

Abstract
Mutation-accumulation experiments are widely used to estimate parameters of spontaneous mutations affecting fitness. In many experiments only one component of fitness is measured. In a previous study involving the diploid yeast Saccharomyces cerevisiae, we measured the growth rate of 151 mutation-accumulation lines to estimate parameters of mutation. We found that an unexpectedly high frequency of fitness-altering mutations was beneficial. Here, we build upon our previous work by examining sporulation efficiency, spore viability, and haploid growth rate and find that these components of fitness also show a high frequency of beneficial mutations. We also examine whether mutation-acycumulation (MA) lines show any evidence of pleiotropy among accumulated mutations and find that, for most, there is none. However, MA lines that have zero fitness (i.e., lethality) for any one fitness component do show evidence for pleiotropy among accumulated mutations. We also report estimates of other parameters of mutation based on each component of fitness.
ADAPTATION can occur from standing genetic variation or from newly arising mutations. The relative importance of these two sources of adaptive mutations is affected by a variety of factors, including those that alter standing levels of genetic variation (see Barrett and Schluter 2008) and those that generate new mutations. Predicting how quickly a population will adapt and the type of beneficial mutations that will fuel that adaptation requires estimates of the additive genetic variance in fitness and of the beneficial mutation rate and the distribution of beneficial effects. While additive genetic variance for fitness has been estimated in a variety of organisms (Mousseau and Roff 1987), the beneficial mutation rate and the distribution of beneficial effects have only been estimated in a few studies (Shaw et al. 2002; Joseph and Hall 2004; Perfeito et al. 2007; Dickinson 2008; Hall et al. 2008). Surprisingly, these studies estimate that between 6 (Joseph and Hall 2004) and 50% (Shaw et al. 2002) of fitness-altering mutations are beneficial. In contrast, most mutation-accumulation (MA) experiments identify few, if any, beneficial mutations. Such wildly different estimates have even been generated from studies of the same species in similar environments (Zeyl and Devisser 2001; Joseph and Hall 2004; Dickinson 2008; Hall et al. 2008). If these estimates are correct, then they would suggest that the genotypes used in these experiments have vastly different evolutionary potential with respect to their capacity to exhibit rapid adaptation from new mutations.
A more likely scenario is that much of the variation in estimates of the beneficial mutation rate is due to methodological differences between studies. One possibility is the fitness component being analyzed. The beneficial mutation rate may be under- or overestimated if the fitness component is under stabilizing selection or subject to antagonistic pleiotropy. Analyses of mutation-accumulation data typically assume that selection is directional. As a result, analyses of phenotypes under stabilizing selection may falsely conclude that mutations that increase a phenotype are beneficial and mutations that lower values are deleterious (see Keightley and Lynch's 2003 criticism of Shaw et al. 2002). Alternatively, the beneficial mutation rate may be over- (or under) estimated if mutations increase fitness in regard to one component, but lower fitness in regard to lifetime fitness or another fitness component (i.e., antagonistic pleiotropy). Here, we explore these possibilities by investigating whether the high beneficial mutation rates estimated from our previous experiments are specific to the fitness component that we examined.
In two previous studies we accumulated mutations in 152 yeast, MA lines and used measures of their effects on diploid growth rate to estimate parameters of beneficial and deleterious mutations. In the first study we estimated that 6% of mutations accumulated during the first 1012 generations of accumulation improved diploid growth (Joseph and Hall 2004). To determine whether this high beneficial mutation rate was due to sampling error, we passaged the lines for an additional 1050 generations and found that 13% of mutations improved diploid growth (Hall et al. 2008). Similarly, another yeast MA experiment (Dickinson 2008) estimated an uncorrected frequency of beneficial mutations of 25%, although correction for within-colony selection reduces this estimate by approximately half. Together, these studies indicate that a substantial proportion of mutations accumulated in these yeast MA lines are beneficial for a single fitness component and that this observation cannot be explained by the chance sampling of a few beneficial mutations.
In this study we return to our yeast MA lines (Joseph and Hall 2004) and examine whether the high beneficial mutation rate that we estimated after 1012 generations is an artifact of the fitness component that we examined. To test this hypothesis we examined whether our MA lines carry mutations that are beneficial across multiple fitness components: diploid growth, sporulation efficiency, spore viability, and haploid growth rate. If our previous results are due to us analyzing a fitness component that is either subject to stabilizing selection or antagonistic pleiotropy, then mutations accumulated in our lines will be conditionally beneficial and analyses of additional fitness components would yield different estimates of the beneficial mutation rate. We found that three of the four fitness components yield high estimates of the beneficial mutation rate. This suggests that multiple MA lines have accumulated beneficial mutations and that the high beneficial mutation rate that we previously estimated is not an artifact of the fitness component that we examined.
Measuring multiple components of fitness also allowed us to examine the pleiotropic effects of beneficial and deleterious mutations. In general, we found that mutations altering one component of fitness have little effect on other components. However, lethal mutations were typically pleiotropic.
MATERIALS AND METHODS
Experimental overview:
A detailed description of the MA procedure can be found in Joseph and Hall (2004). Briefly, we established 152 genetically identical Saccharomyces cerevisiae lines from a diploid ancestor. The ancestor was derived from a haploid strain of genotype ade2, lys2–801, his3–Δ200, leu2–3.112, ura3–52, ho by transforming with a HO plasmid to induce diploidization, after which the plasmid was removed. The ancestor was thus homozygous at all loci except the mating-type locus, which was aα. The ade2 mutation was used to prevent the accumulation of mitochondrial petite mutations (Joseph and Hall 2004), which has been a problem in some previous yeast MA experiments (Korona 1999; Zeyl and Devisser 2001). MA lines were propagated independently via single-cell transfer on YPD solid medium (1% yeast extract, 2% peptone, 2% dextrose, and 2% agar) every 2 days for 100 days, for a total of 1012 cell generations.
After the 50th transfer, we measured the noncompetitive fitness of each diploid MA line and then we induced meiosis (sporulation) and measured sporulation efficiency. For those lines that were able to sporulate, we dissected eight tetrads onto solid medium, scored spore viability, and determined the growth rate of the haploid spores. An overview of the experiment and the components of fitness measured is shown in Figure 1. Figure 1 also lists the number of MA lines scored for each fitness component (see results).
Figure 1.—

Overview of experiment showing fitness components measured (in dashed boxes) and the number of lines that were measured for each component.
Diploid growth rate, sporulation efficiency, spore viability, and haploid growth rate measures were converted to relative fitness by standardizing to the ancestor. For each fitness component, individual MA lines were tested to determine if they were significantly different from the ancestor, using a Kruskal–Wallis test. For each fitness component, a maximum-likelihood (Keightley 1994; Keightley and Ohnishi 1998) and a Bateman–Mukai (Bateman 1959; Mukai 1964) analysis were used to estimate parameters of spontaneous mutations.
Diploid growth rate assays:
The diploid growth rate of each MA line was estimated by comparing its maximum growth rate in liquid medium to that of the ancestor. Maximum growth rates were estimated from optical density measurements obained using a Bioscreen C Microbiological Workstation (Thermo Labsystems). Ten replicates of diploid growth rate were estimated for each MA line. Further details of the diploid growth assay can be found in Joseph and Hall (2004).
Sporulation protocol:
We modified the standard sporulation and tetrad-dissection protocol (Burke et al. 2000) because our ancestral strain exhibited low sporulation efficiency and very low spore viability using that protocol. We streaked samples of each MA line and the ancestor from the freezer onto solid YPD medium and let them grow for 2 days at 30°. We then inoculated 2.5 ml of YPD cultures with individual colonies and incubated them overnight at 25°. The next day we centrifuged 500 μl of each overnight culture and resuspended the cells in water and transferred them into 2.5 ml of fresh, supplemented sporulation medium (1% potassium acetate, 1% yeast extract, 0.05% dextrose, 0.01% adenine sulfate, 0.003% lysine-HCl, 0.002% histidine HCl, 0.01% leucine, 0.002% uracil). These sporulation cultures were incubated for 7 days at 25°, which was enough time to reach maximum sporulation rate (data not shown).
