

Abstract
Proteasome inhibitors induce rapid death of cancer cells. We show that in epithelial cancer cells, such death is associated with dramatic and simultaneous up-regulation of several BH3-only proteins, including BIK, BIM, MCL-1S, NOXA, and PUMA, as well as p53. Elevated levels of these proteins seem to be the result of direct inhibition of their proteasomal degradation, induction of transcription, and active translation. Subsequent cell death is independent of BAX, and probably BAK, and proceeds through the intrinsic mitochondrial apoptosis pathway. We identify the cascade of molecular events responsible for cell death induced by a prototypical proteasome inhibitor, MG132, starting with rapid accumulation of BH3-only proteins in the mitochondria, proceeding through mitochondrial membrane permeabilization and subsequent loss of ΔΨm, and leading to irreversible changes of mitochondrial ultrastructure, degradation of mitochondrial network, and detrimental impairment of crucial mitochondrial functions. Our results also establish a rationale for the broader use of proteasome inhibitors to kill apoptosis-resistant tumor cells that lack functional BAX/BAK proteins.
Introduction
Proteasome inhibitors have emerged as promising chemotherapeutic agents and can induce a range of antitumor activities, including induction of cell cycle arrest, restoration of sensitivity to standard chemotherapy and irradiation, and provocation of apoptosis (1). One of these agents, bortezomib (Velcade), is in clinical trials. It has been proposed that the antitumor activity of proteasome inhibitors mostly resulted from impeded degradation of regulatory proteins such as p21, IκBα, and p53 (1, 2). In addition, several studies have shown that proteasome inhibition leads to apoptosis in various cancer cells (3-6). However, the molecular basis of such apoptotic response is still the subject of intense investigation. In general, it is well established that the complex process of apoptotic cell death is governed by opposing activities of BCL-2-family proteins (see refs. 7, 8 for recent reviews). One group of this family promotes cell survival, which includes BCL-2, BCL-xL, BCL-w, A1, and MCL-1L, whereas the other endorses cell demolition. This proapoptotic group consists of two subgroups: those that share three BCL-2 homology domains (e.g., BAX and BAK) and a diverse group of BH3-only proteins, including but not limited to BIK, BIM, BAD, BID, HRK, NOXA, PUMA, and BNIP3, which share only one BCL-2 homology domain. It is widely accepted that BH3-only proteins are the apical sensors of stress signals and the key initiators of apoptotic cell death. In agreement with this, proapoptotic BH3-only proteins were shown to play a critical role as initiators of the apoptotic program of cell death by some proteasome inhibitors (9-11). The involvement of particular BH3-only protein seems to vary with cell type and p53 status. BH1-3 proteins BAX and BAK are considered to be essential mediators of apoptotic cell death. According to the current widely accepted model, the BH3-only proteins regulate core apoptotic machinery through activation of BAX and BAK, which undergo oligomerization in the outer mitochondrial membrane and cause the release of apoptogenic factors such as cytochrome c. Thus, mitochondria are prominent participants and most likely are obligate organelles in the apoptotic pathway (12). In addition to the release of apoptogenic factors involved in activation of caspase-dependent and caspase-independent cell death mechanisms, mitochondrial damage per se and loss of vital mitochondrial functions play a significant role in apoptotic cell death progression. Mitoptosis was described as a phenomenon of mitochondrial death program (13) and represents a pathway by which mitochondria undergo extensive fragmentation and subsequent elimination during apoptosis (14-16). However, the molecular mechanisms of these processes remain unclear.
Various studies have suggested multiple mechanisms of cancer cell death induced by proteasome inhibitors, and the process is far from being fully understood. It remains important to identify additional downstream targets of proteasome inhibitors and to elucidate the significance of new signaling pathways in cell death by the proteasome inhibitors. In this study, we used cancer cells with different sensitivity to classic antitumor drugs—a set of isogenic HCT116 human colon carcinoma cell lines— which contain targeted disruption of the Bax gene (Bax−/−; ref. 17) or the p53 gene (p53−/−; ref. 18), as well as immortalized bax/bak double-knockout (BAX/BAK DKO) mouse embryonic fibroblasts (MEF), to investigate the molecular mechanisms involved in the antitumor action of proteasome inhibitors. Here, we provide evidence that proteasomal inhibition in epithelial cancer cells activates an apoptotic pathway that occurs in a BAX/BAK–independent manner involving multiple BH3-only proteins (BIK, BIM, MCL-1S, NOXA, and PUMA) and p53 and the mitochondria, suggesting a novel role for these proteins in execution of an apoptotic death paradigm. We show that together several BH3-only proteins and p53 can overcome BAX/BAK dependency to mediate cell death induced by proteasome inhibitors. Thus, our findings reveal an additional level of redundancy between proapoptotic proteins in regulation of life and death of the cell. Such regulation seems to be more complex than it is currently believed to ensure the effective elimination of damaged or excess cells.
Results
p53- and BAX-Independent Cell Death Induced by MG132
We chose to investigate the role of various mammalian apoptosis effectors on cell death induced by a most widely used proteasome inhibitor, MG132. Despite the availability of different proteasome inhibitors, MG132still remains the agent of choice to study proteasome involvement in different cellular processes (19). As expected, treatment with MG132caused significant cell death in wild-type (wt) HCT116 colon cancer cells with characteristic features of rounded detached cells visible as early as 24 hours after the addition of MG132 (Fig. 1A). Cell death was markedly increased during the time (to ∼60% after 48 hours) compared with cells treated with vehicle (DMSO; Fig. 1B). The cell death was attributable to apoptosis because the proteolytic cleavage of two key enzymes involved in apoptosis, caspase-3 and poly(ADP-ribose) polymerase (PARP), was clearly detectable at 24 hours after MG132 treatment (Fig. 1C). Similarly, DNA fragmentation was also observed in MG132-treated cells but not in vehicle-treated cells (Fig. 1D). Analysis of p53-deficient (p53−/−) cells treated with MG132revealed no marked differences in sensitivity to the proteasome inhibition compared with p53-proficient (p53+/+) cells (Fig. 1A and B), although PARP cleavage was somewhat delayed and the extent of DNA fragmentation was reduced compared with wt HCT116 cells (Fig. 1C and D). Thus, cell death by proteasome inhibition in HCT116 colon cancer cells seems to be independent of p53, in contrast to previous results with mammary cancer cells (20). Surprisingly, deletion of the Bax gene (Bax−/−) also had no effect on MG132toxicity (Fig. 1A). Both wt and Bax−/− cell lines underwent rapid cell death, about 60% in 48 hours (Fig. 1B). Consistent with these results, caspase-3 was also activated in Bax-deficient cells after treatment with MG132(Fig. 1C). Interestingly, similar to p53-negative cells, PARP cleavage was delayed and DNA was less fragmentized in Bax−/− cells treated with MG132compared with wt cells (Fig. 1C and D). Previous studies with p53−/− or Bax−/− HCT116 cells clearly showed that p53 and BAX had profound influence on cell responses to therapeutic agents, but the response varied considerably depending on the drug. Thus, the p53-deficient cells were sensitive to the effects of DNA-damaging agents, had the same apoptotic response to sulindac as wt cells, but became resistant to the toxicity of the antimetabolite 5-fluorouracil (17, 21). In contrast, Bax-deficient cells were only partially resistant to the apoptotic effects of 5-fluorouracil, but their apoptotic response to sulindac and other nonsteroidal anti-inflammatory drugs was completely abolished (17). Bax-deficient human colon carcinoma cells were also resistant to apoptosis induced by multiple stimuli such as death-receptor ligands, UV, staurosporine, and thapsigargin (22, 23). Our present results indicate that MG132 has a unique ability to induce colon cancer cell death independently of either p53 or BAX, suggesting that proteasome inhibitors may have a broad therapeutic potential for treating cancers with different genetic abnormalities in the components of the core apoptotic machinery.
FIGURE 1.
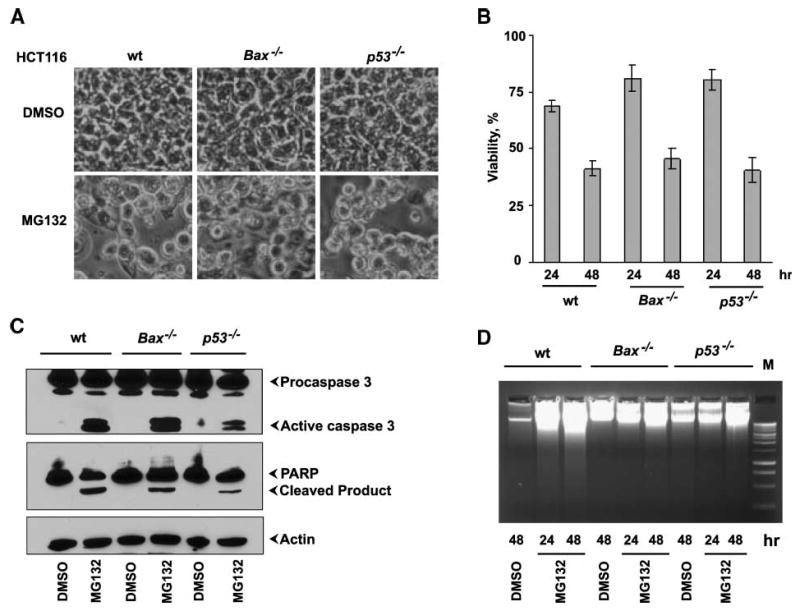
BAX- and p53-independent cell death by MG132. A. Representative light microscopic images (×120) of HCT116 wt, Bax−/−, or p53−/− cells 24 h after treatment with DMSO or MG132. B. The cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Columns, mean from three independent experiments; bars, SD. C. Effect on caspase-3 activation and PARP cleavage. The Western blots were probed with antibodies specific for procaspase-3 or PARP. D. Effect on fragmentation of chromosomal DNA. The low molecular weight DNA isolated from treated cells was analyzed by agarose gel electrophoresis.
