
- 52.Balsamo A, Macchia B, Macchia F, Rossello A, Domiano P. Tetrahedron Lett. 1985;26:4141. [Google Scholar]
- 53.Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas CJ. J. Am. Chem. Soc. 2003;125:2368. doi: 10.1021/ja021304j. [DOI] [PubMed] [Google Scholar]
- 54.(a) Klapper A, Huang X, Buchwald SL. J. Am. Chem. Soc. 2002;124:7421. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]; (b) Klapars A, Antilla JC, Huang X, Buchwald SL. J. Am. Chem. Soc. 2001;123:7727. doi: 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]
- 55.(a) Dunetz JR, Danheiser RL. Org. Lett. 2003;5:4011. doi: 10.1021/ol035647d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kohnen AL, Dunetz JR, Danheiser RL. Org. Syn. 2007;84:88. [PMC free article] [PubMed] [Google Scholar]; (c) Dunetz JR, Danheiser RL. Tetrahedron. 2006;62:3815. doi: 10.1016/j.tet.2005.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dunetz JR, Danheiser RL. J. Am. Chem. Soc. 2005;127:5776. doi: 10.1021/ja051180l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.(a) Zhang Y, Hsung RP, Tracey MR, Kurtz KCM, Vera EL. Org. Lett. 2004;6:1151. doi: 10.1021/ol049827e. [DOI] [PubMed] [Google Scholar]; (b) Zhang X, Zhang Y, Huang J, Hsung RP, Kurtz KCM, Oppenheimer J, Petersen ME, Sagamanova IK, Shen L, Tracey MR. J. Org. Chem. 2006;71:4170. doi: 10.1021/jo060230h. [DOI] [PubMed] [Google Scholar]; (c) Sagamanova IK, Kurtz KCM, Hsung RP. Org. Syn. 2007;84:359. [Google Scholar]; (d) Zhang X, Li H, You L, Tang Y, Hsung RP. Adv. Syn. Catal. 2006;348:2437. [Google Scholar]
- 57.(a) Hirano S, Fukudome Y, Tanaka R, Sato F, Urabe H. Tetrahedron. 2006;62:3896. [Google Scholar]; (b) Fukudome Y, Naito H, Hata T, Urabe H. J. Am. Chem. Soc. 2008;130:1820. doi: 10.1021/ja078163b. [DOI] [PubMed] [Google Scholar]
- 58.Dooleweerdt K, Birkedal H, Ruhland T, Skrydstrup T. J. Org. Chem. 2008;73:9447. doi: 10.1021/jo801935b. [DOI] [PubMed] [Google Scholar]
- 59.Laroche C, Li J, Freyer MW, Kerwin SM. J. Org. Chem. 2008;73:6462. doi: 10.1021/jo801118q. [DOI] [PubMed] [Google Scholar]
- 60.Yao B, Liang Z, Niu T, Zhang Y. J. Org. Chem. 2009;74:4630. doi: 10.1021/jo900595c. [DOI] [PubMed] [Google Scholar]
- 61.(a) Coste A, Karthikeyan G, Couty F, Evano G. Angew. Chem., In. Ed. 2009;48:4381. doi: 10.1002/anie.200901099. [DOI] [PubMed] [Google Scholar]; (b) Coste A, Couty F, Evano G. Org. Lett. 2009;11:4454. doi: 10.1021/ol901831s. [DOI] [PubMed] [Google Scholar]
- 62.Shen W, Wang L. J. Org. Chem. 1999;64:8873. doi: 10.1021/jo991116k. and references therein. [DOI] [PubMed] [Google Scholar]