Sporulation efficiency:
To measure sporulation efficiency we sporulated three replicate colonies from each MA line using the protocol outlined above. After incubating sporulation cultures for 7 days, two 5-μl samples of each replicate were used to estimate sporulation efficiency for that replicate. Each 5-μl sample was placed on a standard microscope slide and examined at 100× magnification. Using a 0.5 × 0.5-mm eyepiece grid to prevent double counting, at least 200 cells were counted and designated as good tetrads (four visible spores), aborted tetrads (three or two spores), or nontetrads. If <20 good tetrads were counted in the sample of 200 cells, additional cells were counted until at least 20 tetrads were recorded. For those cultures in which tetrads were not seen, at least 2000 cells were examined before the culture was determined to have not sporulated. Sporulation efficiency for each replicate was simply the number of good tetrads, divided by the total number of unsporulated cells, plus aborted tetrads, plus good tetrads counted in the two samples. Sporulation efficiency of a MA line was the average of the three replicate efficiencies. Some measures of sporulation efficiency include four-, three-, and two-spore asci (Codón et al. 1995). We chose to focus on four-spore tetrads because they appear to have undergone normal meiosis. Asci containing two or three spores are abnormal and it is not clear how to assign fitness to such asci without knowing how many of the spores were viable and haploid.
Tetrad dissection and spore viability:
After sporulation, the cells were washed by centrifuging 500 μl of the sporulation culture and resuspending them in sterile water. The cells were then centrifuged and resuspended in 45 μl of digestion solution [42.5 μl of a solution of 10 mm KPO4, 10 mm EDTA, 1 m sorbitol, plus 1 μl of 2-mercaptoethanol and 1.5 μl of Zymolase T100 (5 mg/ml)]. The resulting digestion mix was incubated at 30° for 5 min and then gently mixed with 700 μl of sterile, cold water and placed on ice. For each MA line a 3-μl drop of digestion mix was placed on a supplemented YPD plate (YPD + 0.002% adenine sulfate, 0.003% lysine-HCl, 0.002% histidine HCl, 0.01% leucine, 0.002% uracil) and a drop of the digestion mix of the ancestor was placed alongside. Eight tetrads from each MA line and two ancestor tetrads were dissected using a Nikon Eclipse E400 micromanipulator. The four spores from a tetrad were placed in 1 of 10 rows. The spores from the two ancestor tetrads on each plate were placed into the middle 2 rows of the 10-row array.
For all MA lines that sporulated and were dissected, spore viability was determined as the number of spores that grew into colonies. When spore viability was low, complete tetrads were rare or absent. If we did not obtain five complete (four-spore) tetrads from a dissection of a particular MA line, we repeated the sporulation and dissection protocol up to two additional times, unless spore viability was consistent with a haploid-lethal mutation.
Haploid growth:
Haploid growth rate was calculated for MA lines that produced at least five complete tetrads after no more than three dissection attempts. After 3 days of growth we took a digital photograph of the dissection plate with a 20-mm2 grid included for scale. We used NIH ImageJ to measure the area of each colony image on the dissection plate, using the grid to scale the photograph.
We then converted colony area (x) to number of cells (n), using the following relationship, which we determined experimentally:
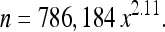 |
(1) |
To obtain Equation 1, we plated a haploid version of the ancestral strain at low density on several petri dishes and allowed growth for varying amounts of time (12–72 hr). We then took pictures of individual colonies, calculated their area using NIH ImageJ, and then scraped those colonies from the plate and used serial dilution and plating to count the number of cells that they contained.
The number of cells was used to determine the number of cell generations (g) as
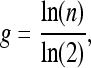 |
(2) |
which we used to calculate the rate of growth (r) in the 48 hr following dissection:
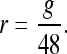 |
(3) |
We also measured the rate of growth for each ancestor colony and calculated an average for the ancestor on a plate. We computed the average relative growth rate of each colony ( ) by dividing its growth rate (r) by the average growth rate of the ancestor on the same plate (
) by dividing its growth rate (r) by the average growth rate of the ancestor on the same plate (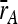 ):
):
 |
(4) |
We used the relative growth rates of the MA line colonies to calculate a measure of haploid fitness for each tetrad. In the simplest scenario, in which a MA line accumulated a single mutation that affected growth rate, a 2:2 pattern would be seen in the tetrad: two colonies would have growth rates equal to the ancestor (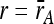 ) and two would have growth rates different from the ancestor [
) and two would have growth rates different from the ancestor [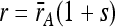 , where s is the selection coefficient of the mutation]. In that case the fitness of the mutant genotype (w = 1 + s) could be obtained as
, where s is the selection coefficient of the mutation]. In that case the fitness of the mutant genotype (w = 1 + s) could be obtained as
 |
(5) |
where 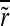 is the average relative growth rate of the four colonies in the tetrad.
is the average relative growth rate of the four colonies in the tetrad.
If no mutations were accumulated, E(w)=1. If a mutation was accumulated that caused an effect in haploids equal to s, then E(w)=1+s. Positive s implies an accumulated mutation is beneficial, while negative s implies it is deleterious. If more than one mutation is accumulated in a MA line, then w will be affected by both the additive and the epistatic effects of the mutations. A value of w > 1 would imply that the combined effects of mutations were beneficial, while a value <1 would indicate a combined deleterious effect.
A spore within a tetrad, or a complete tetrad, occasionally exhibited a growth pattern that was uncharacteristic of the MA line or ancestor. Because we use the ancestral spore colonies to standardize growth on a plate, slow-growing ancestral spores had to be discarded from the analysis. For the 147 dissection plates, 39 of 1176 ancestral spore colonies were discarded. For MA lines, any tetrad containing one or more spores showing an obviously unusual growth pattern relative to the other 7 tetrads of that line was discarded. For the 147 dissected lines, 7 tetrads of 1176 were discarded for this reason. This criterion identified growth rates <90% that of other spores on the same plate.
We found that the position on the dissection plate had an effect on colony size. Specifically, the first and last tetrads on the plate have no neighboring tetrad on one side and thus all four colonies tended to grow slightly faster than colonies in other tetrads. These tetrads cause w values to be >1. To remove this bias, we did not utilize the tetrad in the first or last position on a dissection plate when calculating w values. To obtain MA line fitness, we averaged w across those tetrads that we did utilize. MA line average fitness was thus calculated using three to six tetrads, depending on the number of four-spore tetrads obtained.
Statistical tests:
Statistical tests were performed using JMP statistical software (version 6.0; SAS Institute, Cary, NC). We tested whether particular MA lines were significantly different from the ancestor for diploid growth rate, sporulation efficiency, spore viability, and haploid growth rate using Kruskal–Wallis tests. We used this test because the MA line fitness distributions were not normally distributed (in all cases P < 0.00001, Shapiro–Wilks W) and for all measures but sporulation efficiency, the ancestor fitness distributions were not normally distributed (P < 0.01, Shapiro–Wilks W). We also determined whether the within-line coefficient of variation was significantly different from the ancestor variance (see Baer 2008) using a Levene's test, which is less sensitive to deviations from normality. To achieve the appropriate cutoff for the P-values, we corrected for multiple comparisons using a method that controls the false discovery rate (Benjamini and Hochberg 1995).