Expression of BH3-Only Proteins, MCL-1 and p53
To characterize the components mediating MG132-induced apoptosis, we first determined expression profiles of the BCL-2-family BH3-only members in HCT116 wt, p53−/−, or Bax−/− cells (Fig. 2A). Cells were cultured for 24 hours in the presence of 1 μmol/L MG132 or DMSO, and total lysates were subjected to Western blot analysis. This analysis showed marked enhancement in the levels of several BH3-only proteins including BIK, BIM, and NOXA. The levels of the other proteins, BID, HRK, and BAD (not shown), showed no significant changes, whereas the level of PUMA was marginally increased (Fig. 2A, left). Among the BH3-only proteins, the most pronounced effect was noted in the accumulation of NOXA (Fig. 2A, left). Additionally, we also observed enhanced accumulation of BIK and all major isoforms of BIM (BIM-S, BIM-L, and BIM-EL; Fig. 2A, left). We next examined the levels of expression of BH1-3 proapoptotic members. There were no significant alterations in the expression of BAX and BAK on exposure to MG132 (Fig. 2A, bottom right), suggesting that the effect of MG132 might be primarily specific for BH3-only proteins.
FIGURE 2.
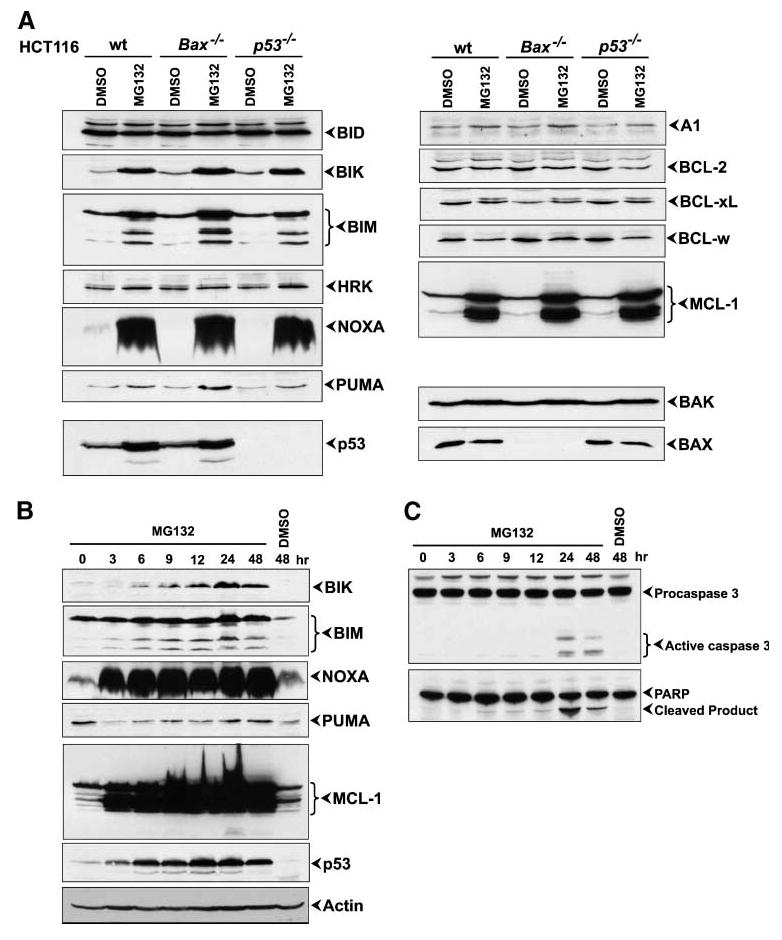
Effect of MG132 on expression of BCL-2-family proteins and p53. A. Western blot analysis of HCT116 wt, Bax−/−, or p53−/− cells 24 h after MG132 treatment. B. Time-course analysis of protein levels during MG132 treatment. C. Time-course analysis of caspase-3 activation and PARP cleavage during MG132 treatment.
We also investigated the effect of MG132on the expression of antiapoptotic members: A1, BCL-2, BCL-xL, BCL-w, and MCL-1. Expression of A1 showed a small increase after MG132treatment whereas the levels of BCL-2, BCL-xL, and BCL-w did not change significantly. We noticed the appearance of an additional, more slowly migrating band in BCL-xL Western blot analysis, probably indicating a modified form of BCL-xL. Among the prosurvival BCL-2-family proteins, the most striking effect was observed with MCL-1. In addition to the antiapoptotic isoform (MCL-1L), a short proapoptotic isoform (MCL-1S) has also been previously identified (24, 25). We observed strong accumulation of both isoforms (Fig. 2A). In addition to the effect on BH3-only proteins and MCL-1, MG132also induced accumulation of p53 in wt HCT116 cells (Fig. 2A).
To further characterize the effect of MG132on proapoptotic proteins induction, we evaluated the kinetics of accumulation of these proteins (Fig. 2B). HCT116 wt cells were treated with 1 μmol/L MG132, and lysates were collected at various time points for immunoblot analysis. Dramatic elevation in the levels of NOXA and MCL-1S was evident at 3 hours after exposure to MG132. Levels of BIM and p53 were also significantly increased at 3 hours after MG132treatment, and accumulation of BIK was noticeable at 6 hours. The levels of all tested proteins continued to increase for several hours, reaching the maximum at 24 hours. In addition, the Western blot analysis showed that activation of caspase-3 was first detectable at 24 hours after MG132 treatment (Fig. 2C), significantly later than induction of proapoptotic proteins, and this activation of caspase-3 was accompanied by increased processing of PARP. Thus, it is conceivable that rapid and sustained accumulation of BH3-only proteins and p53 was the apoptosis initiation event, but not the result of cell death caused by MG132.
Together, our results showed that treatment of colon cancer cells with MG132resulted in simultaneous increase in expression of several proapoptotic proteins, suggesting their synergistic involvement in the potent death-inducing activity of MG132 even in cells deficient in p53 (p53−/−) or BAX (Bax−/−).
We also investigated whether treatment with other major classes of proteasome inhibitors (19) would result in enhanced levels of multiple BH3-only proteins (Supplementary Fig. S1A). Among the tested proteasome inhibitors, PSI, epoxomicin, ALLN, and MG262 induced strong up-regulation of BIK, BIM, NOXA, MCL-1L, MCL-1S, p53, and, to a lesser degree, PUMA (Supplementary Fig. S1A) in a fashion that resembled the pattern induced by MG132. In contrast, AdaAhX3L3VS and α-MOL did not have detectable effect on the accumulation of BH3-only proteins. Interestingly, AdaAhX3L3VS and α-MOL also failed to activate caspase-3 and had no cytotoxic effect in HCT116 cells (Supplementary Fig. S1C). These results suggest a direct correlation between the ability of the proteasome inhibitors to elevate expression of BH3-only proapoptotic proteins and their cell death activity. Elevated levels of multiple BH3-only proteins, MCL-1 (MCL-1L and MCL-1S) and p53, accompanied by significant cell death, were observed in other tumor cell lines such as Saos2, LoVo, H1299, and C-33A (Supplementary Figs. S2 and S3), suggesting that proteasome inhibitors might be effective anticancer drugs for different neoplasms of epithelial origin.
Mode of Accumulation of BH3-Only Proteins
First, we determined whether up-regulation of BH3-only proteins and p53 reflected inhibition of proteasome-mediated protein degradation. We tested whether ubiquitinated forms of endogenous BH3-only proteins accumulated in response to MG132. We used a specific ubiquitin-binding affinity matrix (UbiQapture-Q, Biomol) to capture ubiquitinated proteins, followed by Western blot analysis (Supplementary Fig. S4A). There were marked increases in the levels of ubiquitinated NOXA, MCL-1, p53, BIM, BIK, and, to a lesser extent, PUMA. Thus, up-regulation of BH3-only proteins in HCT116 cells by MG132 is at least partially mediated by inhibition of their proteasomal degradation and accumulation of ubiquitinated forms of these proteins.
We then determined whether up-regulation of the BH3-only members might also be regulated at the level of transcription. We examined the mRNA levels of various BH3-only members by reverse transcription-PCR (RT-PCR) analysis at 20 hours after treatment with MG132(Supplementary Fig. S4B). There was significant induction of Noxa, Mcl-1L, Mcl-1S, Bim, Puma, and p53 mRNAs, whereas Bik mRNA level remained unchanged. Thus, transcriptional activation of several BH3-only proteins and p53 may also contribute to the overall accumulation of the apoptotic effectors in response to treatment with MG132. Because p53 is known to transcriptionally activate BH3-only members PUMA, NOXA (26), and human BIK (27), we determined whether the transcriptional activity of p53 is activated by MG132. HCT116 cells were transiently transfected with a luciferase reporter plasmid containing a p53-responsive element for 24 hours and then treated with MG132 or DMSO. As shown in Supplementary Fig. S4C, there was an activation of luciferase expression at 6 hours following MG132 treatment in p53+/+ cells but not in p53−/− cells, suggesting stabilization of a transcriptionally active form of p53. However, analysis of the mRNA levels of the BH3-only members by RT-PCR analysis of transcripts revealed no major differences between p53+/+ and p53−/− cells (data not shown). The transcription factor E2F1 is also a candidate to mediate up-regulation of BH3-only members such as PUMA, NOXA, BIM, and HRK that have previously been reported to be activated by E2F1 (28). To determine whether MG132 induced E2F1 expression, we examined the level of E2F1 by Western blot analysis. As seen in Supplementary Fig. S4D, E2F1 expression was up-regulated by treatment with MG132. In addition, we carried out a luciferase reporter assay to determine the effect of MG132 on E2F1 transcriptional activity (Supplementary Fig. S4E). The E2F1 promoter activity was increased in cells treated with MG132. These results suggest that E2F1 may function as an additional transcriptional mediator of MG132-induced cell death. Interestingly, E2F1 transcriptional activity as well as protein levels were induced to a greater extent in p53−/− cells than in p53+/+ cells (Supplementary Fig. S4D and E), suggesting that some level of redundancy may exist between the transcription factors involved in the process of cell death.
