

Abstract
A novel method, called linkage disequilibrium multilocus iterative peeling (LDMIP), for the imputation of phase and missing genotypes is developed. LDMIP performs an iterative peeling step for every locus, which accounts for the family data, and uses a forward–backward algorithm to accumulate information across loci. Marker similarity between haplotype pairs is used to impute possible missing genotypes and phases, which relies on the linkage disequilibrium between closely linked markers. After this imputation step, the combined iterative peeling/forward–backward algorithm is applied again, until convergence. The calculations per iteration scale linearly with number of markers and number of individuals in the pedigree, which makes LDMIP well suited to large numbers of markers and/or large numbers of individuals. Per iteration calculations scale quadratically with the number of alleles, which implies biallelic markers are preferred. In a situation with up to 15% randomly missing genotypes, the error rate of the imputed genotypes was <1% and ∼99% of the missing genotypes were imputed. In another example, LDMIP was used to impute whole-genome sequence data consisting of 17,321 SNPs on a chromosome. Imputation of the sequence was based on the information of 20 (re)sequenced founder individuals and genotyping their descendants for a panel of 3000 SNPs. The error rate of the imputed SNP genotypes was 10%. However, if the parents of these 20 founders are also sequenced, >99% of missing genotypes are imputed correctly.
HIGH-DENSITY SNP arrays are currently available for an increasing number of species. QTL mapping, marker-assisted selection (MAS), and other genetic analyses often require or benefit greatly from imputing missing genotypes and from knowing the phase of the SNP genotypes. Although many statistical methods have been developed for phasing in the literature, new methods are needed because of the spectacular improvements in the efficiency of high-throughput genotyping. The older phasing methods often use linkage information (e.g., Sobel and Lange 1996). However, due to the use of increasingly dense marker maps, the use of linkage disequilibrium (LD) information has become increasingly attractive. Moreover, the linkage analysis methods became computationally intractable as the number of SNPs and/or the number of individuals increased. Phasing methods that rely solely on LD, such as Fastphase (Scheet and Stephens 2006), tend to mistakenly introduce recombinations when applied to genotypes covering long genetic distances (Kong et al. 2008). Linkage information and use of LD are not fundamentally different—use of LD may be thought of as linkage analysis based on common ancestors that occur before the known pedigree. Kong et al. suggested a new approach called “long-range phasing (LRP)” that relies on detecting identical-by-descent (IBD) haplotypes in different individuals and can phase large numbers of SNPs. For situations where the pedigree back to the common ancestor is available this may be considered linkage analysis but it can also be used without a pedigree and would use LD information. Long-range phasing may be seen as a set of sensible, heuristic rules to determine the phase using linkage analysis information, without attempting to extract all information in an optimal way. The latter is typical for modern linkage-based phasing methods: they are less concerned about optimally using all information, since there is a surplus of information, and are more concerned with handling high SNP densities over large genetic distances and for many genotyped individuals. The surplus of information is especially large if whole-genome sequence data are used. We define here whole-genome (re)sequence data as all the SNPs in the genome, which ignores information from copy number variation and other non-SNP genetic polymorphisms.
Here we describe a new phasing method, called linkage disequilibrium multilocus iterative peeling (LDMIP), which combines linkage and linkage disequilibrium information and can handle tens of thousands of SNPs per chromosome and thousands of individuals. It was initially developed for the common situation where many individuals at the top of the pedigree are ungenotyped, but the method is general and can be applied in other situations. For instance, we apply it here to the situation where a few individuals have very dense genetic information (e.g., from genome sequencing) while most individuals have sparser or no genotype data. To make optimum use of the known pedigree, family information is used quite extensively in an iterative peeling approach (Elston and Stewart 1971; Van Arendonk et al. 1989; Janss et al. 1995). The use of LD information crudely follows the approach of Meuwissen and Goddard (2001, 2007).
METHODS
Iterative peeling for one locus:
The principles of peeling algorithms are described by Elston and Stewart (1971). The iterative peeling algorithm used here is based on these principles and described in detail by Kerr and Kinghorn (1996), who extended the exact peeling method of Fernando et al. (1993) toward an iterative process, to be able to handle pedigrees with many loops. For the latter reason we also use iterative peeling here, although its results are approximate in nature. For each individual i, four sorted SNP genotypes are considered: ui = 1, 2, 3, or 4, which refers to the genotypes aa, aA, Aa, or AA. The difference between aA and Aa heterozygotes is that in the former case the a allele is inherited from the father and in the latter case from the mother. SNP genotyping reveals only three “unsorted” genotypes: mi = 1, 2, or 3, where mi = 1 denotes homozygous for the a allele; mi = 2 denotes heterozygous, and mi = 3 denotes homozygous for the A allele. The SNP genotypes, mi, are treated as phenotypic records here. Table 1 summarizes the notation that is used.
TABLE 1.