We also calculated pairwise correlations among the four fitness components. These analyses were performed using the PROC MIXED procedure implemented in SAS (version 9.1, SAS Institute). The mixed linear model allows among-line and within-line variances in fitness to differ between fitness components (using TYPE = UNR), thus giving an unbiased estimate of the correlation (Fry 2004). We tested whether each correlation was significantly different from 0, using the appropriate PARMS statement. We calculated these correlations twice: once using the replicate fitness measures for all MA lines and once excluding those lines that had zero fitness for at least one component of fitness.
Estimates of mutational parameters:
We used log likelihood to estimate the proportion of mutations that are beneficial (P), the genome-wide mutation rate to alleles that alter fitness (U), and the absolute value of the mean heterozygous fitness effect of mutations [E(hs or s)]. The maximum-likelihood (ML) estimates were calculated using a program provided by Peter Keightley (Keightley 1994; Keightley and Ohnishi 1998). The program estimates mutation parameters from the fitness values of the MA lines and the ancestor. The program assumes that the number of mutations accumulated in each MA line is Poisson distributed and that the effects of mutations follow a reflected gamma distribution with a fraction P of the mutations having positive (beneficial) effects. The positive and negative parts of the distribution are assumed to have the same scale parameter α and shape parameter β. The mean heterozygous or hemizygous fitness effect, E(hs or s), is equal to β/α.
We separately estimated mutation parameters from measures of the four fitness components. In all analyses, we used average fitness for each line rather than replicate fitness to reduce computer time. We also used the mean fitness of identically sized groups of ancestor replicates (see Joseph and Hall 2004).
For each data set, we performed a search of the parameter space by first choosing values of β and P and then running the program to find the ML values of α and U. After narrowing in on the region of the parameter space in which estimates of β, P, α, and U showed high likelihoods, we performed additional runs of the program in those regions to obtain more accurate estimates of the ML values of the parameters and their 2 log-unit support intervals and ran an equal-effects model for all values of P.
We also performed a Bateman–Mukai analysis to estimate the mutation rate and average effect (Bateman 1959; Mukai 1964). This analysis generates estimates from the change in mean fitness across all MA lines and the among-line variance. There are at least two major problems with this analysis. First, when there is variance in mutational effects, this method underestimates the genome-wide mutation rate and overestimates the average effect (Lynch et al. 1999). Second, the analysis does not allow estimation of the beneficial mutation rate. Despite these problems, we chose to include the Bateman–Mukai analysis because it is standard in MA experiments.
Correcting for selection:
The estimated number of mutations accumulated during our experiment could be affected by selection during colony growth. Our experimental design attempted to minimize the efficacy of selection by maintaining a small effective population size. Even so, deleterious and beneficial mutations are expected to be under- and overrepresented, respectively, in the MA lines relative to their occurrence. This is a problem common to all MA experiments and results in biased estimates of the parameters of mutation. In previous studies we utilized a method developed by Otto and Orive (1995) to correct our parameter estimates. However, this method allows us to correct estimates only of parameters that might have been altered by selection during mutation accumulation. It is not clear how to correct other estimates of mutational parameters for bias caused by selection. If all mutations have the same effect across all fitness components, then the same correction could be employed for all components. However, if the effects of mutations vary across the life cycle, then correcting for selection becomes problematic. For example, haploid-deleterious mutations that have no effect on diploid growth will not be underrepresented among MA lines, while those that do reduce diploid growth will be underrepresented. Given this issue, and coupled with the fact that correcting for selection alters estimates of the proportion beneficial less than twofold when selection coefficients (for diploid growth rate) are <0.1 (see Hall et al. 2008), we have chosen to report uncorrected parameter estimates. To the extent that mutations have pleiotropic effects of similar sign with diploid growth rate, deleterious mutations will be underrepresented and beneficial mutations will be overrepresented.
RESULTS
Generations and effective population size:
From colony size estimates, transfers occurred every 20.3 generations and the mutation-accumulation period was 1012 generations (Joseph and Hall 2004). The harmonic mean population size of our MA lines, which serves as an estimate of the effective population size, was 10.7 cells per line.
Diploid growth rate:
The fitness distributions of the MA lines and the ancestor are shown in Figure 2A and summary statistics are shown in Table 1. The mean diploid growth rate of the MA lines exhibited a small but significant reduction compared to the ancestor (Kruskal–Wallis, P = 0.0015). The within-line coefficient of variation (Baer 2008) was not significantly different (Levene's, P = 0.18) from the ancestor variance. One MA line could not be revived from the freezer, presumably because it had accumulated a cold-sensitive mutation (Hall et al. 2008). Of the 151 MA lines assayed, 19 exhibited a diploid growth rate significantly different from that of the ancestor. Of these, 4 were beneficial and 15 were deleterious (Table 2).
Figure 2.—

Fitness distributions of ancestor groups and MA lines at transfer 50. (A) Diploid liquid growth rate fitness for all 151 MA lines. (B) Sporulation efficiency for all 151 MA lines. (C) Spore viability for the 147 MA lines that sporulated. (D) Haploid solid growth rate fitness for the 130 MA lines that produced five complete tetrads.
TABLE 1.
Summary statistics of the distributions of MA lines and ancestor and estimates of per-generational mutational increase in genetic variance in fitness, σ2m, mutational heritability for fitness, h2m, and the mutational coefficient of variation, CVm
| No. of lines | μL | VT (×103) | VE (×103) | VL (×103) | σ2m (×107) | h2m (×104) | CVm (%) | μA | VA (×103) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Diploid growth rate | 151 | 0.994 | 1.4 | 0.7 | 0.8 | 3.7 | 5.8b | 0.061 | 1.0 | 0.8 |
| Sporulation efficiency (with zero sporulators) | 147 (151) | 0.864 (0.841) | 94.7 (112) | 42.4 (41.2) | 52.5 (70.5) | 259 (348) | 6.1 (8.5) | 0.006 (0.007) | 1.0 | 68 |
| Spore viability | 147 | 0.954 | 56.6 | 31.9 | 24.7 | 122 | 3.8 | 0.004 | 1.0 | 37.9 |
| Haploid growth rate (including 14 lethals) | 130 (144) | 0.988 (0.88) | 3.9 (95.5) | 0.4 (0.4) | 3.5 (95.8) | 17.4 (473) | 43.5 (1180) | 0.0013 (0.0078) | 1.0 | 0.08 |
μL, μA, VT, and VA are the means and (total) variances of the MA lines and the ancestor, respectively. VL and VE are the between- and within-line (error) variances for the MA lines. Data for diploid growth rate are from Joseph and Hall (2004). Haploid growth rate includes only those lines with at least five tetrads. For haploid growth rate only, the within-line coefficient of variation (Baer 2008) was significantly different (larger) from the ancestor variance (Levene's test, P < 0.0001).
TABLE 2.
The number of MA lines that are significantly different at the 5% level after correcting for multiple comparisons while controlling the false discovery rate (Benjamini and Hochberg 1995)
| No. MA lines | Beneficial (%) | Deleterious | |
|---|---|---|---|
| Diploid growth rate | 151 | 4 (21) | 15 |
| Sporulation efficiency | 151 | 2 (10) | 18 |
| Spore viability | 147 | 0 (0) | 15 |
| Haploid growth rate | 142 | 20 (29) | 49 (14 lethal) |
Sporulation efficiency:
The distributions of the MA lines and the ancestor are shown in Figure 2B. The mean sporulation efficiency of the MA lines was significantly smaller than that of the ancestor (Kruskal–Wallis, P = 0.01). The within-line coefficient of variation (Baer 2008) was not significantly different (Levene's, P = 0.08) from the ancestor variance. The ancestral strain exhibited low four-spore sporulation efficiency, with an average of 27%, and very high variation across replicates (Table 1). The substantial variation observed in our ancestor made it essentially impossible to distinguish lines that vary by only a few percent in their sporulation efficiency. Even so, 20 lines were significantly different from the ancestor (Table 2). Eighteen of these showed reduced sporulation (all with values <17% sporulation), 4 did not sporulate at all, and 2 showed significantly higher sporulation than the ancestor (39 and 45% sporulation).