Role of Translation in Up-Regulation of BH3-Only Proteins
We next asked whether active protein synthesis is required for the accumulation of proapoptotic proteins during MG132 treatment. First, we determined the level of protein expression by treating HCT116 Bax−/− cells with translational inhibitor cycloheximide alone or in combination with MG132 (Fig. 3A). Addition of cycloheximide alone resulted in the disappearance (as measured by Western blot analysis) of MCL-1, NOXA, and p53 and a significant reduction of the level of expression of BIK as early as 6 hours after treatment. The level of BIM-EL did not change during this time point. At 24 hours after cycloheximide treatment, the levels of expression of BIK and BIM were also decreased substantially. Combination of MG132and cycloheximide led to some accumulation of BIK, BIM, and MCL-1 compared with the mock treatment, but proteins levels were still significantly lower when compared with MG132 treatment alone. The level of expression of NOXA and p53 (Fig. 3A) remained undetectable during combined treatment of MG132 and cycloheximide, suggesting that these two proteins have the shortest half-life among the proteins analyzed and their levels of expression seem to rely on active translation. In contrast to the effect on BH3-only proteins, the level of BH1-3 proapoptotic protein BAK remained unchanged after cycloheximide treatment alone or in combination with MG132(Fig. 3A). Next, we examined the effect of cycloheximide on MG132-induced apoptosis and showed that cycloheximide considerably inhibited MG132-dependent phosphatidyl-serine externalization by analysis of Annexin V–positive cells by flow cytometry at 48 hours after treatment (Fig. 3B). Thus, the reduction in the levels of BH3-only proteins seems to correlate with the suppression of MG132-dependent cell death despite the presence of additional proapoptotic BH1-3 proteins.
FIGURE 3.
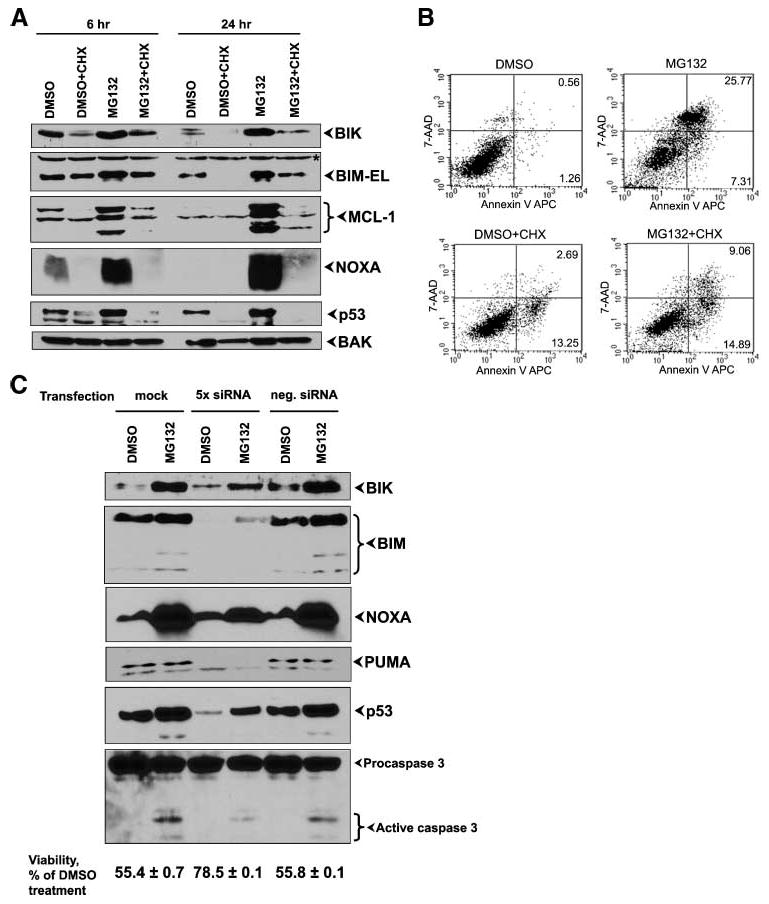
Effect of protein synthesis inhibition and siRNA-mediated knockdown of BH3-only proteins and p53. A. Western blot analysis of protein levels. HCT116 Bax−/− cells were treated with cycloheximide (CHX), DMSO, and MG132 alone or in combination as indicated and proteins were analyzed 6 and 24 h after treatment. B. Effect on apoptosis. The treated cells were stained with Annexin V and 7-amino-actinomycin D (7-AAD) and analyzed by flow cytometry. The percentages of Annexin V–positive (apoptotic) cells within the two right quadrants are indicated. C. Western blot analysis of protein depletion. HCT116 wt cells were transfected with siRNAs against BIK, BIM, NOXA, PUMA, and p53 or with negative control siRNA or mock transfected. Twenty-four hours later, the cells were treated with DMSO or MG132. The protein levels (top) and caspase-3 activation and cell viability (bottom) were determined 24 h after treatment.
Effect of Depletion of BH3-Only Proteins
To show the role of BH3-only proteins in MG132-induced cell death more directly, we performed loss-of-function analysis by knockdown of various BH3-only members and p53 using specific siRNAs. We assessed the levels of the knockdown by Western blot analysis. We found that simultaneous transfection of siRNAs for BIK, BIM, NOXA, PUMA, and p53 caused partial reduction of proteins expression (Fig. 3C, top) in DMSO-treated as well as MG132-treated cells. This partial depletion led to reduction of caspase-3 activation and partial increase in cell viability during exposure to MG132 (Fig. 3C, bottom). Interestingly, omitting p53 siRNA from the mixture of transfected siRNAs abolished the protective effect of siRNAs (not shown). Taken together, our data suggest a strong role for BH3-only proteins and p53 in MG132-induced cell death.
Thus, we conclude that enhanced expression of BH3-only proteins and p53 by the proteasome inhibitor MG132 is mediated by two distinct mechanisms: posttranslational protein stabilization and transcriptional activation. In addition, active protein synthesis is also essential for maintaining high levels of these proteins during proteasome inhibition.
BAX/BAK–Independent Cell Death Induced by MG132
Because the mammalian apoptotic death machinery is critically dependent on the functionally redundant BH1-3 proteins BAX and/or BAK, we undertook a study to determine the requirement of BAX and BAK in MG132-induced cell death. For these studies, we first exploited a HCT116 Bax−/− (BAXKO) cell line (17). We generated derivatives of the BAXKO cell line that are deficient for BAK by stable transfection of vectors that express shRNA against BAK. We choose two cell lines, #5 and #11, with different levels of BAK depletion and a control (C1) cell line from cells transfected with control shRNA for analysis (Fig. 4A). We first performed a short-term cell viability assay to compare MG132 toxicity in different HCT116 cell lines. Strikingly, we found that MG132 induced similar levels of toxicity in various cell lines (Fig. 4B), suggesting that cell death was independent of both BAX and BAK. In addition, analysis of Annexin V–positive cells (Fig. 4C) showed significant apoptosis in BAXKO/BAK↓ #5 cell line in response to MG132. We also examined microscopic images of HCT116 cells treated with MG132 for nuclear morphology characteristic of apoptosis. We observed numerous apoptotic cells showing chromatin condensation and margination with sharply circumscribed, dense crescents, again independently of the presence of BAX and BAK (Fig. 4D, image 1: wt cells, image 2: #5 cell line). Additional electron microscopic analysis revealed that exposure of both cell lines to MG132 resulted, regardless of the presence of BAX and BAK, in extensive cytoplasmic vacuolization clearly visible as early as 6 hours after treatment (Supplementary Fig. S5A). These distended vacuoles were delimited by electron-dense ribosomes (Supplementary Fig. S5B), a morphology consistent with the presence of markedly dilated rough ER. Electron micrographs after treatment of HCT116 cells with MG132 for 24 hours indicate an increase of volume of vacuoles. To determine whether this would result in increase of cellular volumes, we performed particle analysis for cellular sizes in light microscope images at 24 hours. Values for major and minor axes were acquired for individual cells to calculate an average diameter of cells treated with DMSO or MG132. MG132-treated cells (both HCT116 wt and BAXKO/BAK↓) showed marginal (statistically not significant) increase in size compared with control DMSO-treated cells (Supplementary Fig. S5C), arguing that as a population, they are not swollen, but with big deviation for different cells. Thus, the observed morphologic changes were not typical necrotic alterations characterized by nuclear and cytoplasmic swelling. A different proteasome inhibitor, epoxomicin, also efficiently killed the colon cancer cells, independently of their BAX and BAK status, as determined by clonogenic survival assay (Fig. 4E). A short-term dose-response viability study indicated that BAX-positive HCT116 cells were more sensitive to the reversible inhibitor MG132 compared with BAX- and BAX/BAK-negative cells at a low concentration of the inhibitor (0.5 μmol/L; Supplementary Fig. S6A). MG132 and epoxomicin (an irreversible inhibitor) at concentrations of 1 μmol/L and higher induced similar levels of cell death in different HCT116 cell lines (Supplementary Fig. S6A).
FIGURE 4.