Summary of the notation
| Symbol | Description |
|---|---|
| i, s, d | Individual i, its sire, and its dam |
| ui | Genotype of individual i (ui = 1, 2, 3, and 4 denote genotypes aa, aA, Aa, and AA, respectively) |
| m, m(1,…,k), m(−k) | Vector of observed marker genotypes for all L loci, respectively, loci 1, 2, … , k, and all loci except locus k |
| ai(ui) | Anterior probability: probability of genotype ui using only ancestral information |
| ai(ui | us) | Anterior probability: probability of genotype ui using only ancestral information and conditional on the sire having genotype us |
| f(mi | ui) | Penetrance: probability of observing marker genotype mi given genotype ui |
| pij(ui) | Posterior probability: probability of ui using only offspring information with mate j |
| tr(ui | us, ud, m(−k)) | Transmission probability: probability of genotype ui at locus k given the sire and dams genotype are us and ud, resp., and m(−k) |
| trp(ui | us, m(−k)) | Paternal transmission probability: probability that the sire with genotype us has transmitted the paternal allele of ui, given m(−k) |
| trm(ui | ud, m(−k)) | Maternal transmission probability: similar to paternal probability but for the maternally derived allele |
| Si1(k) | Segregation indicator: Si1(k) = 1 (0) if i inherits the paternal (maternal) allele of its sire at locus k |
| Si2(k) | Segregation indicator: Si2(k) = 1 (0) if i inherits the paternal (maternal) allele of its dam at locus k |
| ρ(k−1,k) | Recombination rate between loci k − 1 and k |
Following Fernando et al. (1993), the information about the genotype of individual i, ui, is split in three parts: the anterior probability, ai(ui), which is the information that comes from the parents (i.e., the genotype probability if i had no known phenotypic record or progeny); the penetrance, f(mi|ui), which is the information that comes from its own record (see Table 2); and the posterior probability, pij(ui), which is the information that comes from the offspring that individual i has with mate j. In a pedigree with loops, there are dependencies between the anterior and the posterior information, but these dependencies are assumed negligible here, such that the combined genotype probability given the marker information is
 |
(1) |
where m is the vector of all marker genotypes (which are treated as phenotypes here), Mi is the set of mates of i, and Lik is the likelihood of the marker data:
 |
TABLE 2.
Penetrance probabilities (f(mi | ui): probabilities of observed marker genotypes (mi) conditional on the true genotype (ui) (“–” denotes missing genotype)
| Genotype error rate 0% | Genotype error rate 1% | ||||||
|---|---|---|---|---|---|---|---|
| ui\mi | aa | Aa | AA | – | aa | Aa | AA |
| aa | 1 | 0 | 0 | 1 | 0.99 | 0.005 | 0.005 |
| aA | 0 | 1 | 0 | 1 | 0.005 | 0.99 | 0.005 |
| Aa | 0 | 1 | 0 | 1 | 0.005 | 0.99 | 0.005 |
| AA | 0 | 0 | 1 | 1 | 0.005 | 0.005 | 0.99 |
Two different genotyping error rates are assumed.
If i is an offspring of s and d, the anterior probability is
 |
(2) |
(Fernando et al. 1993), where Csd denotes the set of offspring from the mating pair s and d, and the transmission probability tr(uj | us, ud) is the probability that offspring j has genotype uj given that its parents have genotypes us and ud. The latter way of writing Equation 2 defines ai(ui | us), i.e., the anterior probability of genotype ui given that the sire has genotype us, and will be needed to calculate probabilities of maternal or paternal inheritance using the peeling algorithm.
The posterior probability coming from offspring of i with mate j is (Fernando et al. 1993)
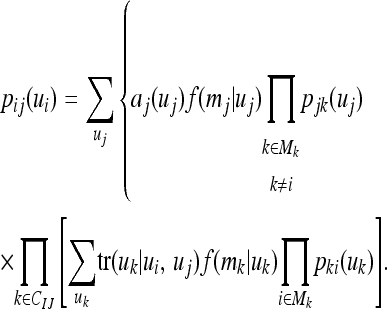 |
The calculation of ai(ui) involves pij(ui), and vice versa. In pedigrees without loops the calculations can be ordered such that all ai(ui) and pij(ui) terms are calculated before they are needed. But in complex pedigrees with many loops, the calculations need to be performed iteratively. At the start all ai(ui) and pij(ui) are set to 1. First the posterior probabilities are calculated working from the youngest to the oldest individual in the pedigree. Next the anterior probabilities are calculated starting from the oldest and working through to the youngest individual. After this the posterior probabilities are updated again, etc., until the sum of squares of the changes of the Pr(ui | m) probabilities is <10−8 times the sum of squares of the Pr(ui | m) values.