Spore viability:
The distributions of the 147 MA lines that sporulated and the ancestor are shown in Figure 2C. The mean spore viability in ancestors was 91%. The mean relative spore viability of the MA lines was not significantly different from that of the ancestor (Kruskal–Wallis, P = 0.52). The within-line coefficient of variation (Baer 2008) was not significantly different (Levene's P = 0.10) from the ancestor variance. Fifteen MA lines had spore viabilities that were significantly different from the ancestor (Table 2) and had relative spore viabilities <55%. Of these 15 MA lines, 9 had a segregation pattern consistent with a single haploid-lethal allele, showing two viable and two inviable spores in each tetrad. Three had a pattern consistent with the segregation of both a lethal and an obviously deleterious allele. Two segregated a borderline lethal allele, with two good spore colonies in each tetrad and two either missing or extremely small. The remaining line showed low spore viability, although the pattern was not consistent with a fully penetrant, haploid-lethal allele. In summary, 14 of the 15 lines appear to have accumulated a lethal allele and were assigned a haploid growth rate of zero, and 1 of these 15 lines was not assigned a haploid growth rate.
Three other lines had low spore viability and were borderline significantly different from the ancestor (P = 0.07). After three dissections, we did not obtain five tetrads on a single plate for any of these lines. For one of them we were able to obtain four complete tetrads that were not in the first or last position on the dissection plate. For this line we were able to obtain an estimate of haploid solid growth rate. The other two lines were not assigned a spore viability or haploid growth rate. Thus, of the 147 MA lines that sporulated, haploid fitness could not be measured in 3 lines, was zero for 14 lines, and was nonzero for 130 lines (Figure 1).
No MA lines exhibited significantly higher spore viability than the ancestor. It may be that no mutation that increased spore viability arose in our accumulation lines. Alternatively, the fact that absolute spore viability is bounded between 0 and 1, and the ancestor is already close to the upper bound, implies that beneficial mutations could not increase spore viability very much. If a beneficial mutation that caused 100% spore viability was fixed in a line, we would have to dissect approximately three times more tetrads per line to conclude it significantly increased spore viability.
Haploid growth rate:
The distribution of the 144 MA lines that could be assigned a haploid growth rate and the ancestor are shown in Figure 2D. The mean haploid growth rate of the MA lines was significantly lower than that of the ancestor when lethal mutations were included (Kruskal–Wallis, P = 0.005) and nonsignificant when lethals are removed (Kruskal–Wallis, P = 0.14). The within-line coefficient of variation (Baer 2008) was significantly larger (Levene's P < 0.0001) than the ancestor variance. Sixty-nine lines were significantly different from the ancestor (Table 2). Of these, 20 had higher fitness and 49 had lower fitness.
Parameters of mutation from MA lines that differ from the ancestor:
Our ability to detect differences between a MA line and the ancestor varied with fitness component. We were able to detect mean differences of ∼2, 35, 17, and 1% for diploid growth, sporulation efficiency, spore viability, and haploid growth, respectively. We found that 84 of the 151 lines displayed evidence of mutation accumulation in the form of at least one component of fitness being significantly different from that of the ancestor (Kruskal–Wallis, α = 0.05). Of these, 20 showed fitness changes that were unconditionally beneficial: these lines show a significantly higher fitness for at least one component of fitness and no significant reduction for any other component, thus suggesting that 24% of accumulated mutations are beneficial. Five MA lines showed fitness changes that were antagonist across fitness components, and the remaining 59 lines showed fitness changes that were unconditionally deleterious: these lines show reduced fitness for at least one fitness component and no significant increase for any other.
In contrast, 67 MA lines escaped (detectable) mutation. Assuming all mutations that affect fitness are detectable, the probability of escaping a mutation is 67/151 = 0.44. To escape a mutation, a MA line must escape mutation at every cell division. The probability that a line escapes mutation for the duration of accumulation is simply (1 − 2U)1012, where U is the genome-wide, haploid mutation rate. We can use this relationship to obtain an estimate of the mutation rate to alleles that alter fitness of 3.9 × 10−4 per haploid genome per generation.
Parameters of mutation from likelihood analysis:
The likelihood program attempts to fit a gamma distribution of effects. For any particular component of fitness, inclusion of MA lines with zero fitness would cause the distribution of effects to have two fitness peaks: one representing zero fitness (lines that have accumulated lethal mutations) and the other representing lines with no lethal mutations. Since the program is not equipped to deal with such a distribution, we elected to remove lines with zero fitness from the likelihood analysis. This is not necessary for diploid growth rate, since a line with zero fitness for this component of fitness could not have accumulated in the experiment.
The diploid growth rate data were analyzed previously (Joseph and Hall 2004) and consist of 151 MA lines and 151 ancestor values. The sporulation efficiency data consist of 147 MA lines and 7 ancestor measures (the 4 MA lines that did not sporulate were not included). The spore viability data consist of 147 MA lines and 37 ancestor measures. The haploid growth rate data consist of 130 MA lines and 42 ancestor values.
The likelihood analyses of these data sets are summarized in Table 3A. For two components of fitness, sporulation efficiency and haploid growth rate, the likelihood continues to increase as the estimates for the genome-wide mutation rate increase and the average effect decreases. We are thus unable to give an estimate for these two parameters for these data sets. The result does imply that a model of many mutations of tiny effect best fits the sporulation efficiency and haploid growth rate data. The other components of fitness yield estimates of the per haploid genome mutation rate that are very similar to one another. Conversely, the estimates of average effect are quite different, in part because we are examining heterozygous effects in diploids and hemizygous effects in haploids. Alleles that are mildly deleterious in diploids might be extremely deleterious in haploids: this is readily seen in the haploid-lethal alleles that accumulated. However, the ML estimates suggest that haploid alleles have smaller effects (Table 2), but this analysis omits lethal alleles. MA lines with large deleterious effects for one fitness component may not be included in the likelihood analysis for later fitness components, which may alter the average effect estimates across stages. For example, the average effect for spore viability is high because haploid-lethal alleles are included in this measure. However, MA lines carrying these mutations are not included in the haploid growth rate likelihood analysis, which causes the average effect to be smaller. On the other hand, the average effect is small for diploid growth rate presumably because of masking of recessive mutations.
TABLE 3.
Estimates of parameters of mutation from four components of fitness, using likelihood (A) and Bateman–Mukai (B) analyses
| No. MA lines | U (×105) | E(hs or s) | P | |
|---|---|---|---|---|
| A. ML estimates | ||||
| Diploid growth rate | 151 | 6.3 (4.6–∞) | 0.061 (0–0.077) | 0.125 (0.008–0.380) |
| Sporulation efficiency without zero sporulators | 147 | → ∞ (25–∞) | → 0 (0–0.07) | 0.2 (0–0.45) |
| Spore viability | 147 | 5.05 (5.0–5.1) | 0.47 (0.44–0.52) | 0 (0–0.13) |
| Haploid growth rate without lethals | 130 | → ∞ (5.0–∞) | → 0 (0–0.13) | 0 (0–0.20) |
| B. Bateman–Mukai estimates | ||||
| Diploid growth rate | 151 | 2.4 | 0.125 | |
| Sporulation efficiency (without zero sporulators) | 151 (147) | 19.3 (22) | 0.70 (0.70) | |
| Spore viability | 147 | 17.2 | 0.282 | |
| Haploid growth rate (without lethals) | 144 (130) | 12.6 (3.7) | 0.79 (0.32) | |
U is the genome-wide mutation rate to alleles that alter fitness, E(hs or s) is the average effect of a mutation on a diploid (hs) or haploid (s) fitness component. For the likelihood analyses, zero sporulators and haploid lethals are omitted because they cause the effect distribution to have two peaks, which violates the assumption of a gamma distribution of effects.