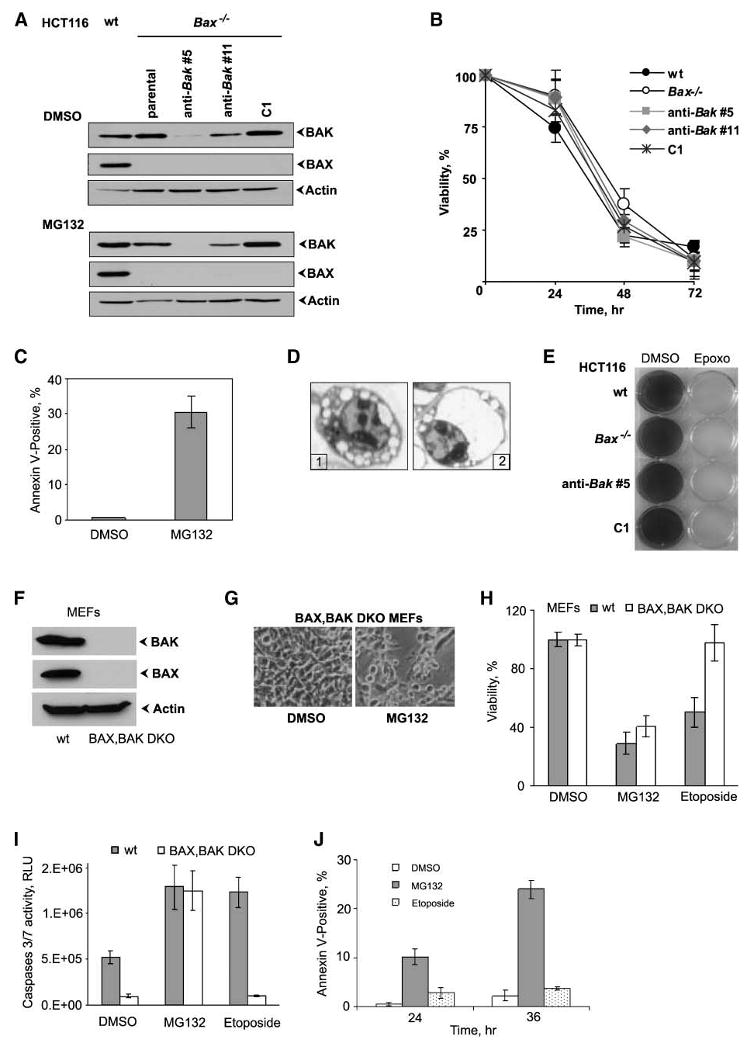
BAX/BAK–independent cell death by MG132. A. Western blot analysis of BAX and BAK levels in different HCT116 cell lines. B. The cell viability was determined by MTT assay. Points, mean percent relative to vehicle-treated cell lines; bars, SD. C. Analysis of apoptosis. HCT116 Bax−/−Bak↓ #5 cells were stained by Annexin V and analyzed by flow cytometry. Columns, mean percentage of Annexin V–positive (apoptotic) cells from three independent experiments; bars, SD. D. Photomicrographs of treated cells. Representative light microscopic images (×750) of HCT116 wt (image 1) and Bax−/−Bak↓ #5 (image 2) cells 24 h after treatment with MG132. Characteristic apoptotic nuclear morphology is seen in both types of cells. E. Colony survival assay. The cells were treated with epoxomicin (Epoxo) or DMSO. Twelve days after the treatment, colonies were stained with crystal violet and photographed. F. Western blot analysis of BAX and BAK proteins in whole-cell extracts of wt and BAX/BAK DKO MEFs. G. Representative light microscopic images (×120) of BAX/BAK DKO MEFs 24 h after treatment with DMSO or MG132. H. Viability of wt and BAX/BAK DKO MEFs. The viability was determined 48 h after exposure to MG132 or etoposide by the MTT assay. Columns, mean percent relative to vehicle-treated cells (n = 3); bars, SD. I. Caspase-3/caspase-7 activity in wt and BAX/BAK DKO MEFs 24 h after treatment with DMSO, MG132, and etoposide as measured with Caspase-Glo 3/7 kit (Promega). J. Analysis of apoptosis in BAX/BAK DKO MEFs. Cells were treated with DMSO, MG132, and etoposide. Annexin V–positive cells were analyzed by flow cytometry. Columns, mean from three independent experiments; bars, SD.
We next extended these studies to wt MEFs and BAX/BAK DKO MEFs (Fig. 4F). Microscopic examination (Fig. 4G) and cell viability assay (Fig. 4H) revealed that, similar to human cancer cells, wt and BAX/BAK DKO MEFs were effectively killed by MG132, whereas only wt MEFs, but not BAX/BAK DKO MEFs, were sensitive to etoposide, a well-known anticancer drug whose activity was shown to be dependent on BAX and BAK proteins in MEFs (29). To further characterize cell death in MEFs by MG132, we assessed the activities of caspase-3/caspase-7 (Fig. 4I). We found that MG132 caused significant increase in caspase-3/caspase-7 activity, comparable to both wt and BAX/BAK DKO MEFs. In contrast, etoposide activated caspase-3/caspase-7 only in wt MEFs. Additionally, MG132, but not etoposide, induced the appearance of Annexin V–positive BAX/BAK DKO MEFs (Fig. 4J).
BCL-2 is up-regulated in various cancers and seems to be a major factor that contributes to chemoresistance in human cancers (30). We asked whether overexpression of BCL-2in HCT116 cells affects the cytotoxicity of MG132. We established HCT116 cell lines that stably overexpressed BCL-2 (Supplementary Fig. S6B) and compared their susceptibility to MG132 with that of the parental cells. Figures S6C and D reveal that constitutive overexpression of BCL-2(in HCT116 BAXKO) did not increase the viability of MG132-treated cells. Similar results were also obtained with HCT116 wt cells (data not shown).
We next asked whether carbobenzoxy-Val-Ala-Asp-fluoromethylketone (zVAD-fmk) would also protect HCT116 cells and MEFs against MG132-induced cell death. Cell viability assay revealed that zVAD-fmk had no significant effect on MG132 toxicity (Supplementary Figs. S6E and S7A). At the same time, zVAD-fmk completely inhibited caspase-3 activation by MG132 as seen in DKO MEFs (Supplementary Fig. S7B) and prevented cleavage and activation of PARP in HCT cells (not shown), but had no effect on the appearance of biochemical markers of apoptosis induced by MG132 in DKO MEFs: mitochondrial outer membrane permeabilization, phosphatidyl-serine externalization, and loss of membrane potential (Supplementary Fig. S7C-E). Taken together, these observations suggested that the mere inhibition of caspase activities by zVAD-fmk is not enough to prevent MG132-induced cell death and that caspase-independent pathways are also involved in the apoptotic process. Thus, we speculate that proteasome inhibitors activate other proteases (e.g., cathepsins and granzyme B) in addition to caspases to promote the cleavage of crucial substrates and ultimate cell death, as many noncaspase proteases can cleave at least some of the classic caspase substrates and might mimic the cellular effects of caspases (31).
Mitochondrial Membrane Permeabilization by BH3-Only Proteins and p53
A key question was how apoptosis occurred in cells lacking two major death effectors—BAX and BAK. As mitochondria play a pivotal role in the regulation of apoptosis, we hypothesized that cell death would be initiated from activation of a cell-intrinsic pathway involving mitochondria resulting in mitochondrial membrane permeabilization (MMP). One of the well-characterized manifestations of MMP is the release of intermembrane proteins such as cytochrome c and apoptosis-inducing factor (AIF) into the cytosol. Therefore, we asked whether treatment of HCT116 cells with MG132 would induce this phenomenon. We isolated cytosolic and mitochondrial fractions from cells treated with MG132 or DMSO and performed Western blot analysis to determine the distributions of mitochondrial proteins. As early as 6 hours after treatment, we found significant amounts of cytochrome c and AIF proteins in the cytosol of MG132-treated cells but not in the cytosol of DMSO-treated cells (Fig. 5A, data are shown for HCT116 Bax−/− cells). Similar results were obtained in different HCT116 cell lines, regardless of the presence of BAX and BAK, as well as in BAX/BAK DKO MEFs (Supplementary Fig. S7D).
FIGURE 5.
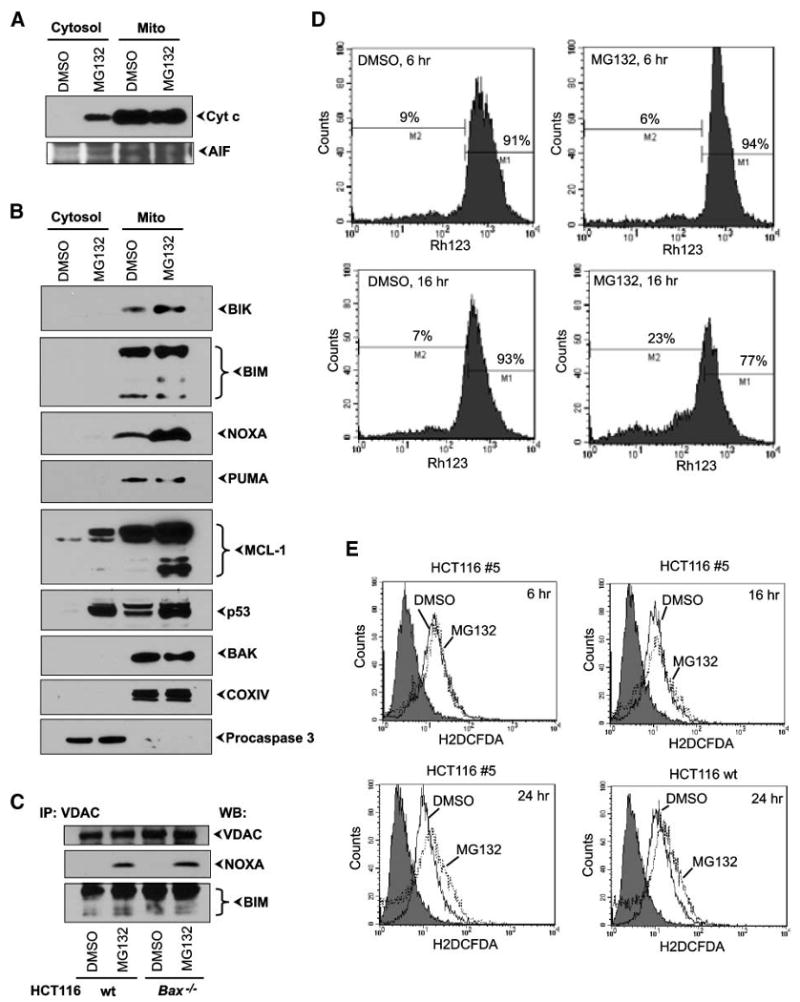
Mitochondrial association of BH3-only proteins and p53 and effect on MMP. A. Western blot analysis of cytochrome c (Cyt c) and AIF. The cytosolic and mitochondrial fractions of DMSO- or MG132-treated HCT116 Bax−/− cells 6 h after exposure were probed with the indicated antibodies. B. Western blot analysis of BH3-only proteins and p53. The above fractions were probed with the indicated antibodies. C. Interaction between VDAC and BH3-only proteins. Indicated HCT116 cell lines were treated as in A, and mitochondrial lysateswere immunoprecipitated with anti-VDAC antibody and analyzed by immunoblotting with anti-NOXA or anti-BIM antibodies. D. Effect on ΔΨm. HCT116 Bax−/−Bak↓ #5 cells were treated with DMSO or MG132, and ΔΨm was measured by flow cytometry using the fluorescent dye Rh123 at 6 and 16 h after treatment. E. Effect on ROS. HCT116 Bax−/−Bak↓ #5 or wt cells were treated with DMSO or MG132, and generation of ROS was measured by flow cytometry using the fluorescent dye 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) at different times.
