

Summary
Chlamydia pneumoniae is a common respiratory pathogen that has been associated with a variety of chronic diseases including asthma and atherosclerosis. Chlamydiae are obligate intracellular parasites that primarily infect epithelial cells where they develop within a membrane-bound vacuole, termed an inclusion. Interactions between the microorganism and eukaryotic cell can be mediated by chlamydial proteins inserted into the inclusion membrane. We describe here a novel C. pneumoniae-specific inclusion membrane protein (Inc) CP0236, which contains domains exposed to the host cytoplasm. We demonstrate that, in a yeast two-hybrid screen, CP0236 interacts with the NFκB activator 1, (Act1) and this interaction was confirmed in HeLa 229 cells where ectopically expressed CP0236 was co-immunoprecipitated with endogenous Act1. Furthermore, we demonstrate that Act1 displays an altered distribution in the cytoplasm of HeLa cells infected with C. pneumoniae where it associates with the chlamydial inclusion membrane. This sequestration of Act1 by chlamydiae inhibited recruitment of the protein to the IL-17 receptor upon stimulation of C. pneumoniae infected cells with IL-17A. Such inhibition of the IL-17 signaling pathway led to protection of Chlamydia-infected cells from NFκB activation in IL-17 stimulated cells. We describe here a unique strategy employed by C. pneumoniae to achieve inhibition of NFκB activation via interaction of CP0236 with mammalian Act1.
Keywords: Inclusion, Inc, IL-17, NFκB
Introduction
Chlamydia pneumoniae is a human respiratory pathogen responsible for approximately 10% of all community-acquired pneumonia and 5% of bronchitis and sinusitis cases (Grayston et al., 1990). Importantly, C. pneumoniae infections have been associated with chronic inflammatory conditions such as atherosclerosis, chronic obstructive pulmonary disease, and asthma (Campbell and Kuo, 2004; Hahn, 1999). Chlamydiae are obligate intracellular pathogens whose developmental cycle occurs entirely within their eukaryotic hosts. Bacteria are internalized by eukaryotic cells into a membrane bound vesicle termed an inclusion that does not fuse with lysosomes (Wolf and Hackstadt, 2001; Heinzen et al., 1996; Schramm and Wyrick, 1995). The inclusion membrane expands throughout the chlamydial developmental cycle and modification of the membrane by insertion of bacterial proteins is one of the critical events required for successful exploitation of the eukaryotic cell. Conversely, inhibition of chlamydial protein synthesis prevents modification of the inclusion membrane, eventually leading to the fusion of vesicle-containing chlamydiae with host lysosomes (Scidmore et al., 1996). An entire family of chlamydial proteins that modify the inclusion membrane has been recognized (Bannantine et al., 2000). Inclusion membrane proteins (Incs) were originally identified in C. psittaci and have subsequently been described in C. trachomatis and C. pneumoniae (Rockey et al., 2002). Inc proteins possess a unique bi-lobed hydrophobic domain of approximately 40–80 amino acids, which is greater than twice the length, predicted to be required to span a typical membrane bilayer. Incs, such as IncA and IncG (Cortes et al., 2007; Scidmore and Hackstadt, 2001; Rockey et al., 1997) contain domains present on the cytoplasmic face of chlamydial inclusion where they mediate interactions with eukaryotic host proteins.
Interleukin 17A (IL-17A), which is primarily produced by a distinct subset of CD4+ T cells termed Th17, coordinates local tissue inflammation via induced release of pro-inflammatory cytokines and neutrophil-mobilizing chemokines from various cell types including epithelial cells (reviewed in (Kolls and Linden, 2004). Although IL-17 was shown to be involved in the pathogenesis of several autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease and asthma, this cytokine also appears to be a key factor contributing to host defense during microbial infections of the lung (reviewed in Tesmer et al., 2008). The stimulation of cells with IL-17A triggers a signaling pathway that initiates from a heteromeric receptor complex consisting of the IL-17A receptor (IL-17RA) and IL-17RC (Toy et al., 2006). One of the essential components in IL-17-mediated signaling is the NFκB activator 1 (Act1) which binds to the SEFIR domain in the cytoplasmic region of the IL-17A receptor (Qian et al., 2007). Act1 is an adaptor protein which lacks apparent enzymatic domains, but contains a helix-loop-helix region at the N-terminus and a coiled-coil region at the C-terminus. The major sites of Act1 expression are epithelial cells of the respiratory and gastrointestinal tract (Qian et al., 2002). Act1-dependent transduction of signaling through IL-17RA leads to NFκB activation (reviewed in (Shen and Gaffen, 2008), expression of IL-17 inducible genes, and production of GCP-2, G-CSF, GRO-α, IL-8, IL-6, HBD-2, MIP-3α, MIP-2 KC, MCP-1 by stimulated cells (reviewed in Kolls and Linden, 2004).
In this study we describe a novel C. pneumoniae-specific inclusion membrane protein designated CP0236 in genome databases. We show that this Inc contains domain(s) exposed to the eukaryotic cytoplasm where it interacts with the host Act1. Furthermore, our data indicate that Act1 is sequestered by CP0236-containing chlamydial inclusions, leading to an altered localization of this adaptor protein in C. pneumoniae infected cells. We also demonstrate that recruitment of Act1 to the IL-17RA is inhibited by chlamydiae in IL-17 stimulated cells, which consequently leads to protection of C. pneumoniae infected cells from IL-17 induced NFκB activation.