Multilocus iterative peeling:
In multilocus iterative peeling we adjust the transmission probabilities for the information from neighboring loci. For example, if at a neighboring locus the paternal allele is transmitted from parent to offspring, then the current locus will also most likely inherit the paternal allele. For this we express the transmission probability in terms of segregation indicators Si1(k). Si1(k) = 1 (0) if individual i inherits the paternal (maternal) allele of its sire at locus k. Similarly, Si2(k) = 1 (0) if individual i inherits the paternal (maternal) allele of its dam at locus k. First we factor the transmission probability tr(ui | us, ud) into probabilities for the paternal and maternal allele of i,
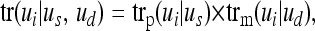 |
where trp(ui | us) is the probability the sire of genotype us transmits the paternal allele of genotype ui. Similarly, trm(ui | ud) is the probability that dam of genotype ud transmits the maternal allele of genotype ui. Now we calculate these transmission probabilities at locus k conditional on all marker genotypes except those at locus k,
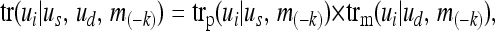 |
(3a) |
where 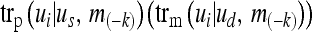 denotes the probability that the sire (dam) transmits the allele that is paternally (maternally) inherited in the ui genotype. m(−k) indicates the marker genotypes of all loci and all individuals except locus k. The paternal and maternal transmission probabilities are
denotes the probability that the sire (dam) transmits the allele that is paternally (maternally) inherited in the ui genotype. m(−k) indicates the marker genotypes of all loci and all individuals except locus k. The paternal and maternal transmission probabilities are
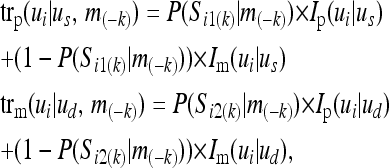 |
(3b) |
where Ip(ui|us) is an indicator variable that is 1 (0) if the inheritance of the paternal allele of the sire can (cannot) give genotype ui given the sire's genotype us, and P(Si1(k) | m(−k)) is the probability that Si1(k) = 1 conditional on the marker genotypes at all loci except k. Similarly, Im(ui|us) indicates that the inheritance of the maternal allele of the sire could give genotype ui. Also, Ip(ui|ud) [Im(ui|ud)] indicates whether inheritance of the paternal [maternal] allele of the dam could give genotype ui.
To calculate Equation 3b, we need to derive the probabilities of the segregation indicators conditional on the information of all other marker loci. For this we use the approach of the Lander–Green algorithm (Lander and Green 1987), which is an implementation of the forward–backward algorithm (Russell and Norvig 2003). Let S(k) denote all segregation indicators of all individuals at locus k; then appendix a shows that
 |
(4) |
where const1 is a constant, and m(i..j) denotes the markers at loci i, i + 1, …, j. Equation 4 writes P(S(k)|m(−k)) in terms of P(S(k) | m(1,...,k−1)), which will be calculated by the forward cycle of the following Lander–Green type of algorithm and P(S(k) | m(k+1,...,L)), which will be calculated by the backward cycle of the algorithm.
For the forward cycle of the algorithm, we start with noting that P(S(1)) = 0.5 for all segregation indicators. Next we calculate P(S(1)|m(1)) using a slightly modified peeling algorithm, which is adapted to calculate the segregation indicator probabilities next to the genotype probabilities (see appendix b for details).
In the absence of a recombination between locus (k − 1) and (k), we have Sij(k) = Sij(k−1), and if there is a recombination we have Sij(k) = 1 − Sij(k − 1), which gives the general equation
 |
(5) |
where ρ(k−1,k) is the recombination rate between locus k − 1 and k. Using Equation 5 we calculate P(Sij(2) | m(1)), which is of the same form as 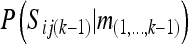 for k = 2. Next we calculate P(Sij(2)|m(1), m(2)) by peeling and applying Equation B1 from appendix b to locus 2, but using transmission probabilities
for k = 2. Next we calculate P(Sij(2)|m(1), m(2)) by peeling and applying Equation B1 from appendix b to locus 2, but using transmission probabilities 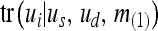 that are obtained from Equation 3 and P(Sij(2)|m(1)).
that are obtained from Equation 3 and P(Sij(2)|m(1)).
Given P(Sij(2)|m(1), m(2)), we calculate P(Sij(3)|m(1), m(2)) using Equation 5, which is of the form 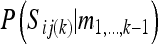 for k = 3, as needed in Equation 4. This cycle of calculations is repeated until all markers are included and until
for k = 3, as needed in Equation 4. This cycle of calculations is repeated until all markers are included and until  is reached, which concludes the forward cycle of the algorithm.
is reached, which concludes the forward cycle of the algorithm.
Next the algorithm is applied in the same way but by starting at the rightmost position L and including markers from the left of the current position, i.e., the backward cycle of the forward–backward algorithm. In the algorithm, this is obtained simply by swapping the indexes of the loci, i.e., counting loci from right to left instead of from left to right.
The algorithm concludes by using Equation 2 to calculate  for every locus k and using these results to calculate the transmission probabilities of Equation 3 followed by an iterative peeling step at each locus k to obtain P(ui|m(1,...,L)), i.e., the probability of the four genotypes for each individual i at each locus k, conditional on the marker genotypes at locus k and all other loci.
for every locus k and using these results to calculate the transmission probabilities of Equation 3 followed by an iterative peeling step at each locus k to obtain P(ui|m(1,...,L)), i.e., the probability of the four genotypes for each individual i at each locus k, conditional on the marker genotypes at locus k and all other loci.