- 63.Hamada T, Ye X, Stahl SS. J. Am. Chem. Soc. 2008;130:833. doi: 10.1021/ja077406x. [DOI] [PubMed] [Google Scholar]
- 64.Ghose S, Gilchrist TL. J. Chem. Soc., Perkin Trans. 1. 1991:775. [Google Scholar]
- 65.Schottelius M, Chen P. Helv. Chim. Acta. 1998;81:2341. [Google Scholar]
- 66.Fromont C, Masson S. Tetrahedron. 1999;55:5405. [Google Scholar]
- 67.Laroche C, Kerwin MS. Tetrahedron Lett. 2009;50:5194. [Google Scholar]
- 68.Mulder JA, Kurtz KCM, Hsung RP, Coverdale H, Frederick MO, Shen L, Zificsak CA. Org. Lett. 2003;5:1547. doi: 10.1021/ol0300266. [DOI] [PubMed] [Google Scholar]
- 69.Kramer S, Dooleweerdt K, Lindhardt AT, Rottländer M, Skrydstrup T. Org. Lett. 2009;11:4208. doi: 10.1021/ol901565p. [DOI] [PubMed] [Google Scholar]
- 70.Mézailles N, Ricard L, Gagosz F. Org. Lett. 2005;7:4133. doi: 10.1021/ol0515917. [DOI] [PubMed] [Google Scholar]
- 71.Oppenheimer J, Johnson WL, Tracey MR, Hsung RP, Yao P-Y, Liu R, Zhao K. Org. Lett. 2007;9:2361. doi: 10.1021/ol0707362. [DOI] [PubMed] [Google Scholar]
- 72.(a) Yasui H, Yorimitsu H, Oshima K. Chem. Lett. 2008;37:40. [Google Scholar]; (b) Kanemura S, Kondoh A, Yasui H, Yorimitsu H, Oshima K. Bull. Chem. Soc. Jpn. 2008;81:506. [Google Scholar]
- 73.Zhang Y. Tetrahedron Lett. 2005;46:6483. [Google Scholar]
- 74.Zhang Y. Tetrahedron. 2006;62:3917. [Google Scholar]
- 75.Witulski B, Buschmann N, Bergsträßer U. Tetrahedron. 2000;56:8473. [Google Scholar]
- 76.Hoffmann RW, Brückner D. New J. Chem. 2001;25:369. [Google Scholar]
- 77.Minière S, Cintrat J-C. Synthesis. 2001:705. [Google Scholar]
- 78.(a) Minière S, Cintrat J-C. J. Org. Chem. 2001;66:7385. doi: 10.1021/jo015824t. [DOI] [PubMed] [Google Scholar]; (b) Timbart L, Cintrat J-C. Chem. Eur. J. 2002;8:1637. doi: 10.1002/1521-3765(20020402)8:7<1637::aid-chem1637>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]; (c) Naud S, Cintrat J-C. Synthesis. 2003:1391. [Google Scholar]
- 79.Buissonneaud D, Cintrat J-C. Tetrahedron Lett. 2006;47:3139. [Google Scholar]
- 80.Chechik-Lankin H, Livshin S, Marek I. Synlett. 2005:2098. [Google Scholar]
- 81.(a) Das JP, Chechik H, Marek I. Nature Chem. 2009;1:128. doi: 10.1038/nchem.131. [DOI] [PubMed] [Google Scholar]; (b) Koester DC, Werz DB. Angew. Chem. Int. Ed. 2009;48:7971. doi: 10.1002/anie.200903773. [DOI] [PubMed] [Google Scholar]
- 82.Charette AB, Marcoux J-F, Molinaro C, Beauchemin A, Brochu C, Isabel E. J. Am. Chem. Soc. 2000;122:4508. [Google Scholar]
- 83.(a) Yasui H, Yorimitsu H, Oshima K. Chem. Lett. 2007;36:32. [Google Scholar]; (b) Yasui H, Yorimitsu H, Oshima K. Bull. Chem. Soc. Jpn. 2008;81:373. [Google Scholar]