Results
CP0236 is a C. pneumoniae-specific Inc exposed to the host cytoplasm
We employed hydropathy plot analysis (Kyte and Doolittle, 1982) of primary amino acid sequences in the C. pneumoniae, AR-39 genome (Read et al., 2000) to screen for proteins containing the bi-lobed hydrophobic domain typical of Inc proteins. These searches identified CP0236 as a putative inclusion membrane protein containing a predicted bilobed hydrophobic domain spanning residues 37-92 (not shown). CP0236 encodes a 279 residue protein with an apparent molecular mass of 31.4 kDa. Subsequent bioinformatic analyses indicate no additional membrane-interactive hydrophobic domains and also revealed CP0236 to be specific to C. pneumoniae, as orthologs were not detected in genomes of other chlamydial species. Since the presence of a bi-lobed domain does not always correlate with protein localization to the inclusion membrane (Li et al., 2008), we used polyclonal antibody specific for the non-hydrophobic region of CP0236 to confirm protein localization by indirect immunofluorescence analysis of Chlamydia-infected HeLa cells. We examined C. pneumoniae inclusions at 64 hrs p.i. or, as a negative control, C. trachomatis cultures at 19 hrs p.i. CP0236 was detected in the inclusion membrane of C. pneumoniae whereas no reactivity was detected with CP0236 antibodies in C. trachomatis cultures (Fig. 1A and D). C. trachomatis cultures at 19 hrs p.i. were used due to differences in chlamydial growth rates (Wolf and Hackstadt, 2001), however, C. trachomatis cultures remained unreactive even at later (40+ hrs) time-points (not shown).
Figure 1.

Localization of CP0236 in C. pneumoniae infected cells. HeLa cells infected with either C. pneumoniae at 64 hrs p.i. or C. trachomatis at 19 hrs p.i. were fixed with methanol and stained with anti-CP0236 antibody (green). CP0236-specific antibody reacts with the inclusion membrane of C. pneumoniae (A), but not with inclusion membrane of C. trachomatis (D). Chlamydial inclusions were counterstained with genus-specific anti-Hsp60 monoclonal antibody (B and E). Composite images (C and F). Scale bar = 10 μm.
In an effort to identify domain(s) of CP0236 exposed to the host cytoplasm, C. pneumoniae infected cells were microinjected with the anti-CP0236 antiserum at 48 hrs p.i. After microinjection, cell cultures were fixed and stained with anti-rabbit secondary antibody. The microinjected cells contained readily detectable rim-like staining surrounding mature C. pneumoniae inclusions (Fig. 2A) which was absent in infected cells microinjected with rabbit pre-immune serum (Fig. 2D). Taken together, these data indicate that the CP0236 is indeed a C. pneumoniae-specific inclusion membrane protein containing a significant domain accessible on the cytoplasmic face of the chlamydial inclusion.
Figure 2.

CP0236 is exposed to the host cytoplasm. C. pneumoniae infected cells were microinjected with either anti-CP0236 or pre-immune serum at 48 hrs p.i. and fixed 3 hrs later prior to the labeling with secondary antibody (green). The anti-CP0236 interacts with C. pneumoniae inclusions (A); no interaction with inclusions is detected in infected cells microinjected with pre-immune serum (D). Chlamydial inclusions were visualized with genus-specific anti-Hsp60 monoclonal antibody (B and E). Merged images (C and F). Scale bar = 10 μm.
CP0236 interacts with the host Act1
Bioinformatic and microinjection data are consistent with CP0236 having a ca.187 residue domain exposed to the host cell cytoplasm and this raises the possibility that CP0236 could interact with host proteins in the cytosol of infected cells. We employed a yeast two-hybrid screen capable of identifying host protein(s) interacting with CP0236 to test this possibility. Since antibodies raised against a recombinant protein containing residues 101-279 of CP0236 reacted with the cytoplasmic face of C. pneumoniae inclusions, this hydrophilic region was used as bait in a yeast two-hybrid screen of a HeLa cell cDNA library. Library plasmids from multiple clones growing on high stringency medium were isolated and retransformed into competent yeast expressing p0236-DBD. Six strains were recovered and sequenced to identify HeLa DNA content. Five of the strains contained cDNA encoding proteins fused out-of-frame with the plasmid-encoded AD. However, one of the positive cDNA clones was fused in frame with the plasmid-encoded AD and encoded the C-terminal 259 residues of NFκB activator 1 or Act1 (Fig. 3A). We further constructed chimeras containing full-length Act1 or portions of the C-terminus containing residues 361-565 or 301-366 (Table 1) to more definitively establish the domain interacting with CP0236. Yeast expressing CP0236-DBD grew on selective medium similarly to the positive control when co-expressing residues 1-565, 306-565, and 301-360 of Act1. No growth was observed in yeast expressing residues 361-565. These data indicate that a domain within residues 306-366 of Act1 is sufficient to mediate interaction with C. pneumoniae CP0236.
Figure 3.

CP0236 interacts with Act1. (A) Yeast grown on synthetic medium (SC) without Leu, Trp, or on SC-TDO (without Leu, Trp, His, + 30 mM 3AT). (1) pCP0236101- 279-DBD + pAct1306-565-AD, (2) pYpkA442-733 -DBD + pRhoA-AD (positive control, (Dukuzumuremyi et al., 2000), (3) pCP0236101- 279-DBD + pDEST22-AD (negative control), (4) pDEST32 + pDEST22 (negative control). (B) Co-immunoprecipitation of FLAG-tagged proteins with endogenous Act1. Act1 was immunoprecipitated from HeLa cells expressing 3xFLAG-CP0236, 3xFLAG-CT694, or empty vector. Purified material was probed with anti-Act1 or anti-FLAG.
Table 1.
| Constructsa | SCb | SC-TDOc |
|---|---|---|
| pYpkA(442-733)d-DBD + pRhoA-AD | +++ | +++ |
| pCP0236(101-279)-DBD + pDEST22 | +++ | −− |
| pCP0236(101-279)-DBD + pAct1(1-565)-AD | +++ | +++ |
| pCP0236(101-279)-DBD + pAct1(306-565)-AD | +++ | +++ |
| pCP0236(101-279)-DBD + pAct1(361-565)-AD | +++ | −− |
| pCP0236(101-279)-DBD + pAct1(301-360)-AD | +++ | +++ |
Respective constructs were created in the binding domain (DBD) vector pDEST32 or activating domain (AD) vector pDEST22.
Growth on vector selective medium lacking leucine and tryptophan.
Growth on high stringency selective medium containing 30 mM 3AT and lacking leucine, tryptophan, and histidine.
Numbers in parentheses indicate residues of respective proteins comprising the chimeric constructs.