Phasing:
The genotype probabilities for the four genotypes aa, aA, Aa, and AA do not provide direct estimates of the phase between the markers (which would require estimates of multilocus genotype probabilities). However, the probabilities distinguish between the paternal or maternal inheritance of the alleles; i.e., the heterozygote Aa differs from aA. If the genotype probabilities are close to certain, this also provides us with the haplotypes; e.g., if at two loci genotypes Aa and bB are almost certain, the phased genotype is almost certainly (Ab)/(aB). However, if Aa and bB are the most probable genotypes at the A and B loci, respectively, this does not imply that (Ab)/(aB) is also the most probable haplotype. Therefore, we require that the genotype probability is >0.95 for both the genotypes before we deduce the haplotype. If genotype probabilities are <0.95, the phased genotype is assumed missing.
For founders, i.e., individuals with unknown parents, the two heterozygous genotypes (Aa and aA) have equal genotype probabilities, P(u2 | m(1,…,L)) = P(u3 | m(1,…,L)) =  , since it cannot be assessed whether the A was inherited from the father or the mother. However, the phases of the markers may still be estimated on the basis of progeny information. To extract the information on the marker phases, one heterozygous marker locus close to the middle of the chromosome segment was arbitrarily set to genotype u2; i.e., the penetrance f(mi | ui = 2) = 1. An arbitrary heterozygous genotype, close to the middle of the segment, was set to ui = 2 for every genotyped founder. The set genotype served as an anchor for the other markers, such that their heterozygous genotype probabilities could deviate from P(u2 | m(1,...,L)) = P(u3 | m(1,...,L)) =
, since it cannot be assessed whether the A was inherited from the father or the mother. However, the phases of the markers may still be estimated on the basis of progeny information. To extract the information on the marker phases, one heterozygous marker locus close to the middle of the chromosome segment was arbitrarily set to genotype u2; i.e., the penetrance f(mi | ui = 2) = 1. An arbitrary heterozygous genotype, close to the middle of the segment, was set to ui = 2 for every genotyped founder. The set genotype served as an anchor for the other markers, such that their heterozygous genotype probabilities could deviate from P(u2 | m(1,...,L)) = P(u3 | m(1,...,L)) =  .
.
Use of LD information:
The algorithm described above uses only linkage analysis information and may result in a lot of missing phased genotypes. Here we use LD information to fill in the missing genotypes, i.e., loci where the algorithm could not achieve a 95% certainty in favor of one of the four possible phased genotypes. For this we crudely approximate the IBD probability estimation methods of Meuwissen and Goddard (2001, 2007). These authors use the number of marker alleles equal to the left (right) of a focal position, before a break occurs (i.e., alleles are not equal), as data for the calculation of the IBD probability of two haplotypes at the focal position. For example, when F denotes the focal position, the haplotypes “111F111” and “110F110” have 2 markers equal to the right before a break occurs and 0 markers equal to the left. Such IBD probability calculations show that IBD probabilities are usually >0.99 if the number of markers equal to the left plus that equal to the right ≥20 and the focal point is not next to a break. In such a situation, it is thus quite safe to assume that the marker alleles at the focal point are the same, and if the marker allele is missing at the focal point at the first haplotype, we can simply assume it is a copy of the allele of the second haplotype. If a missing allele at a haplotype could be copied from several alternative haplotypes (since there are many haplotype pairs), it will only be copied if all alternative haplotypes yield the same allele. However, if the number of alternative haplotypes for filling in the missing allele is large (>20), one of the alternative suggestions is allowed to deviate from all the other alternatives. In the latter case, the allele suggested by the majority of the haplotypes is copied, unless it does not agree with the known genotype. If, when comparing two haplotype pairs, one or both alleles are missing (unknown phase) at a SNP next to the focal position, simply the next SNP is taken, but still the requirement of ≥20 equal SNP alleles is maintained by extending the chromosomal region. Thus, if missing phased genotypes occur, the number of SNPs considered to the left and right of F is extended.
This imputation of missing genotypes means that effectively more marker genotypes become available, and it thus makes sense to rerun the multilocus iterative peeling algorithm. After this more marker loci can be imputed, etc. These two steps are iterated until convergence. The information of an imputed marker genotype, say Aa, is conveyed to the peeling algorithm by setting the penetrance probability of genotype Aa to 0.99 and then setting the penetrance probability of the remaining genotypes, aa, aA, and AA, to 0.01/3. This allows for errors of genotype imputation. The algorithm is considered converged if the number of missing genotypes was not reduced during two iterations. The algorithm is implemented in a software package that is available from the authors upon request.
Testing the algorithm by computer simulation:
Case I:
First, we simulated genome sequence data by forward simulation of a population of Ne = 100 and a chromosome of length 50 cM for 1000 generations. The assumptions about the population model followed those of coalescence theory (Hudson 2002); i.e., the Fisher–Wright idealized population model (Falconer and Mackay 1996) and the infinite-sites mutation model were assumed (Kimura 1969), with a mutation frequency of 2 × 10−8 per nucleotide per generation. Recombinations were sampled according to the Haldane mapping function, assuming a recombination frequency of 10−8 per nucleotide.