Perhaps of most interest is how the proportion of mutations that are beneficial changes across the fitness components. For both diploid growth rate and sporulation efficiency, the frequency of mutations that are beneficial is estimated to be reasonably high (uncorrected estimates are 12.5 and 20%, respectively). However, for spore viability and haploid growth rate, the frequency of beneficial mutations is estimated to be zero, although the confidence intervals are broad. It is not surprising that the ML analysis of spore viability did not identify beneficial mutations given that the direct estimation procedure also failed to identify beneficial mutations that alter spore viability (Table 2). In contrast, the zero estimate generated from measures of haploid growth rate is in stark disagreement with the high estimate obtained from the direct estimation procedure (Table 2). This discrepancy may be due to differences in the effect sizes of beneficial and deleterious mutations altering haploid growth—which is a violation of one of the main assumptions of the model used for this likelihood analysis. If deleterious mutations tend to have larger effect sizes, and are more common, they will tend to dominate the likelihood. In this situation, a fitted gamma distribution with large mean effect will be a good fit for the lines carrying deleterious mutations, but not for the lines carrying beneficial mutations (of small effect). Thus, the likelihood will be greatest when the proportion of beneficials is zero. The fact that deleterious mutations do not dominate the likelihood analysis of diploid growth may be because their effects are partially masked when heterozygous.
Parameters of mutation from Bateman–Mukai analysis:
The Bateman–Mukai analyses give smaller estimates of the genome-wide mutation rates and larger estimates of the average effect of a mutation for all measures except spore viability. This indicates that there is variance in mutational effects across fitness components (Lynch et al. 1999). Results of these analyses are shown in Table 3B.
Pleiotropy:
Single mutations that affect more than one component of fitness are pleiotropic. We identified 32 lines that had at least two components of fitness that were significantly different from that of the ancestor. If such lines carry mutations that are positively pleiotropic (i.e., have similar effects on multiple fitness components), then those fitness components should be positively correlated. When we analyze all MA lines we find that, after correcting for multiple comparisons, there is a significant (α = 0.05), positive, among-line correlation for every pair of fitness components (Table 4).
TABLE 4.
Correlations among fitness components calculated using estimates of within- and between-line variance in fitness
| Sporulation efficiency (no. lines) | Spore viability (no. lines) | Haploid growth (no. lines) | |
|---|---|---|---|
| Diploid growth | 0.45*** (151) | 0.49*** (147) | 0.47*** (144) |
| 0.11 (133) | 0.20 (133) | 0.23 (130) | |
| Sporulation efficiency | 0.2495* (147) | 0.2568** (144) | |
| 0.09 (133) | 0.26 (130) | ||
| Spore viability | 0.9834*** (144) | ||
| 0.14 (130) |
The top values are correlations including all MA lines, and the bottom values exclude those MA lines that exhibit zero fitness for at least one fitness measure. The number of MA lines in each correlation is shown. Numbers in boldface type are significant (α = 0.05) after correcting for multiple comparisons. Only when the zero fitness lines are included in the analysis is there a significant correlation between two fitness components. *P < 0.05, **P < 0.01, ***P < 0.001.
This correlation can be explained either by mutations having pleiotropic effects or by fitness components not being independent. If this latter explanation is correct, then we are actually measuring the same fitness component in different ways. This likely explains the high correlation that we observe between haploid growth rate and spore viability (0.98). The lack of independence between these traits may arise because 14 of the 15 lines that had significantly different spore viability contained a haploid-lethal mutation and because we define haploid-lethal mutations as reducing spore viability by 50% and haploid growth by 100%. As a result, our analyses were set up so that most mutations affecting spore viability had very similar effects on haploid growth rate.
To further examine the independence of traits we used the correlation among traits to calculate the effective number of traits, Neff (Wagner et al. 2008). When an independent trait is incorporated into this analysis, the effective number of traits will increase by ∼1, but addition of a dependant trait will not alter the effective number of traits. This allows us to estimate the number and identity of independent traits.
The effective number of traits is calculated from the variance in the eigenvalues of the correlation matrix, Var(λ), using
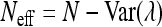 |
We calculated the effective number of traits for all combinations of fitness components measured. Adding haploid growth when spore viability had already been included did not change the effective number of traits. Likewise, adding spore viability when haploid growth had already been included did not change the effective number of traits. This indicates that the high correlation between spore viability and haploid growth is due to their dependence—not pleiotropy. In contrast, when any of the other traits were added to the correlation matrix, the effective number of traits increased by ≥0.66, indicating that the other fitness components are independent and therefore correlations between them are due to pleiotropy.
If the mutations accumulated in our study are highly pleiotropic, we should observe an excess of MA lines having effects for multiple fitness components, relative to the number expected to be caused by multiple, independent mutations. To examine this prediction, we used the genome-wide mutation rate, calculated from the total number of MA lines that did not differ from the ancestor (see previous section), to calculate the expected number of MA lines that should be significantly different for one, two, or three fitness components, assuming no pleiotropy. This expectation is based solely on the probability of a line carrying multiple independent mutations, each affecting a different fitness component. We did not include spore viability because of its dependence on haploid growth. We found a significant difference between the number of lines expected (LE) and observed (LO) to differ from the ancestor in regard to one (LE = 62.5, LO = 58), two (LE = 18.7, LO =22), or three (LE = 1.3, LO = 4) fitness components (χ2 = 6.3, d.f. = 2, P = 0.04). While this finding suggests that some of the mutations accumulated in our study are pleiotropic, it does not reveal whether both deleterious and beneficial mutations are pleiotropic. To determine whether the difference between observed and expected holds when lines are parsed by the effect (beneficial or deleterious) of their altered fitness component, we separately compared the observed and expected number of lines in each category. The nine categories are +, −, + +, + −, − −, + + +, + + −, + − −, and − − −, where the number of pluses or minuses represents the number of affected fitness components and plus (minus) indicates significantly higher (lower) fitness than the ancestor. The expected number of lines in each category depends on both the mutation rate (as above) and the probability that a mutation is beneficial. For example, for lines that are expected to differ from the ancestor for two fitness components, then p2, 2p(1 − p), and (1 − p)2 are expected to be in the + +, + −, and − − categories, where p is the probability that a mutation is beneficial. For values of the proportion beneficial in the range 0–0.3, which covers the range of our estimates, we found a significant difference between the expected and the observed number of lines in each category (χ2 > 24, d.f. = 8, P < 0.005). This suggests that both the beneficial and the deleterious mutations accumulated in our experiment have pleiotropic effects. However, we also noted that lines that differ from the ancestor in regard to three fitness components (e.g., triplets) made a large contribution to the high value of the chi-square statistic.
The fact that only 1.3 of 151 MA lines are predicted to be triplets means that the results described above could be false positives due to the chance accumulation of a few more triplets than expected. To examine this possibility, we repeated the above comparisons, but excluded triplets. We found that there was not a significant difference between the numbers of lines expected and observed to differ from the ancestor in regard to one or two fitness components (Fisher's exact test, P = 0.3). However, when we parsed lines by effect, beneficial or deleterious as before, we found borderline evidence for a difference between the observed and the expected number of lines in each category (χ2 = 8.9, d.f. = 4, P = 0.06). In this comparison, lines that were deleterious for two components displayed an especially large difference between the observation and expectation (observed = 16, expected ≤ 8.4). While excluding triplets helped us avoid false positives (type I error), it reduced our ability to detect pleiotropy (type II error) because it removed the most pleiotropic mutations (triplets) in our study. This information, along with the correlations that we observe between fitness components, strongly suggests that our lines have accumulated deleterious mutations with pleiotropic effects.