To confirm an interaction between CP0236 and Act1, HeLa cell cultures were transfected with either empty vector or a constructs encoding FLAG-tagged CP0236. CT694 is a soluble chlamydial effector protein gaining access to host cytoplasms (Hower and Fields, unpublished) was used as a negative control in addition to empty vector. After 24 hrs, cells were disrupted and endogenous Act1 was immunoprecipitated from lysates with specific antibodies. Immunoprecipitated material was analyzed by immunoblot for the presence of Act1 and Flag-tagged proteins. As expected, Act1 was recovered equally well from all lysates. Ectopically expressed CP0236, but not the control protein CT694, was detected in immunoprecipitated material, indicating co-precipitation with Act1 and confirming that Act1 and CP0236 interact in HeLa cells (Fig. 3B).
Act1 associates with the C. pneumoniae inclusion membrane
Since CP0236 is an inclusion membrane-localized protein, interaction with Act1 should lead to accumulation of Act1 in the vicinity of the chlamydial inclusion. To test this prediction, C. pneumoniae or C. trachomatis infected cells were transfected with full-length FLAG-tagged Act1. Cultures were doubly labeled with anti-FLAG mAb to visualize Act1 and Chlamydia species-specific rabbit antiserum to localize chlamydial inclusions. Ectopically expressed Act1 associated with the inclusion membrane of C. pneumoniae but not with C. trachomatis inclusions (Fig. 4A and D). Act1-specific signal was apparent with only a portion of the inclusion membrane since we specifically focused on cells containing low levels of ectopically expressed proteins to avoid localization artifacts. However, in cells expressing higher levels of protein, Act1 was associated with the entire C. pneumoniae inclusion membrane yet still did not co-localize with C. trachomatis inclusions (not shown). FLAG-tagged C. trachomatis CT847 is a soluble protein and failed to co-localize with the chlamydial inclusion membrane, confirming the specificity of Act1 localization to the C. pneumoniae inclusion membrane (Fig. 4G). Collectively, these data are consistent with an interaction between native CP0236 and Act1 and that this interaction functions to localize Act1 to the C. pneumoniae inclusion membrane.
Figure 4.

Localization of Act1 in C. pneumoniae infected cells. C. pneumoniae infected cells were transfected with FLAG-tagged Act1 at 45 hrs p.i. and cell cultures were fixed with methanol and stained with primary and secondary antibodies after an additional 19 hrs of incubation. (A) Act1 is associated with inclusion membrane of C. pneumoniae (green). (D) Association of Act1 was not detected with C. trachomatis inclusions in cells transfected with FLAG-tagged Act1. (G) Control experiment using C. pneumoniae infected cells transfected with 3xFLAG-CT847. Localization of chlamydial inclusions is depicted by staining with Chlamydia species-specific rabbit antiserum (B, E and H). Composite images (C, F and I). Scale bar = 10 μm.
Partitioning of endogenous Act1 is altered in C. pneumoniae infected cells
We attempted to immunoprecipitate Act1 from cell lysates of mock or C. pneumoniae infected HeLa cells at 64 hrs p.i. to examine whether Act1 can interact directly with endogenous CP0236; however, Act1 was not detectible in immunoprecipitated material derived from C. pneumoniae infected cells (Fig. 5A). This phenomenon clearly required chlamydial protein synthesis since immunoprecipitation of Act1 was unaffected in C. pneumoniae infected cells treated with chloramphenicol (Cmp). In order to address levels of Act1 in infected cells, whole cell lysates of mock and C. pneumoniae infected cells were probed with anti-Act1. In these experiments, abundance of Act1 remained unchanged during infection with C. pneumoniae (Fig. 5B), raising the possibility that Act1 solubility in immunoprecipitation buffer is altered in a Chlamydia-dependent fashion. Interestingly, chlamydial Inc proteins are also typically insoluble in these buffers (unpublished). Since Act1 is capable of interacting with the Inc CP0236, insoluble material obtained after lysis of cell cultures was analyzed for the presence of Act1. Indeed, both Act1 and CP0236 were detected in this insoluble fraction derived from C. pneumoniae infected but not mock-infected cells (Fig. 5C). This change in Act1 solubility in C. pneumoniae infected cells was detected as early as 45 hrs p.i. (data not shown). These results indicate that Act1 appears to be sequestered by C. pneumoniae inclusions and are consistent with an interaction with CP0236 leading to an altered localization of Act1 in infected cells.
Figure 5.

Host Act1 partitioning in C. pneumoniae infected cells. (A) Cell lysates of mock and C. pneumoniae infected cultures at 64 hrs p.i. were immunoprecipitated with anti-Act1 followed by SDS-PAGE and immunoblotting with anti-Act1 antibody. (B) Immunoblots of whole cell lysates from mock and C. pneumoniae infected cells probed with anti-Act1 antibody. (C) Immunoblot of insoluble pellets obtained after lysis of mock and C. pneumoniae infected cultures probed with anti-Act1 or anti-CP0236 antibody. Amount of protein loaded per sample was controlled by probing with anti-Actin antibody.