Second, the 500 SNPs with the highest minor allele frequency (MAF) were selected for genotyping, which resulted in MAFs ranging from 0.245 to 0.5. Genotyping was performed only in the last three generations, while pedigree was recorded for the last five generations (i.e., generations 996–1000), which are called G1, … , G5, respectively. The parents of the individuals of generation G1 were assumed unknown. Thus, the LDMIP algorithm used 500 individuals (five generations × 100), of which the youngest 300 were genotyped. At random some of the marker genotypes within the genotyped individuals were set missing at a rate of 5, 10, or 15%. LDMIP was used to estimate the phase of the markers and to impute the missing genotypes. These estimates were compared to the true values that are known in the simulations and to estimates obtained from FastPhase (Scheet and Stephens 2006) and from LRP. LRP was applied as described by Kong et al. (2008) to the 100-cM region, with an additional step at the end of the LRP algorithm where the surrogate parents were used to impute missing genotypes instead of phase (which had already been imputed). Ungenotyped individuals of generations G1 and G2 were not considered, although some of their genotypes were also imputed by the algorithms.
Case II:
In case II we wanted to test whether the algorithm could be used to impute full genome sequence data. This case considers the situation where a few founder individuals are resequenced, such that their full genome sequence is available, and where later generation descendants are relatively sparsely genotyped for 3000 SNPs. Genome structure and 1000 generations were simulated the same as before, except that generation G1 contained only 10 males and 10 females. Thus, later generations descended from few founders in generation G1. In case IIa, these 20 founders were resequenced and their whole-genome sequence was available and contained 16,722 SNPs. In case IIb, in addition to the 20 founders their parents (40 sires and dams) were resequenced, to facilitate the estimation of the phases of the founders. Generations G4 and G5, but not G2 and G3, were (sparsely) genotyped for a panel of 3000 SNPs, which were randomly selected from among the SNPs with MAF > 0.1. Generations G2 and G3 were not genotyped. The aim here is to reconstruct the genome sequence for the individuals in generations G4 and G5. The simulated data are available in supporting information, File S1.
RESULTS
Case I:
Table 3 shows the number of phased genotypes and the number of erroneously inferred phases in the genotyped generations G3, G4, and G5. The fraction of erroneously inferred phases varied from 1.4 to 1.8%. It is also apparent from Table 3 that almost all the erroneously inferred phases occurred in generation 3, which was the first genotyped generation. Hence, knowing the genotypes of the sires and dams greatly improved the accuracy of phasing, even in a situation where the pedigree of the generation 3 individuals is known and thus some of the generation 2 genotypes could be inferred. The missing genotype rate hardly affected the accuracy of the phase estimates.
TABLE 3.
The number of phased genotypes and erroneously estimated phases for case I
| Missing genotype rate (%) | Unknown phasesa | No. phased | No. of errors |
|---|---|---|---|
| Generations G3, G4, and G5 | |||
| 5 | 64,882 | 64,656 | 1,171 |
| 10 | 61,394 | 61,154 | 1,095 |
| 15 | 57,923 | 57,758 | 823 |
| Generation G3 only | |||
| 5 | 22,200 | 21,974 | 1,171 |
| 10 | 21,020 | 20,781 | 1,094 |
| 15 | 19,802 | 19,638 | 823 |
This equals the number of heterozygous genotypes in generations G3, G4, and G5.
Table 4 shows the error rate of the imputed genotypes is <1%, and nearly all errors occurred in generation 3. Thus, knowledge about the genotypes of the parents greatly improves the imputation of missing genotypes. The fraction of missing genotypes that were imputed by the algorithm was ∼98.6%. Table 5 shows that the number of erroneously imputed genotypes and phases by FastPhase is approximately five times larger than that of LDMIP (Tables 1 and 2), which demonstrates that the use of pedigree information greatly improves the imputation accuracy. FastPhase imputed all missing genotypes and phases. LRP imputed only a limited number of phases and genotypes. The phase imputation error rate of LRP was lower than that of FastPhase, but higher than that of LDMIP. The genotype imputation error rate of LRP was similar to that of LDMIP but about six times fewer genotypes were imputed.
TABLE 4.
The number of imputed genotypes and erroneously imputed genotypes for case I
| Missing genotype rate (%) | Total missinga | No. imputed | No. of errors |
|---|---|---|---|
| Generations G3, G4, and G5 | |||
| 5 | 7,442 | 7,349 | 69 |
| 10 | 14,938 | 14,734 | 143 |
| 15 | 22,391 | 22,080 | 159 |
| Generation G3 only | |||
| 5 | 2,520 | 2,438 | 68 |
| 10 | 4,986 | 4,818 | 132 |
| 15 | 7,550 | 7,294 | 151 |
The expected number of missing genotypes is 5, 10, and 15% of 50,000 (100 animals × 500 SNPs) genotypes per generation.
TABLE 5.