During data analysis, we noted that the 18 lines that had zero fitness for one fitness component seemed to often be affected for a second component of fitness. To examine whether these 18 lines are the primary source of pleiotropy in our data set, we removed them and repeated the analysis described above. We found that with these lines removed, after correcting for multiple comparisons, none of the among-line correlations for pairs of traits is significant (Table 4). There is also no evidence for a difference between the observed vs. expected number of lines exhibiting significant differences for one, two, or three fitness components (χ2 = 0.04, d.f. = 2, P = 1), even when we include whether the fitness effect was beneficial or deleterious (χ2 < 8.7, d.f. = 8, P > 0.4). Again ignoring spore viability, for these 18 lines, 6 are affected for one component of fitness and 12 for two or three components, which is significantly different from the remaining 66 affected lines, where 52 are affected for one component and 14 are affected for two or three (Fisher's exact test, P = 0.0005).
We have been considering haploid and diploid growth as different fitness traits. An alternate view is that haploid growth is equivalent to homozygous diploid growth. Similarities between haploid and diploid growth would then be due to degree of dominance rather than pleiotropy. We do not favor this interpretation for two reasons. First, there are substantial differences between the assays that we use to measure these two growth traits: haploid growth was measured on solid YPD medium supplemented with adeinine, lysine, histidine, leucine, and uracil while diploid growth was measured in liquid YPD medium (materials and methods). These differences suggest that haploid and diploid growth rate will likely be influenced by different physiological processes, implying they are not equivalent. Second, the variance in eigenvalues of the correlation matrix (see above) indicates that these two traits are essentially independent (Neff = 1.78). This is not expected if haploid and diploid growth are equivalent unless the dominance coefficients of accumulated mutations are usually close to zero, which would result in little or no growth effects in heterozygous diploids. While this is possible, previous work in yeast (Phadnis and Fry 2005) indicates that dominance coefficients tend not to be small when selection coefficients are small. Since the vast majority of our selection coefficients for growth rate are small, <0.1 (Figure 2, A and D), small dominance values are not a likely explanation.
In summary, MA lines that accumulate a mutation that is lethal for one component of fitness are the only lines that show strong evidence of pleiotropy. Other MA lines that affect more than one component of fitness are best explained by having accumulated two, or more, independent fitness-altering mutations.
Lethal mutations:
There are 18 MA lines that have accumulated a mutation that is essentially lethal at some point in the life cycle and 3 nearly lethal lines. We observe that 4 of these lines have zero sporulation efficiency that is equivalent to a dominant lethal for sexual fitness. In contrast, 14 show a pattern of spore viability that is consistent with a segregating haploid-lethal allele, such that only half the spores within a tetrad survive. Three other lines show substantially reduced spore viability, although the pattern is not consistent with a fully penetrant, haploid-lethal allele. The greatly reduced, but nonlethal effects seen in these lines could be due to a haploid-expressed, variably penetrant lethal allele or to a diploid-expressed, low-spore viability phenotype with variable expressivity. The number of lines having accumulated a lethal is thus between 18 and 21.
Given that 18–21 lines accumulated a lethal allele, then between 130 and 133 lines escaped a lethal mutation. The probability that a MA line escapes lethal mutation [= (130 or 133)/151] for the duration of accumulation is given by (1 − 2UL)1012, where UL is the haploid-lethal mutation rate (and 1012 is the number of cell generations of accumulation). We can use this relationship to obtain an estimate of the haploid-lethal mutation rate as 6.3–7.4 × 10−5 mutations per haploid genome per generation.
As discussed in the previous section, we find that lethal mutations often show evidence for pleiotropy. Ten of the 18 MA lines that have accumulated a lethal allele have a significantly different diploid growth rate fitness: 9 have lower fitness and 1 has higher. These represent 60% (9 of 15) and 25% (1 of 4) of the lines showing reduced and increased diploid fitness, respectively.
DISCUSSION
The complex relationship between mutational parameters and adaptation, coupled with the lack of estimates of these parameters, makes it extremely difficult to predict how quickly a population will adapt from newly arising beneficial mutations. One might expect higher beneficial mutation rates to speed adaptation, but theory predicts this relationship to be much more complicated. When the beneficial mutation rate is low, the basic Fisher–Muller model of adaptation predicts that asexuals will adapt by a series of sequential sweeps in which a beneficial mutation occurs and is either lost by drift or swept to fixation (Fisher 1930; Muller 1932). In contrast, if the beneficial mutation rate is moderately large, populations may contain multiple beneficial mutations and competition between these mutations may slow adaptation by reducing the substitution rate (i.e., clonal interference) (Gerrish and Lenski 1998). If, however, the beneficial mutation rate is extremely large, the effects of clonal interference may be reduced due to the accumulation of multiple, beneficial mutations in the same genetic background (Kim and Orr 2005). The nonlinear relationship that these theories predict between the beneficial mutation rate and adaptation is further complicated if higher beneficial mutation rates are accompanied by higher deleterious mutation rates (Orr 2000).
While great progress has been made in generating theoretic predictions about the relationships between mutational parameters and adaptation, few empirical estimates of these parameters remain. This is not surprising given how difficult they are to obtain. This difficulty stems largely from the fact that experiments that minimize the effects of natural selection generally do not accumulate sufficient numbers of beneficial mutations from which to estimate these parameters (but see Shaw et al. 2002; Joseph and Hall 2004; Perfeito et al. 2007; Dickinson 2008; Hall et al. 2008) and experiments with efficient natural selection accumulate mutations with a biased distribution of effects.
Here, we return to our yeast MA lines (Joseph and Hall 2004; Hall et al. 2008) and examine how analyses of different fitness components affect estimates of the parameters of beneficial and deleterious mutations. Despite the fact that our MA experiments used an experimental design that greatly reduced the efficiency of selection and used an analytical procedure that corrected for the effects of selection, we previously estimated that 6% (Joseph and Hall 2004) and 13% (Hall et al. 2008) of mutations accumulated during these experiments are beneficial. In the current study we examine whether these extremely high estimates of the proportion of beneficial mutations are peculiar to the fitness component examined. We find that three of the four fitness components show evidence of a high frequency of beneficials on the basis of direct estimation (i.e., using the Kruskal–Wallis test; Table 2) and two yield large, maximum-likelihood estimates of the proportion of beneficial mutations (Table 3). As a result, we conclude that our MA lines accumulated a large proportion of beneficial mutations and that this is not an erroneous finding based on analyses of a single fitness component.
Variation in mutational effects across fitness components:
In this study we compare the estimates of mutational parameters generated from measurements of four fitness components: diploid growth rate, sporulation efficiency, spore viability, and haploid growth rate. We find that the analyses of different fitness components, as well as the use of different estimation procedures (direct estimation from the number of significantly different lines vs. maximum likelihood vs. Bateman–Mukai), yielded radically different parameter estimates. For example, ML estimates from diploid growth and spore viability suggest that the mutation rate is quite low and that mutations have moderate to very large mean effects, while ML estimates from sporulation efficiency and haploid growth indicate that the mutation rate is infinitely large, with mutations having infinitesimal effects (Table 3A). Further, the Bateman–Mukai estimates for all fitness components indicate that the mutation rate is moderately large and that mutations have very large effects (Table 3B).