C. pneumoniae impairs IL-17 signaling pathway leading to NFκB activation
Although Act1 has been implicated in numerous signal transduction pathways, evidence indicates that Act1 is essential and plays a direct role in IL-17 dependent activation of proinflammatory gene transcription in epithelial cells (Qian et al., 2007). Stimulation of cells with pro-inflammatory cytokine IL-17 leads to the recruitment of Act1 to IL-17R triggering a signaling cascade leading to downstream activation of transcription factor NFκB. Phosphorylation and subsequent degradation of the NF-κB inhibitor IκBα is required to allow NFκB nuclear translocation and transcription of IL-17 inducible genes. Given these data and the emerging role of IL-17 in respiratory infections, we chose to test the impact of the C. pneumoniae CP0236 interaction with Act1 by examining effects on Il-17 signaling in epithelial cells. We hypothesized that C. pneumoniae might interfere with IL-17 signaling by impaired recruitment of Act1 to the cytoplasmic portion of the IL-17 receptor. In order to test this hypothesis, mock and C. pneumoniae infected cell cultures were transfected with pORF-hIL-17R encoding the IL-17 receptor and stimulated with IL-17A at 52 hrs p.i. Endogenous Act1 was immunoprecipitated together with the IL-17R from uninfected HeLa cells at 5 min and 20 min after treatment with IL-17, whereas the interaction of Act1 with IL-17R was barely detectable at 20 min post induction in C. pneumoniae infected cells (Fig. 6A). We next addressed the possibility that C. pneumoniae inhibition of Act1 recruitment to the IL-17R is sufficient to prevent NFκB activation. We chose to test this possibility by examination of IκBα phosphorylation in uninfected and infected HeLa cells. In uninfected cells, the stimulation with IL-17 resulted in phosphorylation of IκBα, while in C. pneumoniae infected cells, significant IκBα phosphorylation was not detected. This inhibitory effect required bacterial protein synthesis as the level of IκBα phosphorylation was restored in C. pneumoniae infected cells treated with chloramphenicol (Fig. 6B). The inhibition of IκBα phosphorylation specifically targeted the IL-17 signaling pathway as stimulation of Chlamydia infected cells with TNFα resulted in IκBα phosphorylation comparable to the level of phosphorylation detected in uninfected cells (Fig. 6C). These data are consistent with a lack of Act1 interaction with TNFR in signaling pathways induced by TNFα. Finally, the ability to interfere with IκBα phosphorylation was C. pneumoniae-specific since the response to IL-17 stimulation was not affected in cultures infected with C. trachomatis (Fig. 6D). Collectively, these data indicate that C. pneumoniae infection interferes with IL-17-stimulated NF-κB activation in epithelial cells by preventing Act1 recruitment to the IL-17R.
Figure 6.
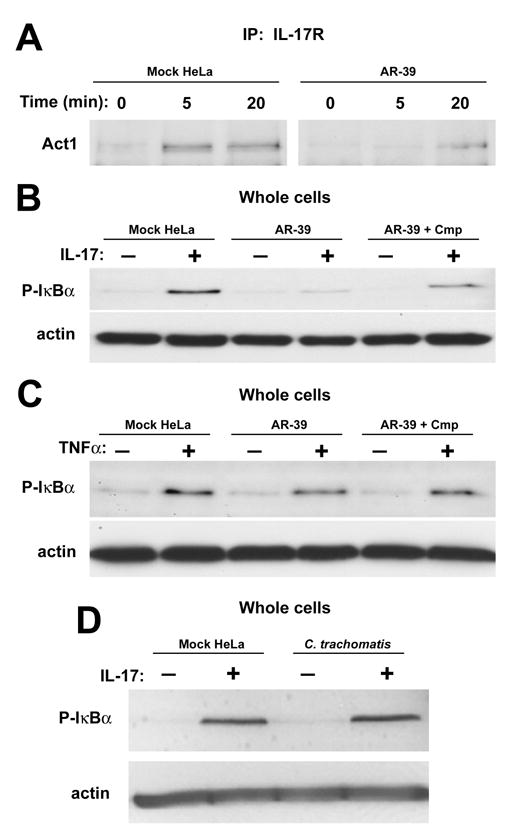
Interference of C. pneumoniae in IL-17 signaling pathway leading to NFκB activation. Mock and C. pneumoniae infected HeLa cells were transfected at 36 hrs p.i. with pORF-hIL-17R and after an additional 16 hrs of incubation cells were either treated with IL-17A (50 ng/ml) for the indicated times or left untreated. (A) Cell cultures were lysed and immunoprecipitated with anti-IL-17R mAb prior to immunoblotting with anti-Act1 antibody. (B) Cell lysates from mock and C. pneumoniae infected cells at 64 hrs p.i. treated with IL-17A (50 ng/ml) for 30 min or untreated were probed in immunoblots with anti-P-IκBα. (C) anti-P-IκBα immunoblot of mock and C. pneumoniae infected cells at 64 hrs p.i. treated with TNFα (10 ng/ml) for 15 min or left untreated. As a specificity control (D), cell lysates from mock and C. trachomatis infected cells at 24 hrs p.i. treated with IL-17A (50 ng/ml) for 30 min or untreated were probed in immunoblots with anti-P-IκBα. All immunoblots were probed with anti-Actin antibody as a loading control. Representative data from three independent experiments are shown.
Discussion
C. pneumoniae is an infectious agent causing a variety of respiratory diseases that occur worldwide. Antibody prevalence to C. pneumoniae increases with age. Seroreactivity to C. pneumoniae reaches 50% at age 20 and continues to rise slowly to 70% to 80% at age 70 (Kuo et al., 1995). Virtually everyone is infected at some point in life and re-infections occur commonly (Campbell et al., 1998). Unlike other chlamydial species causing diseases in humans, C. pneumoniae displays an apparent tissue tropism towards the respiratory tract. The bacterium infects epithelial cells, and the presence of mature chlamydial inclusions has been described in bronchial epithelial cells (Yang et al., 1994). Successful invasion of eukaryotic cells by chlamydiae requires communication between the parasite and host cells. Because chlamydiae physically reside within membrane-bound inclusions during the entire developmental cycle, interactions between microorganism and host are mediated by chlamydial proteins which are either secreted into the eukaryotic cytoplasm or localized to the inclusion membrane. We have identified a novel C. pneumoniae-specific inclusion membrane protein CP0236 which contains domain(s) exposed to the eukaryotic cytoplasm where it interacts with host Act1.