- 84.(a) Gourdet B, Lam HW. J. Am. Chem. Soc. 2009;131:3802. doi: 10.1021/ja900946h. [DOI] [PubMed] [Google Scholar]; (b) Gourdet B, Rudkin ME, Watts CA, Lam HW. J. Org. Chem. 2009;74:7849. doi: 10.1021/jo901658v. [DOI] [PubMed] [Google Scholar]
- 85.(a) Marion F, Courillon C, Malacria M. Org. Lett. 2003;5:5095. doi: 10.1021/ol036177q. [DOI] [PubMed] [Google Scholar]; (b) Marion F, Coulomb J, Servais A, Courillon C, Fensterbank L, Malacria M. Tetrahedron. 2006;62:3856. [Google Scholar]
- 86.Sato A, Yorimitsu H, Oshima K. Synlett. 2009:28. [Google Scholar]
- 87.Bergman RG. J. Am. Chem. Soc. 1973;6:25. [Google Scholar]
- 88.(a) Kraka E, Cremer D. J. Mol. Structure. 2000;506:191. [Google Scholar]; (b) Kraka E, Cremer D. J. Am. Chem. Soc. 2000;122:8245. [Google Scholar]
- 89.Li H, Hsung RP. Org. Lett. 2009;11:4462. doi: 10.1021/ol901860b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clark TB, Woerpel KA. Organometallics. 2005;24:6212. [Google Scholar]
- 91.Couty S, Meyer C, Cossy J. Synlett. 2007:2819. [Google Scholar]
- 92.Al-Rashid ZF, Johnson WL, Hsung RP, Wei Y, Yao P-Y, Liu R, Zhao K. J. Org. Chem. 2008;73:8780. doi: 10.1021/jo8015067. [DOI] [PMC free article] [PubMed] [Google Scholar]; For our preliminary disclosure of this work, see: Al-Rashid ZF, Hsung RP, Antoline JE, Ko C, Wei Y, Yang J. 40th ACS National Organic Symposium; Durham, NC. June 3, 2007; Abstract No. A-11..
- 93.Al-Rashid ZF, Hsung RP. Org. Lett. 2008;10:661. doi: 10.1021/ol703083k. [DOI] [PubMed] [Google Scholar]
- 94.(a) Riddell N, Villeneuve K, Tam W. Org. Lett. 2005;7:3681. doi: 10.1021/ol0512841. [DOI] [PubMed] [Google Scholar]; (b) Cockburn N, Karimi E, Tam W. J. Org. Chem. 2009;74:5762. doi: 10.1021/jo9010206. [DOI] [PubMed] [Google Scholar]
- 95.(a) Villeneuve K, Riddell N, Tam W. Tetrahedron. 2006;62:3823. [Google Scholar]; (b) Riddell N, Villeneuve K, Tam W.J. Org. Chem 200974576219572573 [Google Scholar]
- 96.Kohnen AL, Mak XY, Lam TY, Dunetz JR, Danheiser RL. Tetrahedron. 2006;62:3815. doi: 10.1016/j.tet.2005.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hsung RP, Zificsak CA, Wei L-L, Douglas CJ, Xiong H, Mulder JA. Org. Lett. 1999;1:1237. [Google Scholar]
- 98.Kurtz KCM, Hsung RP, Zhang Y. Org. Lett. 2006;8:231. doi: 10.1021/ol052487s. [DOI] [PubMed] [Google Scholar]
- 99.You L, Al-Rashid ZF, Figueroa R, Ghosh SK, Li G, Lu T, Hsung RP. Synlett. 2007:1656. [Google Scholar]
- 100.Shindoh N, Takemoto Y, Takasu K. Chem. Eur. J. 2009;15:7026. doi: 10.1002/chem.200901103. [DOI] [PubMed] [Google Scholar]
- 101.IJsselstijn M, Cintrat J-C. Tetrahedron. 2006;62:3837. [Google Scholar]