What are the mitochondria-specific cell-death mediators in MG132-treated cells? Because we observed dramatic overexpression of several BH3-only proteins, we decided to examine whether the various BH3-only proteins activated by MG132 treatment were localized in the mitochondria. Western blot analysis of the mitochondrial fraction from Bax−/− HCT116 cells revealed that BH3-only proteins BIK, BIM, NOXA, PUMA, and MCL-1S, as well as MCL-1L and p53, were accumulated in the mitochondrial fraction, and dramatic differences in mitochondria from DMSO and MG132-treated cells were easily detectable as early as 6 hours after treatment (Fig. 5B). In contrast, there was no significant difference in the level of mitochondrial localization of BAK (Fig. 5B, bottom).
Multiple signaling pathways converge to cause MMP (32). We explored the interaction between BCL-2-family apoptosis regulators and mitochondrial proteins involved in vital cellular functions (e.g., VDAC, hexokinase, or glucokinase). VDAC proteins are attractive candidates for interactions with BH3-only proteins because they are constitutively anchored at the outer mitochondrial membrane and are essential components of the permeability transition pore complex. Moreover, it seems that Ca2+-induced permeability transition can occur efficiently in mitochondria from BAX/BAK deficient cells (33). To test whether complexes between VDAC and BH3-only proteins exist in our model system, we carried out coimmunoprecipitation analysis of mitochondrial fractions from DMSO- or MG132-treated cells. Specific endogenous complexes of VDAC/NOXA and VDAC/BIM-L were detected when extracts from MG132-treated, but not DMSO-treated, cells were precipitated with anti-VDAC and blotted with anti-NOXA or anti-BIM antibodies (Fig. 5C). In contrast, no complexes between VDAC and BIK, PUMA, MCL-1, or p53 were detected (data not shown). It is worth noting that the observed interaction between VDAC and BIM agrees with a previous report (34), whereas the VDAC/NOXA interaction is novel.
We then examined the effect of MG132on the mitochondrial inner transmembrane potential (ΔΨm) in different HCT116 cells and MEFs. There were no significant differences in ΔΨm at 6 hours after treatment (data not shown), but at 16 hours, we observed significant decreases in ΔΨm (Fig. 5D). Importantly, MG132-induced changes in ΔΨm were very similar in all tested HCT116 cell lines (Fig. 5D and data not shown) as well as in wt and Bax/Bak DKO MEFs (Supplementary Fig. S7E) regardless of the presence of BAX and BAK, supporting BAX/BAK–independent mode of apoptosis by MG132. Loss in ΔΨm occurs before significant changes in cell viability induced by MG132 but after mitochondrial accumulation of BH3-only proteins and p53 and release of cytochrome c and other apoptogenic factors from mitochondria (Fig. 5; Supplementary Fig. S7). In addition, we examined if the mitochondrial permeability transition pore inhibitor cyclosporin A was able to inhibit loss in ΔΨm during MG132-induced apoptosis in BAX/BAK DKO MEFs. We found that cyclosporin A was ineffective against MG132-dependent loss in ΔΨm (Supplementary Fig. S7E) and failed to inhibit release of cytochrome c and AIF from mitochondria of MG132-treated BAX/BAK DKO MEFs (Supplementary Fig. S7D) and cell death by MG132(Supplementary Fig. S7A).
Generation of reactive oxygen species (ROS) by mitochondria is also believed to contribute to mitochondria-dependent apoptosis. Therefore, we examined whether MG132treatment would result in up-regulation of ROS levels. We measured the ROS level using the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate in HCT116 cells with different BAX and BAK backgrounds that were treated with MG132 or DMSO. Elevation of ROS was detected only at 24 hours (Fig. 5E). HCT1116 wt cells and BAXKO/BAK↓ cells showed similar patterns of ROS generation on treatment with MG132 (Fig. 5E and data not shown). Thus, our results are consistent with the interpretation that in HCT116 cells exposed to MG132, ROS generation might be a consequence of apoptotic cascade (cytochrome c release and loss of ΔΨm).
Taken together, our observations provide evidence for a sequence of events leading to a mitochondrial apoptotic pathway following proteasome inhibition by MG132. We suggest that MG132 induces BAX/BAK–independent apoptosis through activation of multiple BH3-only proteins and p53 and their accumulation at mitochondria leading to the release of apoptogenic factors into the cytosol, followed by loss of ΔΨm and, consequently, increased generation of ROS.
MG132-Induced Mitochondrial Degradation
To determine whether the structural changes to mitochondria accompanied the observed loss of MMP, we collected transmission electron microscopic images of HCT116 wt and BAXKO/BAK↓ cells treated with MG132 or DMSO. We used two time points after addition of MG132 or DMSO: 6 hours (stage I, characterized by dramatic accumulation of proapoptotic proteins in mitochondria, release of cytochrome c, but no changes in ΔΨm) and 24 hours (stage II, after the release of cytochrome c and loss of ΔΨm). Mitochondria from DMSO-treated HCT116 wt and BAXKO/BAK↓ cells were of normal morphology at all time points of observation (Fig. 6A, images a and f). However, after the addition of MG132, mitochondria were seen to undergo dramatic changes. At 6 hours of MG132treatment, most mitochondria had normal morphology but some of them appeared in a transition form, similar to the “normal-vesicular” transition described by Sun et al. (ref. 35; Fig. 6A, images b and g). At 24 hours, the majority of the mitochondria in MG132-treated cells exhibited either very distinctively altered “vesicular” ultrastructure (ref. 35; Fig. 6A, images c and h) or swollen ultrastructure (Fig. 6A, images e and j). In addition, an intermediate form, “vesicular-swollen,” was also observed in which one part of a mitochondrion appeared to be swollen, whereas another part appeared vesicular (Fig. 6A, images d and i). The above-mentioned mitochondrial changes resembled the recently described structural transformations of mitochondria during etoposide-induced apoptosis (35). However, we have observed such transformations during MG132-induced apoptosis independently of the presence of BAK and BAX.
FIGURE 6.
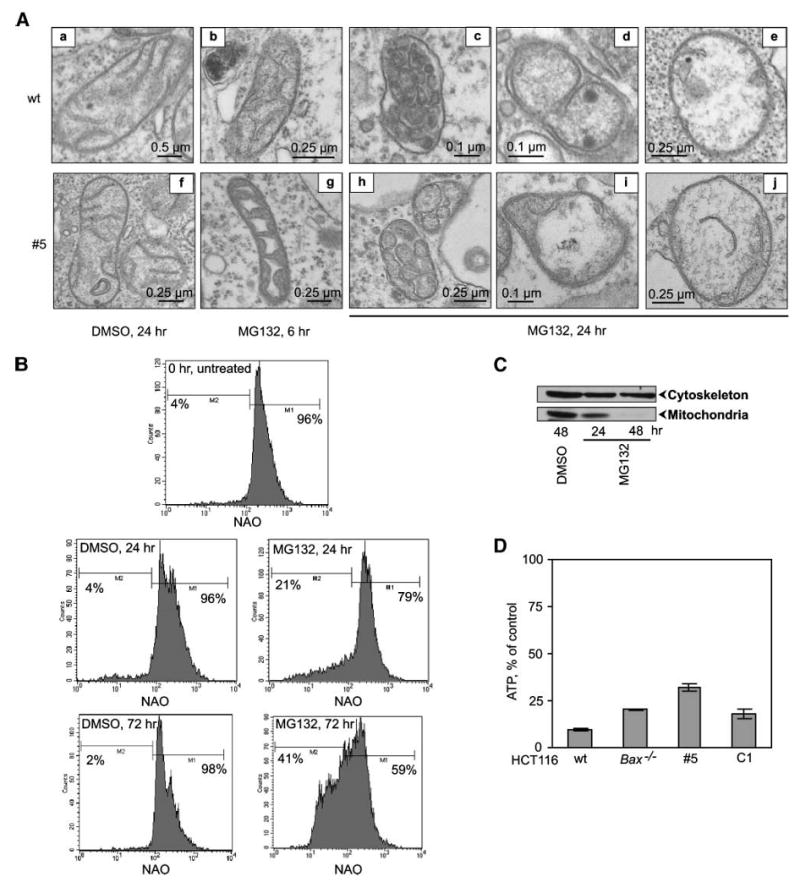
Analysis of mitochondrial ultrastructure and functions induced by MG132 in HCT116 cells. A. Representative electron microscopic images of mitochondria from HCT116 wt and Bax−/−Bak↓ #5 cells treated with MG132 or DMSO. B. The effect on mitochondrial mass was determined by staining with nonyl-acridine orange at different times after treatment with MG132 or DMSO and analyzed by flow cytometry. C. Analysis of mitochondrial and cytoskeletal markers. The total cell lysates from treated cells were analyzed by immunoblotting using anti-CoxIV and anti-actin antibodies. D. ATP levels in MG132-treated cells relative to control DMSO-treated cells at 48 h. Columns, mean; bars, SD.