Act1 or CIKS (connection to IKK and SAPK/JNK) is an adaptor protein that plays an important role in several cellular processes including stress response, inflammation, and protection from apoptosis. Act1 is able to form oligomers and has been shown to interact with NEMO/IKKγ where it participates in regulation of the IKK complex. Overexpression of Act1 leads to constitutive activation of NFκB and SAPK/JNK (Mauro et al., 2003; Leonardi et al., 2000; Li et al., 2000). Act1 forms a complex with IKK through its helix-loop-helix domain and leads to the activation of NFκB (Li et al., 2000). Furthermore, it has been demonstrated that endogenous Act1 is recruited to the CD40 receptor after stimulation with CD40L, mediating CD40L-induced NFκB activation and protection against CD40L-induced apoptosis in epithelial cells (Qian et al., 2002). In B lymphocytes, Act1 recruited to the CD40 receptor or the BAFF receptor through its interaction with TRAF3 negatively regulates B cell survival (Qian et al., 2004). We did not address whether C. pneumoniae infections impact TRAF-dependent pathways but predict that the capacity may exist. Interestingly, our yeast two-hybrid data indicated that a domain of Act1 spanning residues 301-360 was sufficient to interact with CP0236. This domain contains one of the two TRAF binding sites (residues 333-337) present in Act1 (Li, 2008). The role of Act1 in TRAF signaling is complex and alternative cascades are likely involved that lead to NFκB activation. Therefore, additional manifestations arising from the interaction of CP0236 with Act1 represent long-term investigations that are underway.
Importantly, Act1 has also been shown in epithelial cells to interact with the cytoplasmic portion of the IL-17 receptor through a SEFIR domain (Qian, et al., 2007). IL-17R is ubiquitously expressed by various cell types and its activation by IL-17 leads to production of inflammatory cytokines and recruitment of leukocytes, especially neutrophils, thus creating a link between innate and adaptive immunity. Although IL-17 has been connected to pathogenesis of several human and animal autoimmune diseases, this pro-inflammatory cytokine is also important in defense against several microorganisms that infect the lung, such as Klebsiella pneumoniae, Mycoplasma pneumoniae and Bordetella pertussis (Wu et al., 2007; Higgins et al., 2006; Happel et al., 2003). In mice infected with Klebsiella pneumoniae, TLR4 expression in the lung leads to production of IL-23 by DCs which stimulates CD4+ as well as CD8+ lymphocytes to release IL-17 (Happel et al., 2003). Similarly in Mycoplasma pneumoniae infected mice, infiltration of the lung by neutrophils is dependent on IL-23 induced upregulation of IL-17 (Wu et al., 2007). Interestingly, protection of mice by the pertussis vaccine requires production of IL-23, IL-1 and IL-17 though TLR4 present on APCs. IL-17 then activates killing of Bordetella pertussis by macrophages. It seems that a Th1 as well as Th17 responses are necessary for optimal immunity to Bordetella pertussis (Higgins et al., 2006). A variety of microbial products can lead to IL-17 production through both TLR-dependent but also through TLR-independent pathways (Hofstetter et al., 2006).
We report here that Act1 displayed an altered distribution in the cytoplasm of HeLa cells infected with C. pneumoniae where it was associated with the chlamydial inclusion membrane. Furthermore, we demonstrate that by sequestering Act1, chlamydiae inhibited recruitment of this signaling molecule to the IL-17RA in cells stimulated by IL-17A. Consequently, C. pneumoniae infected cells were protected from NFκB activation upon IL-17A induction. It is very likely that this impaired recruitment of Act1 in epithelial cells infected with C. pneumoniae affects more than just the IL-17 signaling pathway, but that remains to be determined. A variety of microbial products activate macrophages and DCs leading to production of chemokines and cytokines including IL-1β, IL-6 and IL-23, which are necessary in humans for Th17 differentiation and IL-17 production (reviewed in (Tesmer et al., 2008). IL-17 coordinates local tissue inflammation which results in recruitment of PMNs to the site of inflammation. It has been demonstrated that in mice infected with C. pneumoniae, significant numbers of PMN leukocytes infiltrate the bronchiolar lumen and interstitial and alveolar space (Yang et al., 1994). However, in studies using this animal model of infection, often 107 –108 of C. pneumoniae is administered per 4–6 week old mouse causing an acute pneumonia within 2 days post infection (Yang et al., 1993). In humans, however, C. pneumoniae causes an atypical pneumonia often leading to relatively mild illness where onset of symptoms takes several weeks, the course of infection is protracted and elevation of white blood cell counts is uncommon (Grayston et al., 1990). Thus, natural C. pneumoniae infections of humans are consistent with delayed inflammatory response and inhibition of NFκB signaling.
In the present study we have also confirmed a previous observation that productive infection of epithelial cells with chlamydiae does not lead to NFκB activation since no phosphorylation of IκBα was detected in C. pneumoniae or C. trachomatis infected cells. In fact, it seems that chlamydiae exploit NFκB-dependent pathways to regulate innate immune responses. All Chlamydia species possess a tail-specific protease (Tsp) which selectively cleaves the p65/RelA subunit of NFκB to potentially interfere with the host inflammatory response (Lad et al., 2007). Moreover, C. trachomatis, but not C. pneumoniae, express ChlaDub1 which binds to IκBα and impairs its ubiquitination and subsequent degradation in infected cells (Le Negrate et al., 2008). Based on C. pneumoniae transcriptome profiling (Maurer, et al., 2007), CP0236 expression occurs mid to late cycle. This is consistent with our RT-PCR analyses which indicate that de novo CP0236 expression occurs after 24 hrs p.i. (not shown). Interestingly, Tsp (Cpn0555) was also found to exhibit increasing expression toward the end of the C. pneumoniae developmental cycle (Maurer, et al., 2007). We could detect CP0236 protein via indirect immunofluorescence as early as 38 hrs p.i. but could not resolve whether the protein localized to the inclusion membrane due to the small size of inclusions (not shown). However, our observations that Act1 partitions to an insoluble fraction as early as 45 hrs p.i. suggests that CP0236 could interfere with Act1 activity for ca. the second half of the C. pneumoniae developmental cycle.