Results from genotype and phase imputation by FastPhase (FP) and long-range phasing (LRP) for generations G3, G4, and G5 in case I
| Missing genotype rate (%) | No. imputeda |
No. of errors |
||
|---|---|---|---|---|
| FP | LRP | FP | LRP | |
| Phase imputation | ||||
| 5 | 64,882 | 56,435 | 5,490 | 3,297 |
| 10 | 61,394 | 52,603 | 5,096 | 3,313 |
| 15 | 57,923 | 49,284 | 5,166 | 3,066 |
| Genotype imputation | ||||
| 5 | 7,442 | 1,325 | 340 | 6 |
| 10 | 14,938 | 2,222 | 671 | 15 |
| 15 | 22,391 | 1,962 | 1,029 | 34 |
FastPhase imputes all genotypes (phases); i.e., the number of imputed genotypes equals the number of missing genotypes (phases).
Case II:
Table 6 shows the results on the imputation of missing genome sequence data of generations G4 and G5, using the genome sequence of the generation G1 individuals. Without resequencing the parents of the founders, ∼96% of the missing SNP genotypes were imputed, and 10.5% of the missing SNP genotypes were erroneously imputed. For the missing phases, these numbers were 97 and 3.3%, respectively. When, in addition to the founders, their parents were resequenced, 99.7% of the missing genotypes were imputed and 0.3% of the missing genotypes were erroneously imputed. For the missing phases these numbers were ∼100 and 0.09%, respectively. Hence, the resequencing of the parents of the founders improved the imputation of the genome sequence of generation G4 and G5 animals dramatically.
TABLE 6.
Numbers of imputed genotypes and phases for cases IIa and IIb
| Total | No. imputed | No. of errors | |
|---|---|---|---|
| Case IIa: generations G1, G4, and G5 | |||
| Missing genotypesa | 2,744,400 | 2,630,051 | 287,287 |
| Phases | 307,345 | 297,195 | 10,150 |
| Case IIb: generations G1, G4, and G5 and the parents of G1 | |||
| Missing genotypesa | 2,744,400 | 2,736,016 | 8,384 |
| Phases | 464,223 | 464,132 | 432 |
There are missing genotypes only in generations G4 and G5. Their number equals (16,722 − 3000) × 100 × 2 genotypes.
The analysis of case I, with 500 individuals and 500 SNPs, ran within a few minutes on a PC type of computer. Case II, which comprised 16,722 SNPs and 520 or 560 individuals, took ∼4 hr on a PC without parallel processing. It is possible to parallelize the computations by splitting the chromosome into, e.g., four large, equally sized segments with, say, 100 SNPs overlap between the segments. This would reduce the time required to perform this analysis to ∼1 hr on a computer that can handle four simultaneous jobs.
DISCUSSION
A novel method, LDMIP, for the phasing of genotype data and the imputation of missing genotypes was developed that combined linkage and linkage disequilibrium information and could handle tens of thousands of SNPs per chromosome. LDMIP was especially developed for situations where extensive pedigree is available, as well as the LD information that can always be used. The use of pedigree as well as the LD information (i) helps to impute missing genotypes and marker phases, which became particularly evident when comparing LDMIP results to those of FastPhase, (ii) detects genotyping errors that are inconsistent with the pedigree (this was found when analyzing real data using LDMIP), and (iii) ensures that the imputed genotypes and phases are consistent with the pedigree. The method was tested in two cases where the total number of individuals was ∼500 and there were up to 16,722 SNPs, but we have also used the method in situations where the pedigree consisted of up to 20,000 individuals. It has been demonstrated before that iterative peeling can handle large complex pedigrees (e.g., Szydlowski and Gengler 2008). In a situation with randomly missing genotypes, the error rate of the imputed genotypes was <1%, and ∼99% of the missing genotypes were imputed. For the phase estimates these numbers were 1–2% and >99%, respectively. Thus, LDMIP seems to yield accurate and quite completely phased genotypes, even in situations where up to 15% of the SNP genotypes are missing.
In case II, LDMIP was used for whole-genome sequence imputation. Here, some founder individuals had complete genome sequence data available, while later generation descendants were only relatively sparsely genotyped. In this case, LDMIP imputed the missing sequence data of the sparsely genotyped individuals with an error rate of ∼10%. This error rate was reduced to 0.3% when the parents of the founders were also resequenced. The resequencing of the parents greatly improves the estimation of the phases and thus haplotypes of the founder individuals, which then greatly improves the imputation of the whole-genome sequence of sparsely genotyped descendants. The error rate of the genome sequence imputation is similar to that of the resequencing technology, so there is no need to resequence the descendants. Few populations will have gone through such a severe bottleneck as the population in case II, which may imply that many more founder individuals need to be resequenced to obtain similar accuracies of sequence imputation. More research is needed on the effects of population structure and experimental designs for whole-genome sequence imputation. However, Table 6 clearly shows that it is highly advisable to also resequence the parents of the founders in addition to the founder individuals. More generally, the individuals to be sequenced must be chosen so that the sequence data can be phased, and it is preferable to have the densest genotypes on the ancestors instead of the descendants, because the ancestors are less related and thus their genotypes provide more information.