There are at least three potential causes for the discrepancies in the estimates of mutation rate and effect across fitness components. First is the inherent difficulty in distinguishing many mutations of small effect from fewer mutations of larger effect (Keightley 1998). For three of the fitness measures, the confidence intervals are broadly overlapping and include an infinite mutation rate and infinitesimal effect size (Table 3A). While our analyses cannot rule out the possibility that our yeast strain has an extremely large mutation rate, sequence data from a recent study (Lynch et al. 2008) suggest that the haploid genome-wide mutation rate in yeast is between 0.03 and 0.32. Assuming that the vast majority (99–99.9%, Lynch et al. 2008) of mutations do not affect fitness at a level that can be measured in the lab, the results of this study place an upper bound on the mutation rate to alleles that alter fitness and bring our estimates across fitness components closer together. Second is the difference between measuring diploid fitness in which accumulated mutations are heterozygous, such that dominance may mask their effects, and measuring haploid fitness in which recessive mutations are not masked. Third, our data violate assumptions of the ML analysis if beneficial and deleterious have different effect sizes (see results). Despite these discrepancies, we are able to make the following conclusions:
A large proportion of the mutations accumulated in our MA lines are beneficial. This conclusion is supported by parameter values estimated from multiple fitness components, using a variety of estimation procedures. We reason that if many beneficial mutations have accumulated in our MA lines, then those mutations will be reflected in the parameters estimated from different fitness components. Consistent with this prediction, three components yielded large estimates (0.10, 0.21, and 0.29) of proportion of the beneficial mutations using the direct estimation procedure (Table 2), and two fitness components yielded large estimates (0.125 and 0.20) using the likelihood analysis (Table 3A). We therefore conclude that a large proportion of mutations accumulated in our lines are, in fact, beneficial.
Pleiotropy seems to be restricted to lethal mutations. In our experiment, most nonlethal mutations are not pleiotropic, and the few putative examples of pleiotropy are best explained by the accumulation of multiple independent mutations in the same MA line. In contrast, 12 of 18 lethal mutations were pleiotropic, 2 of which displayed antagonistic pleiotropy. Both of the mutations displaying antagonistic pleiotropy were lethal for haploid growth, but improved a diploid fitness measure: either diploid growth rate or sporulation efficiency. It should be noted that our methodology for estimating the degree of pleiotropy among fitness components is conservative because we assumed that a MA line that is not significantly different from the ancestor was in fact not different from the ancestor.
The complex life history of yeast may allow antagonistically pleiotropic mutations to play a major role in yeast evolution. One way of generating antagonistic pleiotropy is for mutations to be beneficial when heterozygous, but deleterious when homo- or hemizygous (overdominance). Like the pattern observed in our study, these overdominant mutations would be beneficial in the diploid phase and deleterious in the haploid phase. Alternatively, adaptation to the diploid phase may pose inherent trade-offs with haploid fitness. Regardless of its source, antagonistic pleiotropy across life-history stages may be extremely important in natural populations where yeast tend to be diploid asexuals that occasionally undergo sexual reproduction. In these populations a mutation that improves fitness in the diploid/asexual phase could reach a high frequency even if it is highly deleterious in the sexual phase.
Factors influencing the proportion of beneficial mutations:
The large proportion of beneficial mutations estimated by this and our two previous studies (Joseph and Hall 2004; Hall et al. 2008) contradicts the commonly held belief that the vast majority of mutations are deleterious (Keightley and Lynch 2003). We previously proposed the following explanations for these observations:
The ancestral strain used to establish our MA lines may be far from its fitness optimum. This is consistent with Fisher's geometric model, which predicts that the farther a genotype is from the fitness optimum, the larger the proportion of beneficial mutations (Fisher 1930).
The mutations accumulated in our lines may be deleterious or neutral in some environments or for some life-history traits, but beneficial in our diploid fitness assay.
Deleterious mutations may be more recessive than beneficial mutations, causing many of the deleterious mutations accumulated in our MA lines to be masked in the diploid fitness assay, while the beneficial mutations would have been expressed.
By examining differences among our MA lines with respect to multiple components of fitness from across the yeast life cycle, we are able conclude that our large estimates of the proportion of beneficial mutations are not peculiar to the fitness component that we analyzed, but we cannot rule out the possibility that these elevated estimates are in part due to the ancestor being far from its fitness optimum or our lines accumulating recessive, deleterious mutations. In fact, our observation that many more lines displayed low haploid growth than low diploid growth (49 and 15, respectively; Table 2) strongly suggests that the accumulation of recessive deleterious mutations may have inflated our previous estimate of the proportion of beneficial mutations. In addition, it is possible that all of our apparently beneficial mutations might be deleterious in another environment, or for another unmeasured, but important, component of fitness (ethanol tolerance or spore resistance to dessication, for example).
Lethal mutations:
Lethal mutations may be critically important in evolution. In particular, they may make a large contribution to inbreeding depression and genetic load if they tend to be recessive. Lethal mutations that have little or no deleterious effect in the heterozygous state are effectively neutral when rare in sexual diploids, unless they are expressed during the gametic phase of the life cycle (Joseph and Kirkpatrick 2004). As a result, recessive lethals could drift to intermediate frequency and then make important contributions to inbreeding depression and load. Lethal mutations that are actually beneficial when heterozygous, as we observed in two cases, could make even larger contributions to inbreeding depression and load because they will be driven to high frequencies by selection.
Lethal mutations are often overlooked in MA experiments. The reason is a technical one: in many experiments, mutations are accumulated in haploids or in inbred diploids, which quickly become homozygous. Thus, lethal alleles are exposed to selection, even in the MA framework. Our experimental protocol, like experiments in Drosophila that accumulate mutations using a balancer chromosome (reviewed by Simmons and Crow 1977), allows mutations to accumulate in the heterozygous state. Thus, lethal mutations can accumulate as long as they are not too dominant.
We estimate the haploid-lethal mutation rate as 6.3–7.4 × 10−5 mutations per haploid genome per generation. One other MA study (Wloch et al. 2001) has also examined the lethal mutation rate in yeast. In this study, Wloch et al. (2001) found 20 lethal mutations in 508 lines, examined after 64 generations of diploid accumulation. Using the same calculation as above, we estimate that that the lethal mutation rate in their study was 3.1 × 10−4 mutations per haploid genome per cell generation. This is four times larger than the lethal mutation rate in our study and would have resulted in our MA lines accumulating ∼70 lethal mutations. The actual number of lethal mutations observed in our experiment was significantly lower (Poisson, P < 10−10), thus suggesting that the two studies have very different lethal mutations rates. The true difference between these rates may be even larger because our estimate is based on the rate of haploid- and diploid-lethal (zero sporulation) mutations, while theirs is based solely on haploid-lethal mutations. Differences between these estimates may reflect differences between the strains used in the two experiments and/or it may indicate that their lethal mutations are not independent of one another. Their MA protocol involved generating replicate colonies after 34 generations of growth as a single colony. If a mutation occurred during the first 34 generations, it would be present in several clones and would thus be overrepresented relative to its true mutation frequency.
Like Wloch et al. (2001), we find that lethal mutations affecting haploid-growth rate generally do not alter growth of heterozygous diploids. Eight of 14 MA lines with these lethals do not have significantly different diploid growth rates. Of the 6 that do, 1 shows increased diploid growth rate and the other 5 show a mean decrease in fitness of 7%. Assuming that haploids and homozygous diploids show similar growth, which has been observed for several isogenic strains (D. W. Hall, personal observation), our data suggest that lethal mutations are partially or fully recessive. It is possible that there is a class of lethal alleles that tend to be additive or dominant and would thus be underrepresented or missed completely in either experiment because of selection during colony growth. Such lethals are of little importance in natural populations, because they will be immediately removed by selection in all but the smallest populations. Alternatively, the recessivity of lethal mutations may be explained by the hyperbolic relationship between enzyme concentration and flux through a metabolic pathway (Kacser and Burns 1981). If (as predicted) enzymatic flux of the wild-type homozygote is on the plateau of this curve, then a heterozygous mutation that substantially reduces enzyme concentration will lower flux by only a small amount, thus making the mutation recessive. While designed to explain the recessive nature of enzymatic mutations, this theory may also explain the recessivity of nonenzymatic mutations (Kacser and Burns 1981; Phadnis and Fry 2005). Thus, both natural selection and the physiological theory may explain the recessive nature of lethal mutations accumulated in our study and in Wloch et al. (2001).