We described here a novel strategy employed by C. pneumoniae to accomplish inhibition of NFκB activation triggered by IL-17A through an interaction of CP0236 with host Act1 (Fig. 7). In addition, our data indicate that this impaired NFκB activation by chlamydiae depends on the activating stimulus as C. pneumoniae infection did not efficiently protect HeLa cells from NFκB activation upon TNFα induction. Concomitantly it seems evident that sequestering of Act1 by C. pneumoniae inclusions did not impair the interaction of Act1 with IKK complex as treatment of C. pneumoniae infected cells with TNFα resulted in IκBα phosphorylation. Since activation of the innate immune system is critical for host defense against bacteria, this Chlamydia-dependent disruption of innate immunity by interfering with NF-κB activation may significantly delay activation of adaptive immune responses. This may allow infections to proceed for a sufficient period of time to ensure transmission to new hosts.
Figure 7.

A working model for C. pneumoniae-mediated disruption of Act1-dependent signaling through the IL-17 receptor. In the absence of chlamydial infection (left), IL-17 stimulation of cells through plasma-membrane localized IL-17 receptors (IL-17RA, IL-17RC) recruits Act1, enabling activation of cytoplasmic IKK complex and subsequent degradation of IκBα. This leads to nuclear translocation of active NFκB. In the presence of chlamydial infection (right), Act1 is sequestered by the chlamydial inclusion and is unable to act as a signal transducer upon stimulation of the IL-17 receptor.
Materials and Methods
Cell culture and organisms
C. pneumoniae AR-39, purchased from American Type Culture Collection (Manassas, VA) was propagated in HeLa 229 cells as previously described (Wolf et al., 2000). Sub-confluent monolayers of HeLa cells were infected with density gradient purified C. pneumoniae AR-39 at an MOI of 2. HeLa cells were infected by incubation with chlamydial inoculum in a 37 °C incubator for 2 hrs with occasional agitation. Chlamydia-infected and mock-infected cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA) supplemented with 10% FBS and in the presence of 1 μg/ml cycloheximide for the first 24 hrs. No antibiotics were present in the culture medium after 24 hrs p.i. Cultures were incubated at 37°C in an atmosphere of 5% CO2/95% humidified air. Growth and purification of C. trachomatis, L2 was performed as previously described (Caldwell et al., 1981).
Antibodies
The coding sequence for the non-hydrophobic, C-terminal region (amino acids 101-279) of CP0236 was PCR amplified from C. pneumoniae, AR-39 genomic DNA and inserted into the donor vector pDONR221 creating a p0236 Entry Clone using Gateway Technology Kit (Invitrogen) according to the manufacturer’s instructions. The CP0236 insert was then transferred from the p0236 Entry Clone into the pDEST17 expression vector (Invitrogen) and expressed as a HIS-tagged fusion. The purified fusion protein was used as immunogen for production of specific polyclonal antibody in rabbits and mice by Proteintech Group, (Chicago, IL). The rabbit CP0236 antiserum as well as the rabbit pre-immune serum was purified using a Protein A Purification Kit (Sigma, St. Louis, MO) according to the manufacturer’s instructions. The rabbit polyclonal antibodies recognizing either C. pneumoniae, AR-39 or C. trachomatis, L2 were kindly provided by T. Hackstadt (Rocky Mountain Laboratories, Hamilton, MT). The anti-FLAG mAb and anti-Actin antibody were from Sigma. Anti-IL-17R mAb was purchased from R&D systems (Minneapolis, MN) and the anti-Act1 polyclonal antiserum from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-Phospho-IκB alpha (phosphorylated on serine residues 32 and 36) mAb was obtained from US biological (Swampscott, MA). The genus-specific monoclonal anti-Hsp60 antibody was a generous gift from H. Caldwell (Rocky Mountain Laboratories, Hamilton, MT).
Microscopy and microinjection
HeLa cells grown on 12 mm glass coveslips were infected with C. pneumoniae or C. trachomatis at an MOI of ~1. At different times post infection cells were fixed with methanol at room temperature for 3 min and processed for immuno-fluorescent staining. After incubation with appropriate primary antibody in PBS containing 3% BSA (Sigma) cells were stained with either AlexaFluor 546- or 488-conjugated anti-rabbit or anti-mouse IgG (Invitrogen). For microinjection experiments, coverslips with C. pneumoniae infected cells were transferred to a 100-cm diameter tissue culture dish containing complete medium. An Eppendorf FemtoJet/Transferman NK2 was used with Femtotips (Eppendorf, Westbury, NY) to microinject the cytoplasm of infected cells with purified rabbit antiserum recognizing CP0236 or pre-immune serum diluted in microinjection buffer (48 mM K2HPO4, 14 mM NaH2PO4, KH2PO4, pH 7.2). Microinjection was monitored on a Nikon TE2000 fluorescent microscope. Microinjected cells were then incubated for additional 3 hrs and then fixed with methanol and processed for staining with secondary antibody. Coverslips were then mounted on glass slides using Mowiol (http://altervista.org/confocal_protocols/mowiol.html). All fluorescent images were taken with a Zeiss Axiovert Zoom LSM 510 confocal microscope using a 63x, 1.4 NA objective.
Yeast two-hybrid assay
The CP0236 insert from p0236 Entry Clone was transferred into the GAL4 DBD destination vector pDEST32 according to the manufacturer’s instructions (Invitrogen) generating p0236-DBD. The MaV203 yeast strain was transformed with p0236-DBD plasmid using S.c. Easy Comp. Kit (Invitrogen) and competent yeast cells were then used to screen the pre-made HeLa cell cDNA library (Invitrogen) constructed in pPC86 GAL4 AD. Possible interacting clones were selected by the ability of transformants to grow on SC-TDO plates (without Leu, Trp, His, + 30 mM 3AT) or on SC-QDO (without Leu, Trp, His, Ura). Library plasmids were extracted with EZ Yeast Plasmid Miniprep (G-biosciences, St. Louis, MO) and processed for sequencing (Genewiz, South Plainfield, NJ). For domain interaction studies, the coding sequence for either the full-length Act1 (amino acids 1-565) or C-terminal regions of the gene containing residues 301-360 or 361-565 were PCR amplified from the HeLa cell cDNA library using primer sets IP2-Sense: 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGAACCGAAGCATTCCTGTGGAG-3′ and IP2-Antisense: 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTATCACAAGGGAACCACCTGAAGGGT-3′ (codons 1-565), IP2-60aa-Sense: 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGGAGCCAGTGTGAGAGGCCTG-3′ and IP2-60aa-Antisense: 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTACTCCAAGGACTCCCCAGGAGC-3′ (codons 301-360), or IP2-C-term-Sense: 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCTGCCCTGCAGAGCTGAGACCACAG-3′ and IP2-Antisense (codons 361-565). PCR products were inserted into the donor vector pDONR221 creating Entry Clones using Gateway Technology Kit. The generated chimeras were then transferred from Entry Clones to pDEST22 creating pAct1(1-565)-AD, pAct1(301-360)-AD and pAct1(361-565)-AD for two-hybrid studies.