Kong et al. (2008) recently described a phasing method called LRP. It outperformed local phasing methods such as PHASE (Stephens and Scheet 2005) and fastPHASE (Scheet and Stephens 2006) both in speed and accuracy, because the local phasing methods use only the strong correlations between alleles within LD blocks. Across LD blocks, over long distances, the accuracy of local phasing reduced quickly. LRP was found accurate for regions of ∼10 Mb. If larger regions need to be processed, as was the case here, it is more difficult to find “surrogate parents” for the LRP algorithm that are identical by state (IBS) ≥ 1 for the entire region, which are then considered IBD for the entire region. For many marker positions the algorithm failed to find informative surrogate parents for the entire 100-cM region due to the large region and the smaller number of genotyped individuals, which explains the low imputation rates of LRP (Table 5). Also the region may not be entirely IBD despite being IBS ≥ 1 because our marker density was more than one order of magnitude lower than that considered by Kong et al. (2008). The latter may explain the higher imputation error rate in Table 5 than that found by Kong et al. Thus, the LRP algorithm requires more genotyped individuals, a higher marker density than LDMIP, and that the regions considered should not be too long. Otherwise the region needs to be broken down into subregions that are treated by LRP separately, which leaves the problem of deciding where the boundaries are. The multilocus peeling algorithm of LDMIP naturally models the boundaries between IBD regions, i.e., the boundary between a region that is IBD with one surrogate parent and a region that is IBD with another. A detailed comparison of these and other methods is needed to guide the choice between alternative methods for genotype imputation under various circumstances.
Within the category of family-based haplotyping methods, SIMWALK2 (Sobel and Lange 1996) is probably the most used and well known. SIMWALK2 uses a Monte Carlo Markov chain (MCMC) algorithm that in principle considers all possible haplotypes, although the Markov chain concentrates on the most likely haplotypes. The algorithm becomes, however, computationally too demanding with more than a few hundred individuals in the pedigree. In comparison to SIMWALK2, LDMIP (i) can handle larger pedigrees and (ii) uses LD information next to linkage analysis.
The computational costs per iteration of the multilocus peeling increase approximately linearly with the number of SNPs and the number of individuals in the pedigree. However, the use of LD information requires the comparison of every haplotype of every genotyped individual with every other haplotype of a genotyped individual and computations increase thus quadratically with the number of genotyped individuals. The latter computation is, however, relatively fast and computation time thus increases generally linearly with the number of individuals in the pedigree, although the number of iterations may also increase. The number of iterations of the algorithm, i.e., number of cycles of using LD and linkage analysis, is usually <10. Whether LD information is used or not is optional, and if LD is not used, no iteration is needed to obtain the final solution. The algorithm can be extended to multiallelic markers such as microsatellites in a straightforward manner. However, computational costs per iteration and storage requirements increase quadratically with the number of alleles.
The presented methodology can easily account for genotyping errors by adjusting the penetrance probabilities f(mi | ui) (Gorjanc et al. 2010). Table 2 shows how to adjust penetrance probabilities if a genotyping error rate of 1% is assumed, and other error rates can be accommodated in a similar way. If the true genotype is AA, an erroneous call of Aa may be more likely than aa, which may also be reflected in the penetrance probabilities, although this was not done in Table 2. Genotyping errors can be detected by scanning the results for discrepancies between the ultimate genotype probabilities P(ui|m(1), …, m(k), …, m(L)) and the marker genotype data mi(k). The penetrance probabilities allow for a quite flexible modeling of the observed information and can for instance also be used to model dominant genetic markers.
LDMIP relies heavily on extensive family data to accurately phase over long regions. Family structure seems to be important to achieve accurate long-range phase estimates. In data without strong family structures, the long-range phases need to rely on many small phased, LD blocks coupled together, with the inherent problem that errors accumulate across the blocks. Many of the applications of dense SNP genotyping such as QTL (fine) mapping, haplotype-based genomic selection, the estimation of effective population sizes (Tenesa et al. 2007), population admixture, and the estimation of signatures of selection (Voight et al. 2006) rely on accurate estimates of phase and missing genotypes and their utility is thus greatly improved by phase and genotype imputation methods such as LDMIP.
APPENDIX A: THE SEGREGATION INDICATOR PROBABILITIES AT LOCUS K CONDITIONAL ON ALL OTHER LOCI
In the main text, Equation 3b requires the probabilities of the segregation indicators at locus k conditional on the information of all other marker loci, e.g.,  . If S(k) denotes all segregation indicators of all individuals at locus k, and using an approach similar to that of the Lander–Green algorithm,
. If S(k) denotes all segregation indicators of all individuals at locus k, and using an approach similar to that of the Lander–Green algorithm,
 |
(A1) |
where m(i..j) denotes the markers at loci i, i + 1, …, j, const0 and const1 are different constants, and P(S(k)) is constant since, a priori, all segregation indicators have probability 0.5. The third line in Equation A1 follows, because conditional on the segregation indicators at locus k, the markers to the left and to the right of locus k are independent. Equation A1 writes P(S(k) | m(−k)) in terms of which 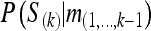 will be calculated by the forward cycle of the following Lander–Green type of algorithm, and
will be calculated by the forward cycle of the following Lander–Green type of algorithm, and  will be calculated by the backward cycle of the algorithm.
will be calculated by the backward cycle of the algorithm.