Mitochondrial damage was further determined by flow cytometric analysis of total mitochondrial mass. We found that at 24 hours of MG132 treatment, a significant subpopulation of HCT116 cells displayed a reduced mitochondrial mass whereas control DMSO-treated cells showed no such alterations (Fig. 6B). MG132-induced loss of total mitochondrial mass progressed over time and, more importantly, the process was BAX/BAK independent (Fig. 6B). In addition, the mitochondrial content after exposure to MG132 in HCT116 cells was assessed in whole-cell extracts by probing for cytochrome c oxidase subunit IV (CoxIV), an inner mitochondrial membrane protein in comparison with actin (Fig. 6C). After MG132 treatment, CoxIV levels were greatly dimished whereas actin levels were unaffected. Mitochondrial disappearance was not detectable in DMSO-treated cells (Fig. 6C).
We also determined the general metabolic activity of mitochondria by determining the levels of ATP in MG132-treated cells at different time points. Intracellular ATP levels of control cells and MG132-treated cells remained comparable at 6, 16, and 24 hours (not shown) but dropped dramatically at 48 hours, indicating almost complete metabolic catastrophe in cells treated with MG132 (Fig. 6D). Sudden loss of ATP level (70-90%) was observed in all tested cell lines, although some differences were noticeable between wt and BAXKO/BAK↓ cells, that is, the remaining level of ATP was higher in BAXKO/BAK↓ cells than in wt MG132-treated cells, and the remaining ATP levels in BAXKO or C1 cells were intermediate.
Importantly, analysis of mitochondrial damage by MG132 in BAX/BAK DKO MEFs revealed a similar pattern (Fig. 7). Thus, mitochondria from BAX/BAK DKO MEFs exposed to MG132 underwent similar ultrastructural alterations (Fig. 7A). MG132, but not etoposide, caused loss of total mitochondrial mass in BAX/BAK DKO MEFs (Fig. 7B). In accordance with the previous results, significant decrease in ATP levels was observed in wt and BAX/BAK DKO MEFs treated with MG132(Fig. 7C). It is also important to note that zVAD-fmk and cyclosporin A failed to prevent alterations in mitochondrial ultrastructure and mitochondrial degradation induced by MG132(Fig. 7).
FIGURE 7.
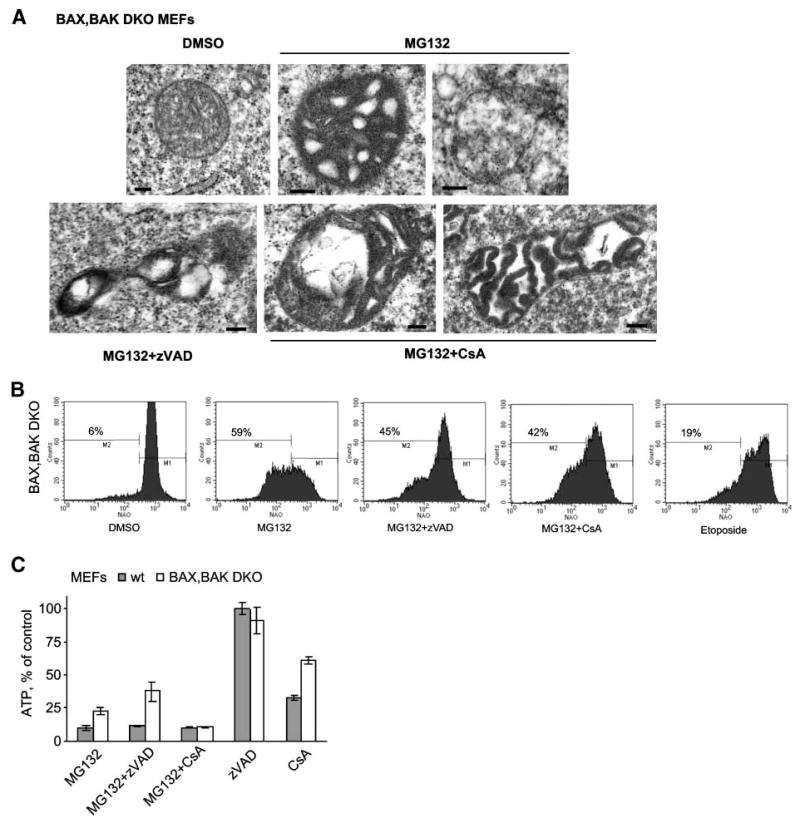
Analysis of mitochondrial ultrastructure and functions induced by MG132 in BAX/BAK DKO MEFs. A. Representative electron microscopic images of mitochondria from BAX/BAK DKO MEFs treated with DMSO or MG132 alone or in combination with zVAD-fmk (zVAD) or cyclosporin A (CsA). B. Mitochondrial mass was determined by staining with nonyl-acridine orange 72 h after treatment with DMSO or MG132 alone or in combination with zVAD-fmk or cyclosporin A and then analyzed by flow cytometry. C. ATP levels in wt or BAX/BAK DKO MEFs treated with MG132 and/or zVAD-fmk and/or cyclosporin A relative to control DMSO-treated cells at 48 h. Columns, mean; bars, SD.
Thus, taken together, our results indicate that MG132-induced cell death proceeds through a mitochondrial death program and loss of essential mitochondrial functions. The ultrastructural transformation and the ultimate functional demise of mitochondria observed here seem to be consistent with the features associated with mitochondrial apoptosis—fragmentation and disruption of the mitochondrial network, leading to mitochondrial “death” or mitoptosis (13, 16).
Cell Death by Simultaneous Expression of Several BH3-Only Proteins and p53 in the Absence of BAX and Reduced BAK Expression
BH3-only proteins, when acting individually, require proapoptotic BH1-3 proteins to release cytochrome c and induce cell death (29, 36) in MEFs or only BAX in HCT116 cells (23). Our results indicate that MG132-induced cell death occurs in a BAX/BAK–independent manner, but in the presence of overexpression of multiple BH3-only proteins and p53. Therefore, we hypothesized that simultaneous expression of several BH3-only proteins and p53 would result in death of cells deficient in BAX and BAK. Hence, we asked whether coexpression of several BH3-only proteins and p53 could induce cell death in HCT116 cells. For this purpose, we used a clonogenic survival assay of HCT116 cells with different backgrounds for BAX and BAK expression. Cotransfection of various BH3-only members and p53 resulted in significant reduction in colony formation compared with cells that were transfected with individual expression constructs (Fig. 8A). As expected, multiple BH3-only proteins and p53 induced cell death in wt HCT116 as well as in Bax−/− and #5 cell lines, although to a lesser extent than in wt. To ascertain expression of various proteins coded by the transfected constructs in surviving colonies, the cells from pooled colonies were expanded and analyzed by Western blotting. Interestingly, cells derived from wt HCT116 cells did not express any of the transfected genes (Fig. 8B), showing efficient cell killing by various proapoptotic proteins. Cells survived after transfection and selection of BAXKO/BAK↓ HCT116 cells expressing BIK, BIM-S, MCL-1S, and PUMA, but not NOXA or additional p53 (Fig. 8B). These results suggest that whereas engagement of these proteins is required for efficient cell-death activity in cells lacking BAX and reduced BAK, NOXA and p53 are probably the most critical proteins for this process. Thus, expression of several BH3-only proteins without p53 resulted in significant cell death in wt but not in BAXKO/BAK↓ HCT116 cells (Fig. 8C).
FIGURE 8.
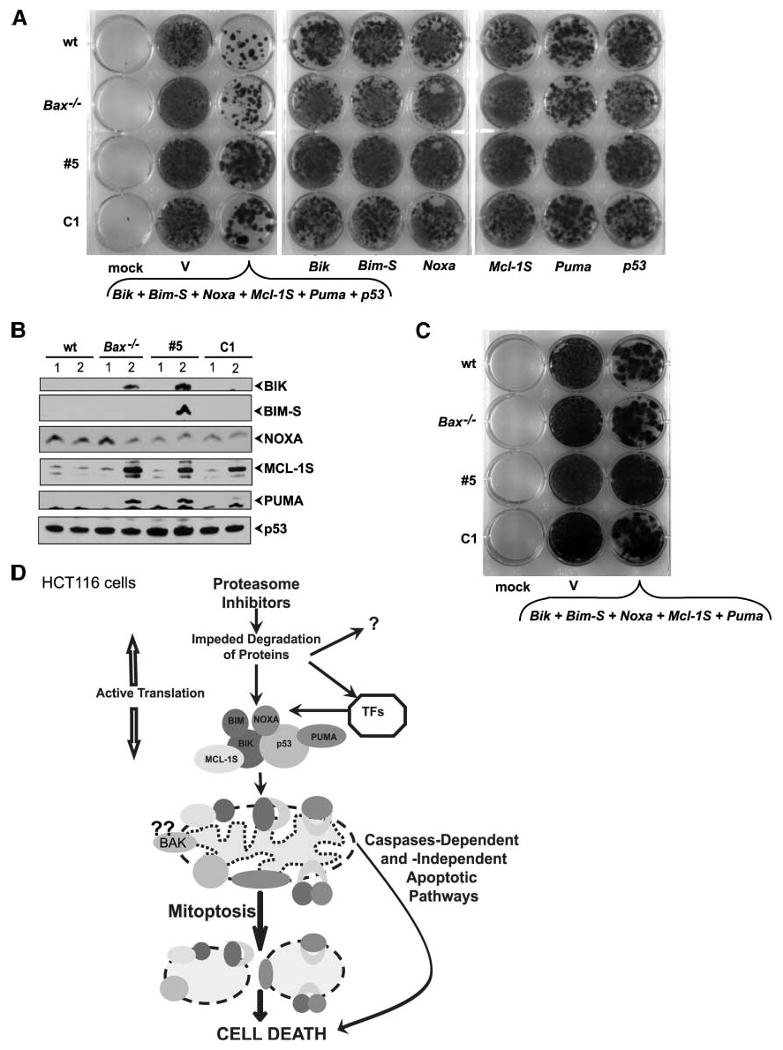
Effect of coexpression of BH3-only proteins and p53 on cell death in HCT 116 cells. A. Colony survival assay. Indicated HCT116 cell lines were either mock transfected or transfected with empty vector or vectors expressing various proteins together or individually. Cells were then selected with G418 for 14 d, stained with crystal violet, and photographed. B. Western blot analysis of BH3-only proteins and p53 in surviving cells. The transfected cells were expanded after G418 selection and analyzed by Western blotting. C. Colony survival assay. Indicated HCT116 cell lines were either mock transfected or transfected with empty vector or vectors expressing indicated proteins. Cells were then selected with G418 for 14 d, stained with crystal violet, and photographed. D. Model for the MG132-induced cell death in HCT 116 cells. Exposure to proteasome inhibitors results in fast and dramatic up-regulation of BH3-only proteins and p53 through inhibition of degradation and activation of transcription by different transcription factors (TFs). Proapoptotic proteins accumulate at the mitochondria and induce structural and functional mitochondrial demise—mitoptosis and, consequently, cell death. Additional death signaling pathways (e.g., caspase-dependent as well as caspase-independent cell death pathways) are also activated.