- 102.Zhang X, Hsung RP, You L. Org. Biomol. Chem. 2006;4:2679. doi: 10.1039/b606680a. [DOI] [PubMed] [Google Scholar]
- 103.Zhang X, Li H, You L, Tang Y, Hsung R. Adv. Synth. Catal. 2006;348:2437. [Google Scholar]
- 104.Zhang X, Hsung RP, Li H. Chem. Commun. 2007:2420. doi: 10.1039/b701040k. [DOI] [PubMed] [Google Scholar]
- 105.Kim JY, Kim SH, Chang S. Tetrahedron Lett. 2008;49:1745. [Google Scholar]
- 106.Oppilliart S, Mousseau G, Zhang L, Jia G, Thuéry P, Rousseau B, Cintrat J-C. Tetrahedron. 2007;63:8094. [Google Scholar]
- 107.Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao H, Lin Z, Jia G, Fokin VV. J. Am. Chem. Soc. 2008;133:8923. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- 108.Li H, You L, Zhang X, Johnson WL, Figueroa R, Hsung RP. Heterocycles. 2007;74:553. [Google Scholar]
- 109.Grecian S, Fokin VV. Angew. Chem. Int. Ed. 2008;47:8285. doi: 10.1002/anie.200801920. [DOI] [PubMed] [Google Scholar]
- 110.Zhang X, Hsung RP, Li H, Zhang Y, Johnson WL, Figueroa R. Org. Lett. 2008;10:3477. doi: 10.1021/ol801257j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.(a) Martínez-Esperón MF, Rodríguez D, Castedo L, Saá C. Org. Lett. 2005;7:2213. doi: 10.1021/ol050609a. [DOI] [PubMed] [Google Scholar]; (b) Martínez-Esperón MF, Rodríguez D, Castedo L, Saá C. Tetrahedron. 2008;64:3674. [Google Scholar]
- 112.Movassaghi M, Hill MD, Ahmad O. J. Am. Chem. Soc. 2007;129:10096. doi: 10.1021/ja073912a. [DOI] [PubMed] [Google Scholar]
- 113.Buzas A, Istrate F, Le Goff XF, Odabachiam Y, Gagosz F. J. Organomet. Chem. 2009;694:515. [Google Scholar]
- 114.Witulski B, Gößmann M. Synlett. 2000;12:1793. [Google Scholar]
- 115.Domínguez G, Casarrubios L, Rodríguez-Noriega J, Pérez-Castells J. Helv. Chim. Acta. 2002;85:2856. [Google Scholar]
- 116.Shen L, Hsung R. Tetrahedron Lett. 2003;44:9353. [Google Scholar]
- 117.Gupta AK, Park DI, Oh CH. Tetrahedron Lett. 2005;46:4171. [Google Scholar]
- 118.Witulski B, Stengel T. Angew. Chem. Int. Ed. 1999;38:2426. [PubMed] [Google Scholar]
- 119.Witulski B, Stengel T, Fernández-Hernández J. Chem. Commun. 2000:1965. [Google Scholar]
- 120.(a) Tanaka K, Takeishi K, Noguchi K. J. Am. Chem. Soc. 2006;128:4586. doi: 10.1021/ja060348f. [DOI] [PubMed] [Google Scholar]; (b) Tanaka K, Takeishi K. Synthesis. 2007;18:2920. [Google Scholar]
- 121.Tracey M, Oppenheimer J, Hsung R. J. Org. Chem. 2006;71:8629. doi: 10.1021/jo061683p. [DOI] [PubMed] [Google Scholar]