Transfections
Coding sequence for the C-terminal region of CP0236 (101-279 aa) was PCR amplified from C. pneumoniae, AR-39 genomic DNA and ligated into p3XFLAG-CMV-10 expression vector (Sigma) generating 3xFLAG-CP0236. Full length act1 was PCR amplified from a HeLa cell cDNA library (Invitrogen) and ligated into the p3XFLAG-CMV-10 expression vector generating 3xFLAG-Act1. Sub-confluent monolayer of HeLa cells in 6 well plates was transfected with either 3xFLAG-CP0236 or 3xFLAG-Act1 using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The pORF-hIL-17R vector, purchased from InvivoGen (San Diego, CA), was used for transfection of mock and C. pneumoniae infected cells at 45 hrs p.i. using Lipofectamine LTX (Invitrogen) and cell cultures were incubated in RPMI 1640 supplemented with 5% FBS for additional 19 hrs.
Co-immunoprecipitation and Immunoblotting
C. pneumoniae and mock infected HeLa cells at 64 hrs p.i. were washed with cold PBS and lysed in immunoprecipitation buffer (Qian et al., 2002) containing 0.5% Triton-X-100, 20 mM Hepes (pH 7.4), 150 mM NaCl, 12.5 mM β–glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 2 mM DTT 2 mM EGTA, 1 mM sodium orthovanadate and protease inhibitors (Roche, Indianapolis, IN). In experiments where chlamydial protein synthesis was inhibited, C. pneumoniae infected cells were incubated in the presence of 200 μg/ml Chloramphenicol (Sigma). Pre-cleared cell lysates were incubated with either anti-Act1 or anti-IL-17R antibody overnight at 4°C followed by incubation with 50% slurry of protein A-sepharose (Sigma). After incubations, the protein A-bound immunocomplexes were washed with immunoprecipitation buffer and resuspended in Laemmli buffer (Laemmli, 1970). Protein extracts were separated by SDS-PAGE electrophoresis and blotted onto PVDF membrane (Millipore Corp., Bedford, MA) in Tris-Glycine buffer (25 mM Tris and 192 mM Glycine) containing 10% methanol. Membranes were blocked in PBS plus 5% non-fat dried milk for 1 h and incubated with primary antibody diluted in PBS-5% milk overnight at 4°C. Membranes were washed in 0.1% Tween 20 in PBS and incubated with either anti-mouse or anti-rabbit IgG secondary antibody conjugated to peroxidase (Sigma). Polypeptide bands were visualized by development with ECL Plus Western Blotting Detection Reagents (GE Healthcare, Piscataway, NJ) according to the manufacturer’s instructions.
IκBα phosphorylation experiments
IκBα phosphorylation was induced in mock and C. pneumoniae infected HeLa cultures at 64 hrs post infection by treatment with either 10 ng/ml of human TNF-α for 15 min or with 50 ng/ml of human IL-17A (R&D systems) for indicated time points. After induction, cells were washed with cold PBS and either lysed with immunoprecipitation buffer or harvested with hypotonic solution (50 mM Tris/pH 7.4/, 20 mM NaF, 5 mM MgCl2, 1 mM EGTA). Protein samples in hypotonic solution were concentrated by addition of trichloroacetic acid (Sigma) to 10% (v/v) and collected protein pellets were resuspended in Laemmli buffer (Laemmli, 1970).
Acknowledgments
We gratefully acknowledge the technical assistance of C. Linton and thank Drs. T. Hackstadt and H. Caldwell for the kind gift of Chlamydia-specific antibodies. We thank Drs. L. Plano and H. Betts for critical review of the manuscript. This work was supported by the Public Health Service Grant AI072126 from the National Institutes of Health to K.A.F.