Summarizing, these experiments established proof-of-principle that several BH3-only proteins and p53, when expressed simultaneously, can instigate apoptosis in the absence of BAX and with significantly reduced BAK in HCT116 cells. Thus, in addition to the well-established and accepted role of BH3-only proteins and p53 as initiators of mammalian apoptosis program, we propose that these proteins might also act as effectors of the apoptotic cascade modulating MMP as well as other mitochondrial functions essential for cell survival independently of BAX and, probably, BAK.
Discussion
Initially, a major rationale for the therapeutic use of the proteasome inhibitor bortezomib was its ability to inhibit nuclear factor-κB activation (37-39). Later, the use of these agents was advanced on their ability to induce apoptotic cell death (4, 40). Additionally, nonapoptotic mechanisms were also suggested, and it was further suggested that BAX might be the switch between apoptotic nonapoptotic death (41). Our results clearly show that MG132induces apoptosis independently of both BAX and BAK. More strikingly, our data implicate several BH3-only proteins, BIK, BIM, NOXA, PUMA, and MCL-1S, and p53 as initiators and mediators of cell death by MG132. Several observations support a major role for these proteins in directly triggering mitochondrial proapoptotic action of MG132in the absence of BAX and BAK: (a) MG132 induces dramatic up-regulation in the level of expression of these proteins very early after exposure; (b) only proteasome inhibitors that provoke an increase in the expression of BH3-only proteins and p53 cause cell death; (c) down-regulation of expression of BH3-only proteins and p53, but not BH1-3 proteins, by cycloheximide leads to the reduction of MG132-dependent cell death; (d) specific reduction of expression BIK, BIM, NOXA, PUMA, and p53 by siRNAs results in reduction in MG132-induced cell death; (e) BH3-only proteins and p53 rapidly accumulate at the mitochondria, resulting in (f) MMP and (g) pronounced changes in mitochondrial ultrastructure. We also show that the initial mitochondrial alterations induced by proteasome inhibitors progress over time and result in structural and functional death of mitochondria. These impairments have detrimental effects on cell metabolism and lead to cell death. A mitochondrial death program or “mitoptosis” has been described in various experimental systems (14, 16) and was hypothesized to take place in cells undergoing apoptosis (13).
Only limited data about the mechanism of mitoptosis are available at present. Our results show that mitoptosis is an essential pathway of MG132-induced apoptosis regardless of the presence BAX and BAK. Moreover, experiments with zVAD-fmk reveal that mitoptosis may proceed in the absence of caspase activation. During cell death by proteasome inhibitors, mitoptosis is initiated by multiple BH3-only proteins and p53. The precise role of p53 in apoptosis by MG132is not clear at present. Our data show that p53−/− HCT116 cells are as sensitive to proteasome inhibitors as their wt counterparts. It should be noted that p53−/− and wt cells express BAX, and activation of even a single BH3-only protein by MG132would be enough for apoptosis to proceed. In the absence of BAX and BAK, however, p53 is an important member of proapoptotic team. As shown recently, mitochondrial p53 is able to disrupt the integrity of both outer and inner mitochondrial membranes in vivo, functionally resembling a “super” BH3-only protein (42). Furthermore, the predominant form of p53 at the mitochondria is a dimer or a higher-order oligomer (43). This interesting observation led us to speculate that, similar to oligomerization of BAX and BAK, this might contribute to the pore-forming activity of p53 besides its interaction with BCL-2-family proteins at mitochondrial membrane. Therefore, mitochondrial p53 might functionally substitute for BAX and BAK. It is not clear how BH3-only proteins and p53 cause changes in mitochondrial ultrastructure and ultimately mitoptosis. Previous publications have suggested that changes in ultrastructure occur and lead to mitochondrial fragmentation and then mitoptosis (14). It was also shown that mediators involved in mitochondrial fission (44) and fusion (45) may regulate mitochondrial fragmentation during apoptosis due to an increase in fission, decrease of fusion, or both. A recent study in C. elegans showed that EGL-1 (worm BH3-only protein) induced mitochondrial fragmentation that was dependent on CED-9 and mediated by dynamin-related protein DRP1, which is one of the mediators of mitochondrial fission (46). In mammalian cells, activation of DRP1 and its recruitment to the mitochondria can be induced by the release of Ca2+ from endoplasmic reticulum stores and subsequent uptake by mitochondria. Interestingly, the BH3-only protein BIK was shown to activate this pathway (47). In addition, BH3-only proteins and p53 may also promote mitochondrial fragmentation through direct binding to antiapoptotic BCL-2-family proteins and thus inhibition of mitochondrial fusion. Support for this mechanism came from recent studies with CED-9 and EGL-1 proteins, which showed that expression of CED-9 (as well as BCL-xL) in mammalian cells promoted mitochondrial fusion, and that EGL-1 antagonized this function by binding to CED-9 (48). The possibility that mammalian BH3-only proteins and p53 may inhibit the fusion-promoting activity of BCL-xL and result in mitochondrial fragmentation remains to be investigated. Several studies have also shown that following MMP, other intermembarane space proteins in addition to cytochrome c, such as DDP/TIMM8a (15) and OPA1 (45), were released, resulting in mitochondrial fission and inhibition of fusion, respectively. Thus, it seems that by causing MMP, BH3-only proteins and p53 may also promote the release of these proteins leading to mitochondrial fragmentation and subsequent elimination during apoptosis by the proteasome inhibitors.
Various mechanisms for the apoptosis-induced release of proapoptotic proteins from the mitochondrial intermembrane space may exist (32). In a more well-known MEFs model, the BH3-only proteins have been proposed to initiate this process through activation of multidomain proapoptotic BAX and BAK (29) either through direct binding or through interaction with the antiapoptotic BCL-2-family members (49, 50). Cells from BAX/BAK DKO mice fail to undergo MMP in response to a wide range of apoptotic signals, including individual BH3-only proteins (29, 36). Our results show that MG132induces MMP independently from BAX and BAK, most likely through activation of multiple BH3-only proteins and p53 simultaneously. We found that two BH3-only proteins, BIM and NOXA, bound to VDAC, and this interaction may promote MMP. However, the results with mitochondrial permeability transition pore inhibitor, cyclosporin A, argue that this is the only mechanism in MG132-induced MMP because cyclosporin A was ineffective against MG132-induced MMP. We propose that direct insertion of multiple BH3-only proteins into the outer mitochondrial membrane (in the absence of interaction with other BCL-2-family proteins) may also induce MMP. A precedent for this scenario was suggested by a recent report that mutants of BIM-S competent for mitochondrial insertion but not for interaction with BCL-2-family members induced MMP and cell death (51).
Taken together, we suggest that proteasomal inhibition by MG132in epithelial cancer cells activates factors involved in the intrinsic apoptotic pathway, including p53 and the BH3-only proteins BIK, BIM, MCL-1S, NOXA, and PUMA. Subsequent cell death occurs through mitoptosis, the irreversible deterioration of mitochondrial ultrastructure, and final organelle demolition leading to the metabolic collapse (Fig. 8D). Concurrently, MMP and the release of intermembrane space proteins (such as cytochrome c, AIF, etc.) into the cytosol might cause cell death through overlapping caspase-dependent and caspase-independent pathways.
Materials and Methods
Cells and Plasmids
HCT116 wt, Bax−/−, and p53−/− cell lines were gifts from Dr. B. Vogelstein. C-33A and LoVo cell lines were obtained from American Type Culture Collection. SV40-transformed wt and bak−/−, bax−/− DKO MEFs were gifts from Dr. S. Korsmeyer. Plasmids used in this study were pcDNA3HABIK, pcDNA3p53, pcDNA3N-HAhNOXA, pcDNA3FTBbc3, pE-FEEBIM-S, and pcDNA3MCL-1L. pcDNA3MCL-1S plasmid was constructed from pMIG-MCL-1 (a gift from E. Cheng) by introducing a deletion, Δ690-937, using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) and recloning into pcDNA3 vector.
Proteasome Inhibitors and Antibodies
The following inhibitors obtained from Calbiochem were used: AdaAhX3L3VS, ALLN, epoxomicin synthetic [Ac (Me)-IITL-EX], Hdm2 E3 ligase inhibitor, clasto-lactastatin-β-lactone, α-methylmuralide, MG132(Z-LLL-CHO), MG262 [Z-LLL-B(OH)2], and proteasome inhibitor I [Z-IE(OtBu)AL-CHO]. The antibodies were obtained from the following commercial sources: A1, actin, BAD, BIK, and PUMA from Santa Cruz Biotechnology; BCL-2, BCL-w, cytochrome c, BID, and BIM from Pharmingen; BCL-xL from Chemicon International; MCL-1 and caspase-3 from Stressgen; BAK, BAX, and E2F-1 from Upstate; HRK from MBL International; p53 from Oncogene; CoxIV from Molecular Probes; and NOXA, VDAC, and PARP from Calbiochem.
Cell Viability and Apoptosis Assays
Cell viability was determined using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). Cell death was calculated as percent decrease of absorbance in proteasome inhibitor-treated cells compared with vehicle-treated cells. The data represent the mean ± SD from eight microwells from each of at least two independent experiments. The percentage of apoptotic cells was determined by Annexin V-allophycocyanin (Pharmingen) staining and by flow cytometric analysis. The viability stain 7-amino-actinomycin D (Pharmingen) was also included in the assay and used as an indicator of membrane structural integrity. DNA fragmentation assay was done as described (52). Colony survival assay was carried out by transfection of HCT116 cells with vectors expressing BIK, BIM-S, NOXA, PUMA, p53, and MCL-1S using FuGene transfection reagent (Roche); cells were then selected for 14 d with G418, stained with crystal violet, and photographed.