- 122.(a) Oppenheimer J, Hsung R, Figueroa R, Johnson W. Org. Lett. 2007;9:3969. doi: 10.1021/ol701692m. [DOI] [PubMed] [Google Scholar]; (b) Oppenheimer J, Johnson W, Figueroa R, Hayashi R, Hsung R. Tetrahedron. 2009;65:5001. doi: 10.1016/j.tet.2009.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tanaka D, Sato Y, Mori M. J. Am. Chem. Soc. 2007;129:7730. doi: 10.1021/ja071954t. [DOI] [PubMed] [Google Scholar]
- 124.Garcia P, Moulin S, Miclo Y, Leboeuf D, Gandon V, Aubert C, Malacria M. Chem. Eur. J. 2009;15:2129. doi: 10.1002/chem.200802301. [DOI] [PubMed] [Google Scholar]
- 125.Friedman RK, Rovis T. J. Am. Chem. Soc. 2009;131:10775. doi: 10.1021/ja903899c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.(a) Marion F, Coulomb J, Courillon C, Fensterbank L, Malacria M. Org. Lett. 2004;6:1509. doi: 10.1021/ol049530g. [DOI] [PubMed] [Google Scholar]; (b) Marion F, Coulomb J, Servais A, Courillon C, Fensterbank L, Malacria M. Tetrahedron. 2006;62:3856. [Google Scholar]
- 127.Soriano E, Marco-Contelles J. J. Org. Chem. 2005;70:9345. doi: 10.1021/jo0514265. [DOI] [PubMed] [Google Scholar]
- 128.Zhang Y, Hsung RP, Zhang X, Huang J, Slater BW, Davis A. Org. Lett. 2005;7:1047. doi: 10.1021/ol0473391. [DOI] [PubMed] [Google Scholar]
- 129.Poloukhtine A, Popik VV. J. Am. Chem. Soc. 2007;129:12062. doi: 10.1021/ja073614d. [DOI] [PubMed] [Google Scholar]
- 130.(a) Buzas A, Gagosz F. Org. Lett. 2006;8:515. doi: 10.1021/ol053100o. [DOI] [PubMed] [Google Scholar]; (b) Buzas A, Istrate F, Gagosz F. Tetrahedron. 2009;65:1889. [Google Scholar]
- 131.Hashmi AS, Salathie R, Frey W. Synlett. 2007;11:1763. [Google Scholar]
- 132.Istrate FM, Buzas AK, Jurberg ID, Odabachian Y, Gagosz F. Org. Lett. 2008;10:925. doi: 10.1021/ol703077g. [DOI] [PubMed] [Google Scholar]
- 133.Lin G-Y, Li C-W, Hung S-H, Liu R-S. Org. Lett. 2008;10:5059. doi: 10.1021/ol802047g. [DOI] [PubMed] [Google Scholar]
- 134.Hashmi AS, Rudolph M, Bats J, Frey W, Rominger F, Oeser T. Chem. Eur. J. 2008;14:6672. doi: 10.1002/chem.200800210. [DOI] [PubMed] [Google Scholar]
- 135.Yao P-Y, Zhang Y, Hsung R, Zhao K. Org. Lett. 2008;10:4275. doi: 10.1021/ol801711p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dooleweerdt K, Ruhland T, Skrydstrup T. Org. Lett. 2009;11:221. doi: 10.1021/ol802477d. [DOI] [PubMed] [Google Scholar]
- 137.(a) Couty S, Meyer C, Cossy J. Angew. Chem., Int. Ed. 2006;45:6726. doi: 10.1002/anie.200602270. [DOI] [PubMed] [Google Scholar]; (b) Couty S, Meyer C, Cossy J. Tetrahedron. 2009;65:1809. [Google Scholar]
- 138.Mulder JA, Hsung RP, Frederick MO, Tracey MR, Zificsak CA. Org. Lett. 2002;4:1383. doi: 10.1021/ol020037j. [DOI] [PubMed] [Google Scholar]