Literature Cited
- Bannantine JP, Griffiths RS, Viratyosin W, Brown WJ, Rockey DD. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell Microbiol. 2000;2:35–47. doi: 10.1046/j.1462-5822.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun. 1981;31:1161–1176. doi: 10.1128/iai.31.3.1161-1176.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LA, Kuo CC. Chlamydia pneumoniae--an infectious risk factor for atherosclerosis? Nat Rev Microbiol. 2004;2:23–32. doi: 10.1038/nrmicro796. [DOI] [PubMed] [Google Scholar]
- Campbell LA, Kuo CC, Grayston JT. Chlamydia pneumoniae and cardiovascular disease. Emerg Infect Dis. 1998;4:571–579. doi: 10.3201/eid0404.980407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C, Rzomp KA, Tvinnereim A, Scidmore MA, Wizel B. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infect Immun. 2007;75:5586–5596. doi: 10.1128/IAI.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukuzumuremyi JM, Rosqvist R, Hallberg B, Akerstrom B, Wolf-Watz H, Schesser K. The Yersinia protein kinase A is a host factor inducible RhoA/Rac-binding virulence factor. J Biol Chem. 2000;275:35281–35290. doi: 10.1074/jbc.M003009200. [DOI] [PubMed] [Google Scholar]
- Grayston JT, Campbell LA, Kuo CC, Mordhorst CH, Saikku P, Thom DH, Wang SP. A new respiratory tract pathogen: Chlamydia pneumoniae strain TWAR. J Infect Dis. 1990;161:618–625. doi: 10.1093/infdis/161.4.618. [DOI] [PubMed] [Google Scholar]
- Hahn DL. Chlamydia pneumoniae, asthma, and COPD: what is the evidence? Ann Allergy Asthma Immunol. 1999;83:271–288. 291. doi: 10.1016/S1081-1206(10)62666-X. quiz 291–272. [DOI] [PubMed] [Google Scholar]
- Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun. 1996;64:796–809. doi: 10.1128/iai.64.3.796-809.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins SC, Jarnicki AG, Lavelle EC, Mills KH. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T cells. J Immunol. 2006;177:7980–7989. doi: 10.4049/jimmunol.177.11.7980. [DOI] [PubMed] [Google Scholar]
- Hofstetter HH, Luhder F, Toyka KV, Gold R. IL-17 production by thymocytes upon CD3 stimulation and costimulation with microbial factors. Cytokine. 2006;34:184–197. doi: 10.1016/j.cyto.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Jackson LA, Campbell LA, Grayston JT. Chlamydia pneumoniae (TWAR) Clin Microbiol Rev. 1995;8:451–461. doi: 10.1128/cmr.8.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lad SP, Li J, da Silva Correia J, Pan Q, Gadwal S, Ulevitch RJ, Li E. Cleavage of p65/RelA of the NF-kappaB pathway by Chlamydia. Proc Natl Acad Sci U S A. 2007;104:2933–2938. doi: 10.1073/pnas.0608393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Le Negrate G, Krieg A, Faustin B, Loeffler M, Godzik A, Krajewski S, Reed JC. ChlaDub1 of Chlamydia trachomatis suppresses NF-kappaB activation and inhibits IkappaBalpha ubiquitination and degradation. Cell Microbiol. 2008 doi: 10.1111/j.1462-5822.2008.01178.x. [DOI] [PubMed] [Google Scholar]
- Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc Natl Acad Sci U S A. 2000;97:10494–10499. doi: 10.1073/pnas.190245697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Act1 modulates autoimmunity through its dual functions in CD40L/BAFF and IL-17 signaling. Cytokine. 2008;41:105–113. doi: 10.1016/j.cyto.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Li X, Commane M, Nie H, Hua X, Chatterjee-Kishore M, Wald D, et al. Act1, an NF-kappa B-activating protein. Proc Natl Acad Sci U S A. 2000;97:10489–10493. doi: 10.1073/pnas.160265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chen C, Chen D, Wu Y, Zhong Y, Zhong G. Characterization of fifty putative inclusion membrane proteins encoded in the Chlamydia trachomatis genome. Infect Immun. 2008;76:2746–2757. doi: 10.1128/IAI.00010-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäurer AP, Mehlitz A, Mollenkopf HJ, Meyer TF. Gene expression profiles of Chlamydiophila pneumoniae during the developmental cycle and iron depletion-mediated persistence. PLoS Pathogens. 2007;3:752–769. doi: 10.1371/journal.ppat.0030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro C, Vito P, Mellone S, Pacifico F, Chariot A, Formisano S, Leonardi A. Role of the adaptor protein CIKS in the activation of the IKK complex. Biochem Biophys Res Commun. 2003;309:84–90. doi: 10.1016/s0006-291x(03)01532-8. [DOI] [PubMed] [Google Scholar]
- Qian Y, Zhao Z, Jiang Z, Li X. Role of NF kappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci U S A. 2002;99:9386–9391. doi: 10.1073/pnas.142294499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Qin J, Cui G, Naramura M, Snow EC, Ware CF, et al. Act1, a negative regulator in CD40- and BAFF-mediated B cell survival. Immunity. 2004;21:575–587. doi: 10.1016/j.immuni.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28:1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockey DD, Scidmore MA, Bannantine JP, Brown WJ. Proteins in the chlamydial inclusion membrane. Microbes Infect. 2002;4:333–340. doi: 10.1016/s1286-4579(02)01546-0. [DOI] [PubMed] [Google Scholar]
- Rockey DD, Grosenbach D, Hruby DE, Peacock MG, Heinzen RA, Hackstadt T. Chlamydia psittaci IncA is phosphorylated by the host cell and is exposed on the cytoplasmic face of the developing inclusion. Mol Microbiol. 1997;24:217–228. doi: 10.1046/j.1365-2958.1997.3371700.x. [DOI] [PubMed] [Google Scholar]
- Schramm N, Wyrick PB. Cytoskeletal requirements in Chlamydia trachomatis infection of host cells. Infect Immun. 1995;63:324–332. doi: 10.1128/iai.63.1.324-332.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore MA, Hackstadt T. Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol. 2001;39:1638–1650. doi: 10.1046/j.1365-2958.2001.02355.x. [DOI] [PubMed] [Google Scholar]
- Scidmore MA, Rockey DD, Fischer ER, Heinzen RA, Hackstadt T. Vesicular interactions of the Chlamydia trachomatis inclusion are determined by chlamydial early protein synthesis rather than route of entry. Infect Immun. 1996;64:5366–5372. doi: 10.1128/iai.64.12.5366-5372.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen F, Gaffen SL. Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine. 2008;41:92–104. doi: 10.1016/j.cyto.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- Wolf K, Hackstadt T. Sphingomyelin trafficking in Chlamydia pneumoniae-infected cells. Cell Microbiol. 2001;3:145–152. doi: 10.1046/j.1462-5822.2001.00098.x. [DOI] [PubMed] [Google Scholar]
- Wolf K, Fischer E, Hackstadt T. Ultrastructural analysis of developmental events in Chlamydia pneumoniae-infected cells. Infect Immun. 2000;68:2379–2385. doi: 10.1128/iai.68.4.2379-2385.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007;9:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZP, Kuo CC, Grayston JT. A mouse model of Chlamydia pneumoniae strain TWAR pneumonitis. Infect Immun. 1993;61:2037–2040. doi: 10.1128/iai.61.5.2037-2040.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZP, Cummings PK, Patton DL, Kuo CC. Ultrastructural lung pathology of experimental Chlamydia pneumoniae pneumonitis in mice. J Infect Dis. 1994;170:464–467. doi: 10.1093/infdis/170.2.464. [DOI] [PubMed] [Google Scholar]