If the percentage of fitness-altering mutations with recessive, haploid-lethal effects is as high as observed in our (12–20%) or Wloch et al.'s (30%) experiment, then we expect that they will be at reasonably high frequencies in populations. The few studies that have estimated the number of segregating lethals in a population have observed only one or two per individual (Halligan and Keightley 2003)—much lower than our study would predict. This discrepancy suggests that either the frequency of recessive lethals is much higher in yeast than in other organisms or lethals are efficiently removed from populations via haploid selection (Joseph and Kirkpatrick 2004) and/or inbreeding. Further research is clearly needed.
Conclusions:
We find that for three of four fitness components examined, a high frequency of spontaneous, fitness-altering mutations in diploid yeast is beneficial. Further, we do not detect pleiotropy of small-effect mutations, while lethal mutations show high levels of pleiotropy. In most cases, pleiotropy is positive. Two lines show evidence of antagonistic pleiotropy, indicating trade-offs, although heterozygote advantage cannot be ruled out.
Acknowledgments
We thank three anonymous reviewers for comments that substantially improved the manuscript. We also thank Mark Kirkpatrick for laboratory space and guidance. This work was supported in part by National Science Foundation, dissertation improvement grant DEB-0309372 (to S.B.J.), and by The University of Georgia Research Foundation (D.W.H.).
References
- Baer, C. F., 2008. Quantifying the decanalizing effects of spontaneous mutations in rhabditid nematodes. Am. Nat. 172 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, R. D. H., and D. Schluter, 2008. Adaptation from standing genetic variation. TREE 23 38–44. [DOI] [PubMed] [Google Scholar]
- Bateman, K. G., 1959. The viability of near-normal irradiated chromosomes. Int. J. Radiat. Biol. 2 170–180. [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57 289–300. [Google Scholar]
- Burke, D., D. Dawson and T. Stearns, 2000. Methods in Yeast Genetics (A Cold Spring Harbor Laboratory Manual). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Codón, A. C., J. M. Gasent-Ramírez and T. Benítez, 1995. Factors which affect the frequency of sporulation and tetrad formation in Saccharomyces cerevisiae baker's yeasts. Appl. Environ. Microbiol. 61 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson, W. J., 2008. Synergistic fitness interactions and a high frequency of beneficial changes among mutations accumulated under relaxed selection in Saccharomyces cerevisiae. Genetics 178 1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A., 1930. The Genetical Theory of Natural Selection. Oxford University Press, Oxford.
- Fry, J. D., 2004. Estimation of genetic variances and covariances by restricted maximum likelihood using proc mixed, pp. 11–34 in Genetic Analysis of Complex Traits Using SAS, edited by A. M. Saxton. SAS Institute, Cary, NC.
- Gerrish, P. J., and R. E. Lenski, 1998. The fate of competing beneficial mutations in an asexual population. Genetica 103 127–144. [PubMed] [Google Scholar]
- Hall, D. W., R. Mahmoudizad, A. Hurd and S. B. Joseph, 2008. Spontaneous mutations in diploid Saccharomyces cerevisiae: another thousand cell generations. Genet. Res. 90 229–241. [DOI] [PubMed] [Google Scholar]
- Halligan, D. L., and P. D. Keightley, 2003. How many lethal alleles? Trends Genet. 19 57–60. [DOI] [PubMed] [Google Scholar]
- Joseph, S. B., and D. W. Hall, 2004. Spontaneous mutation in Saccharomyces cerevisiae: more beneficial than expected. Genetics 168 1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph, S. B., and M. Kirkpatrick, 2004. Haploid selection in animals. Trends Ecol. Evol. 19 592–597. [Google Scholar]
- Kacser, H., and J. Burns, 1981. The molecular basis of dominance. Genetics 97 639–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., 1994. The distribution of mutation effects on viability in Drosophila melanogaster. Genetics 138 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., 1998. Inference of genome-wide mutation rates and distributions of mutation effects for fitness traits: a simulation study. Genetics 150 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., and M. Lynch, 2003. Toward a realistic model of mutations affecting fitness. Evolution 57 683–685. [DOI] [PubMed] [Google Scholar]
- Keightley, P. D., and O. Ohnishi, 1998. EMS–induces polygenic mutation rates for nine quantitative characters in Drosophila melanogaster. Genetics 148 753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and H. A. Orr, 2005. Adaptation in sexuals vs. asexuals: clonal interference and the Fisher–Muller model. Genetics 171 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korona, R., 1999. Genetic load of the yeast Saccharomyces cerevisiae under diverse environmental conditions. Evolution 53 1966–1971. [DOI] [PubMed] [Google Scholar]
- Lynch, M., J. Blanchard, D. Houle, T. Kibota, S. Schultz et al., 1999. Perspective: spontaneous deleterious mutation. Evolution 53 645–663. [DOI] [PubMed] [Google Scholar]
- Lynch, M., W. Sung, K. Morris, N. Coffey, C. R. Landry et al., 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. USA 105 9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousseau, T. A., and D. A. Roff, 1987. Natural selection and the heritability of fitness components. Heredity 59 181–197. [DOI] [PubMed] [Google Scholar]
- Mukai, T., 1964. The genetic structure of natural populations of Drosophila melanogaster. I. Spontaneous mutation rate of polygenes controlling viability. Genetics 50 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, H. J., 1932. Some genetic aspects of sex. Am. Nat. 66 118–128. [Google Scholar]
- Orr, H. A., 2000. The rate of adaptation in asexuals. Genetics 155 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, S. P., and M. E. Orive, 1995. Evolutionary consequences of mutation and selection within an individual. Genetics 141 1173–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito, L., L. Fernandes, C. Mota and I. Gordo, 2007. Adaptive mutations in bacteria: high rate and small effects. Science 317 813–815. [DOI] [PubMed] [Google Scholar]
- Phadnis, N., and J. D. Fry, 2005. Widespread correlations between dominance and homozygous effects of mutations: implications for theories of dominance. Genetics 171 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, F. H., C. J. Geyer and R. G. Shaw, 2002. A comprehensive model of mutations affecting fitness and inferences for Arabidopsis thaliana. Evolution 56 453–463. [DOI] [PubMed] [Google Scholar]
- Simmons, M. J., and J. F. Crow, 1977. Mutations affecting fitness in Drosophila populations. Annu. Rev. Genet. 11 49–78. [DOI] [PubMed] [Google Scholar]
- Wagner, G. P., J. P. Kenney-Hunt, M. Pavlicev, J. R. Peck, D. Waxman et al., 2008. Pleiotropic scaling of gene effects and the ‘cost of complexity’. Nature 452 470–472. [DOI] [PubMed] [Google Scholar]
- Wloch, D. M., K. Szafraniec, R. H. Borts and R. Korona, 2001. Direct estimate of the mutation rate and the distribution of fitness effects in the yeast Saccharomyces cerevisiae. Genetics 159 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyl, C., and J. DeVisser, 2001. Estimates of the rate and distribution of fitness effects of spontaneous mutation in Saccharomyces cerevisiae. Genetics 157 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]