- 139.(a) Saucy G, Chopard-dit-Jean LH, Guex W, Ryser G, Isler O. Helv. Chim. Acta. 1958;41:160. [Google Scholar]; (b) Marbet R, Saucy G. Chimia. 1960;14:361. [Google Scholar]; (c) Saucy G, Marbet R. Helv. Chim. Acta. 1967;50:1158. [Google Scholar]; (d) Jones ERH, Loder JD, Whiting MC. Proc. Chem. Soc. 1960:180. [Google Scholar]
- 140.Frederick M, Hsung R, Lambeth R, Mulder J, Tracey M. Org. Lett. 2003;5:2663. doi: 10.1021/ol030061c. [DOI] [PubMed] [Google Scholar]
- 141.Kurtz K, Frederick M, Lambeth R, Mulder J, Tracey M, Hsung R. Tetrahedron. 2006;62:3928. [Google Scholar]
- 142.Zhang Y, DeKorver K, Lohse A, Zhang Y-S, Huang J, Hsung R. Org. Lett. 2009;11:899. doi: 10.1021/ol802844z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Saito N, Sato Y, Mori M. Org. Lett. 2002;4:803. doi: 10.1021/ol017298y. [DOI] [PubMed] [Google Scholar]
- 144.Mori M, Wakamatsu H, Saito N, Sato Y, Narita R, Sato Y, Fujita R. Tetrahedron. 2006;62:3872. [Google Scholar]
- 145.Huang J, Xiong H, Hsung R, Rameshkumar C, Mulder J, Grebe T. Org. Lett. 2002;4:2417. doi: 10.1021/ol020097p. [DOI] [PubMed] [Google Scholar]
- 146.Rosillo M, Domínguez G, Casarrubios L, Amado U, Pérez-Castells J. J. Org. Chem. 2004;69:2084. doi: 10.1021/jo0356311. [DOI] [PubMed] [Google Scholar]
- 147.Mori M, Tanaka D, Saito N, Sato Y. Organometallics. 2008;27:6313. [Google Scholar]
- 148.Saito S, Uchiyama N, Gevorgyan V, Yamamoto Y. J. Org. Chem. 2000;65:4338. doi: 10.1021/jo000171m. [DOI] [PubMed] [Google Scholar]
- 149.Rodriguez D, Castedo L, Saa C. Synlett. 2004:377. [Google Scholar]
- 150.Martinez-Esperon MF, Luis Castedo DR, Saa C. Tetrahedron. 2006;62:3843. [Google Scholar]
- 151.Tracy MR, Zhang Y, Frederick MO, Mulder JA, Hsung RP. Org. Lett. 2004;6:2209. doi: 10.1021/ol0493251. [DOI] [PubMed] [Google Scholar]
- 152.Lindhardt AT, Mantel MLH, Skrydstrup T. Angew. Chem. Int. Ed. 2008;47:2668. doi: 10.1002/anie.200705558. [DOI] [PubMed] [Google Scholar]
- 153.Witulski B, Alayrac C, Tevzadze-Saeftel L. Angew. Chem. Int. Ed. 2003;42:4257. doi: 10.1002/anie.200351977. [DOI] [PubMed] [Google Scholar]
- 154.Couty S, Liegault B, Meyer C, Cossy J. Org. Lett. 2004;6:2511. doi: 10.1021/ol049302m. [DOI] [PubMed] [Google Scholar]
- 155.Tanaka R, Hirano S, Urabe H, Sato F. Org. Lett. 2003;5:67. doi: 10.1021/ol027209x. [DOI] [PubMed] [Google Scholar]
- 156.Hirano S, Tanaka R, Urabe H, Sato F. Org. Lett. 2004;6:727. doi: 10.1021/ol036396b. [DOI] [PubMed] [Google Scholar]
- 157.Tanaka R, Yuza A, Watai Y, Suzuki D, Takayama Y, Sato F, Urabe H. J. Am. Chem. Soc. 2005;127:7774. doi: 10.1021/ja050261e. [DOI] [PubMed] [Google Scholar]