APPENDIX B: CALCULATION OF SEGREGATION INDICATOR PROBABILITIES USING THE PEELING ALGORITHM
Here we need to calculate the segregation indicator probabilities given the marker data at a locus, i.e., P(S | m) (the locus subscript is omitted). For the paternal allele of individual i, this can be written as
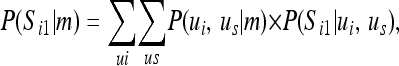 |
where P(Si1 | ui, us) is the probability that Si1 = 1 given the genotypes of the sire s and offspring i, P(Si1 | ui, us) = 0, 1, or 0.5, where the latter occurs if the sire is homozygous and thus paternal or maternal inheritance is equally likely. P(S | m) can be approximated from the peeling results by
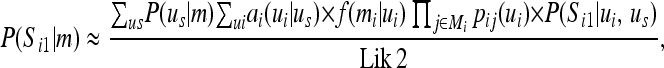 |
(B1) |
where ai(ui | us) is from Equation 2 in the main text, and the approximation is due to the fact that P(us | m) also contains information about i, which is thus double counted in determining the segregation indicator probablities. The approximation is needed because otherwise the summations could not be factored in the efficient peeling way. Due to this approximation,  , but equals
, but equals 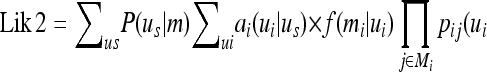 ). Similarly, P(Si2 | m) is calculated by the above Equation B1, but with the sire, s, being replaced by the dam, d, of individual i.
). Similarly, P(Si2 | m) is calculated by the above Equation B1, but with the sire, s, being replaced by the dam, d, of individual i.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.113936/DC1.
References
- Elston, R.C., and J. Stewart, 1971. A general model for the genetic analysis of pedigree data. Hum. Hered. 21 523–542. [DOI] [PubMed] [Google Scholar]
- Falconer, D., and T. Mackay, 1996. Introduction to Quantitative Genetics. Longman, London, UK. [DOI] [PMC free article] [PubMed]
- Fernando, R. L., C. Stricker and R. C. Elston, 1993. An efficient algorithm to compute the posterior genotypic distribution for every member of a pedigree without loops. Theor. Appl. Genet. 87 89–93. [DOI] [PubMed] [Google Scholar]
- Gorjanc, G., M. Kovača and D. Kompan, 2010. Inference of genotype probabilities and derived statistics for PrP locus in the Jezersko–Solcava sheep. Livest. Sci. 129 232–236. [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18 337–338. [DOI] [PubMed] [Google Scholar]
- Janss, L. L. G., J. A. M. Van Arendonk and J. H. J. VanderWerf, 1995. Computing approximate monogenic model likelihoods in large pedigrees with loops. Genet. Sel. Evol. 27 567–579. [Google Scholar]
- Kerr, R. J., and B. P. Kinghorn, 1996. An efficient algorithm for segregation analysis in large populations. J. Anim. Breed. Genet. 113 457–469. [Google Scholar]
- Kimura, M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, A., G. Masson, M. L. Frigge, A. Gylfason, P. Zusmanovich et al., 2008. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 40 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and P. Green, 1987. Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. USA 84 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2001. Prediction of identity by descent probabilities from marker-haplotypes. Genet. Sel. Evol. 33 605–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2007. Multipoint identity-by-descent prediction using dense markers to map quantitative trait loci and estimate effective population size. Genetics 176 2551–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, S., and P. Norvig, 2003. Artificial Intelligence: A Modern Approach, Ed. 2. Pearson Education, Harlow, Essex, UK.
- Scheet, P., and M. Stephens, 2006. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel, E., and K. Lange, 1996. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker sharing statistics. Am. J. Hum. Genet. 58 1323–1337. [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., and P. Scheet, 2005. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 76 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szydlowski, M., and N. Gengler, 2008. Sampling genotype configurations in a large complex pedigree. J. Anim. Breed. Genet. 125 330–338. [DOI] [PubMed] [Google Scholar]
- Tenesa, A., P. Navarro, B. J. Hayes, D. L. Duffy, G. M. Clarke et al., 2007. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 17 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Arendonk, J. A. M., C. Smith and B. W. Kennedy, 1989. Method to estimate genotype probabilities at individual loci in farm livestock. Theor. Appl. Genet. 78 735–740. [DOI] [PubMed] [Google Scholar]
- Voight, B. F., S. Kudaravalli, X. Wen and J. K. Pritchard, 2006. A map of recent positive selection in the human genome. PLoS Biol. 4 e72. [DOI] [PMC free article] [PubMed] [Google Scholar]