- 158.Saito N, Katayama T, Sato Y. Org. Lett. 2008;10:3829. doi: 10.1021/ol801534e. [DOI] [PubMed] [Google Scholar]
- 159.Barbazanges M, Meyer C, Cossy J. Org. Lett. 2007;9:3245. doi: 10.1021/ol0711725. [DOI] [PubMed] [Google Scholar]
- 160.Barbazanges M, Meyer C, Cossy J. Tetrahedron Lett. 2008;49:2902. [Google Scholar]
- 161.(a) Bae I, Han H, Chang S. J. Am. Chem. Soc. 2005;127:2038. doi: 10.1021/ja0432968. [DOI] [PubMed] [Google Scholar]; (b) Cho SH, Yoo EJ, Bae I, Chang S. J. Am. Chem. Soc. 2005;127:16046. doi: 10.1021/ja056399e. [DOI] [PubMed] [Google Scholar]
- 162.Cassidy MP, Raushel J, Fokin VV. Angew. Chem., Int. Ed. 2006;45:3154. doi: 10.1002/anie.200503805. [DOI] [PubMed] [Google Scholar]
- 163.Yoo EJ, Ahlquist M, Bae I, Sharpless KB, Fokin VV, Chang S. J. Org. Chem. 2008;73:5520. doi: 10.1021/jo800733p. [DOI] [PubMed] [Google Scholar]
- 164.Whiting M, Fokin VV. Angew. Chem. Int. Ed. 2006;45:3157. doi: 10.1002/anie.200503936. [DOI] [PubMed] [Google Scholar]
- 165.Xu X, Cheng D, Li J, Guo H, Yan J. Org. Lett. 2007;9:1585. doi: 10.1021/ol070485x. [DOI] [PubMed] [Google Scholar]
- 166.Yoo EJ, Bae I, Cho SH, Han H, Chang S. Org. Lett. 2006;8:1347. doi: 10.1021/ol060056j. [DOI] [PubMed] [Google Scholar]
- 167.(a) Cui S-L, Lin X-F, Wang Y-G. Org. Lett. 2006;8:4517. doi: 10.1021/ol061685w. [DOI] [PubMed] [Google Scholar]; (b) Cui S-L, Wang J, Wang Y-G. Org. Lett. 2007;9:5023. doi: 10.1021/ol702241e. [DOI] [PubMed] [Google Scholar]; (c) Cui S-L, Wang J, Wang Y-G. Org. Lett. 2008;10:1267. doi: 10.1021/ol800160q. [DOI] [PubMed] [Google Scholar]
- 168.Jin Y, Fu H, Yin Y, Jiang Y, Zhao Y. Synlett. 2007:901. [Google Scholar]
- 169.Bendikov M, Duong HM, Bolanos E, Wudl F. Org. Lett. 2005;7:783. doi: 10.1021/ol0477327. [DOI] [PubMed] [Google Scholar]
- 170.Egi M, Yamaguchi Y, Fujiwara N, Akai S. Org. Lett. 2008;10:1867. doi: 10.1021/ol800596c. [DOI] [PubMed] [Google Scholar]
- 171.(a) Couty S, Liegault B, Meyer C, Cossy J. Tetrahedron. 2006;62:3882. [Google Scholar]; (b) Couty S, Meyer C, Cossy J. Tetrahedron Lett. 2006;47:767. [Google Scholar]
- 172.Alayrac C, Schollmeyer D, Witulski B. Chem. Commun. 2009:1464. doi: 10.1039/b820291e. [DOI] [PubMed] [Google Scholar]
- 173.DeKorver KA, Hsung RP, Lohse AG, Zhang Y. Org. Lett. 2010;12 doi: 10.1021/ol100446p. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yamasaki R, Terashima N, Sotome I, Komagawa S, Saito S. J. Org. Chem. 2010;75:480. doi: 10.1021/jo902251m. [DOI] [PubMed] [Google Scholar]
- 175.Burley GA, Davies DL, Griffith GA, Lee M, Singh K. J. Org. Chem. 2010;75:980. doi: 10.1021/jo902466f. [DOI] [PubMed] [Google Scholar]