Analysis of ΔΨm, Mitochondrial Mass, Caspase Activity, and Cellular ATP Levels
Mitochondrial dysfunction was assessed using the cationic lipophilic green fluorochrome Rh123 (Invitrogen), as previously described (53). Disruption of ΔΨm was measured by flow cytometric analysis of cells incubated with Rh123 (0.5 μg/mL) for 30 min. The mitochondrial mass was determined by flow cytometric analysis using 10-N-nonyl acridine orange (Molecular Probes) as described (54). The activity of caspase-3/caspase-7 was measured using Caspase-Glo 3/7 kit from Promega. ATP levels were analyzed using the ATP Bioluminescence assay (Promega).
Subcellular Fractionation, Western Blot Analysis, and Immunoprecipitation
Cells (5 × 106 per dish) were plated in 100-mm-diameter dishes and, on the next day, treated with DMSO or MG132 for 6 h, and the mitochondrial and cytosolic fractions were isolated as described earlier (52). After proteasome inhibitor treatment, the cell extracts were prepared; immunoprecipitation and immunoblotting were carried out as previously described (52).
RT-PCR Analysis
mRNA was isolated from HCT116 wt and p53−/− cells treated with MG132or DMSO using the QuickPrep micro mRNA Purification Kit (Amersham Biosciences). cDNA was synthesized using Protoscript II RT-PCR Kit and oligo-dT primers (New England Biolabs) following the manufacturer's instructions. PCR amplification was done using Taq polymerase (New England Biolabs) as recommended by the manufacturer. The following primers were used: Gapdh, TGAAGGTCGGAGTCAACGGATTTGGT (forward) and CATGTGGGCCATGAGGTCCACCAC (reverse); Bik, CTCAGGGCCCCGCGCCTGGCCCAGCTC (forward) and CGCCGGGGCCTCACTTGAGCAGCAGGT (reverse); Bim, ATGAGAAGATCCTCCCTGCT (forward) and AATGCATTCTCCACACCAGG (reverse); Mcl-1, GAGGAGGAGGAGGACGAGTT (forward) and ACATTCCTGATGCCACCTTC (reverse); Noxa, GAGATGCCTGGGAAGAAGGCGCGCAAG (forward) and AAAGTGGTTTTATAATGTTCTCTCATC (reverse); p53, CCTCACCATCATCACACTGG (forward) and CCTCATTCAGCTCTCGGAAC (reverse); and Puma, CAGACTGTGAATCCTGTGCT (forward) and ACAGTATCTTACAGGCTGGG (reverse). The specificity of PCR amplification of each primer pair was confirmed by analyzing the PCR products by gel electrophoresis. Each primer pair was also tested with a logarithmic dilution of a cDNA mix to generate a linear curve. The PCR products were then amplified in the linear range, separated by 6% PAGE gel electrophoresis, and visualized by Vistra Green (Amersham Biosciences). The images of the stained bands were obtained using Storm 860 system (Amersham Biosciences) and quantified using ImageQuant software.
Luciferase Reporter Assay
Luciferase reporter plasmids p53-Luc (Stratagene) or pGL2-E2F/luc (a gift from S. Chellappan), as well as the respective negative control plasmids (pCis-CK and pGL2-E2Fmut/luc), and pRL-TK (Promega) were cotransfected into HCT116 wt or p53−/− cells. Twenty-four hours after transfection, cells were treated with MG132or vehicle (DMSO) and, 6 h later, assayed for luciferase activity using the Dual Luciferase Reporter Assay System (Promega).
Ubiquitin Pull-Down Assay
Ubiquitinated proteins were isolated using UbiQapture-Q Kit (Biomol) according to the manufacturer's instructions. HCT116 cells were plated at 5 × 106 per dish in 100-mm dishes and, on the next day, treated with DMSO or MG132for 16 h. Ubiquitinated proteins were isolated from the total cell lysates with 150 μL UbiQapture-Q matrix by rotating samples for 3 h at 4°C. After washing four times, captured proteins were eluted with 2× SDS-PAGE loading buffer and analyzed by Western blotting using specific antibodies.
Stable and Transient Protein Knockdown
HCT116 Bax−/− cells were transfected with a vector expressing BAK-specific shRNA (pRS-shBAK, OriGene) and selected with puromycin (1 μg/mL; Invitrogen). Puromycin-resistant clones were isolated and expanded. Clones with significant down-regulation of BAK were identified by Western blotting. Transient depletion of BH3-only proteins and p53 was carried out by transfection of siRNAs (Qiagen) targeted against various proteins—BIK (Hs_BIK_4), BIM (Hs_BCL2L11_5), NOXA (Hs_PMAIP1_1), PUMA (Hs_BBC3_2), and p53 (Hs_TP53_9). The siRNAs were transfected into HCT116 wt cells with HiPerFect transfection reagent (Qiagen). After 24 h, cells were treated with 1 μmol/L MG132or DMSO for 24 h and analyzed for protein expression, caspase-3 activation, and cell viability.
Transmission Electron Microscopy
Cells were harvested by trypsinization and washed once in PBS, then the cell pellets were fixed with 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer (pH 7.25) containing 2% sucrose and 2mmol/L calcium chloride for 24 h at 4°C. The following steps up to polymerization were at room temperature. Cell pellets were washed in 0.1 mol/L sodium cacodylate buffer containing 5% sucrose and postfixed in 1% osmium tetroxide in 0.1 mol/L sodium cacodylate buffer containing 2% sucrose for 24 h. The pellets were washed twice in distilled water, dehydrated through graded ethanols to 100%, rinsed twice in propylene oxide, and infiltrated with a 1:1 mixture of Polybed resin (Polysciences, Inc.) and propylene oxide in capped microcentrifuge tubes for 24 h. The cell pellets were then incubated in fresh Polybed resin for 6 h, transferred to BEEM capsules filled with fresh resin, and polymerized overnight at 75°C. Thick (0.25 μm) sections were heat-attached to glass microscope slides, stained with toluidine blue, coverslipped in Permount mounting medium (Fisher Scientific), and photographed using a Zeiss research light microscope equipped with an Olympus digital camera. Ultrathin sections (0.05 μm) were cut with a diamond knife using a Reichert Ultracut E ultramicrotome, collected on 200-mesh copper grids, post-stained with uranyl acetate and lead citrate, and viewed and photographed with a JEOL 100CX transmission electron microscope.
Supplementary Material
Acknowledgments
We thank Drs. S. Chellappan (H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL), E. Cheng (Washington University, St. Louis, MO), the late S. Korsmeyer (Dana-Farber Cancer Institute, Boston, MA), B. Vogelstein (Johns University, Baltimore, MD), David Huang (Water and Eliza Hall Institute of Medical Research, Melbourne, Australia), and E. White (Rutgers University, Piscataway, NJ) for the generous gifts of plasmids and cell lines, and Joy Eslick and Sherri Koehm for flow cytometry analysis.
Grant support: National Cancer Institute grants CA-33616 and CA-73803.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–60. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 2.Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P. Proteasome inhibitors: antitumor effects and beyond. Leukemia. 2007;21:30–6. doi: 10.1038/sj.leu.2404444. [DOI] [PubMed] [Google Scholar]
- 3.Lopes UG, Erhardt P, Yao R, Cooper GM. p53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem. 1997;272:12893–6. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- 4.Chauhan D, Catley L, Li G, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib. Cancer Cell. 2005;8:407–19. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Nawrocki ST, Carew JS, Dunner K, Jr, et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–9. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- 6.Concannon CG, Koehler BF, Reimertz C, et al. Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway. Oncogene. 2007;26:1681–92. doi: 10.1038/sj.onc.1209974. [DOI] [PubMed] [Google Scholar]
- 7.Youle RJ, Strasser A. The BCL-2protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 8.Chipuk JE, Green DR. How do BCL-2proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–64. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu H, Zhang L, Dong F, et al. Bik/NBK accumulation correlates with apoptosis-induction by bortezomib (PS-341, Velcade) and other proteasome inhibitors. Oncogene. 2005;24:4993–9. doi: 10.1038/sj.onc.1208683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin JZ, Ziffra J, Stennett L, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65:6282–93. doi: 10.1158/0008-5472.CAN-05-0676. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez Y, Verhaegen M, Miller TP, et al. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 2005;65:6294–304. doi: 10.1158/0008-5472.CAN-05-0686. [DOI] [PubMed] [Google Scholar]
- 12.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–12. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 13.Skulachev VP. Programmed death phenomena: from organelle to organism. Ann N Y Acad Sci. 2002;959:214–37. doi: 10.1111/j.1749-6632.2002.tb02095.x. [DOI] [PubMed] [Google Scholar]
- 14.Skulachev VP, Bakeeva LE, Chernyak BV, et al. Thread-grain transition of mitochondrial reticulum as a step of mitoptosis and apoptosis. Mol Cell Biochem. 2004:256–7. 341–58. doi: 10.1023/b:mcbi.0000009880.94044.49. [DOI] [PubMed] [Google Scholar]
- 15.Arnoult D, Rismanchi N, Grodet A, et al. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–8. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 16.Tinari A, Garofalo T, Sorice M, Esposti MD, Malorni W. Mitoptosis: different pathways for mitochondrial execution. Autophagy. 2007;3:282–4. doi: 10.4161/auto.3924. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–92. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 18.Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 19.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–58. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 20.MacLaren AP, Chapman RS, Wyllie AH, Watson CJ. p53-dependent apoptosis induced by proteasome inhibition in mammary epithelial cells. Cell Death Differ. 2001;8:210–8. doi: 10.1038/sj.cdd.4400801. [DOI] [PubMed] [Google Scholar]
- 21.Bunz F, Hwang PM, Torrance C, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–9. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LeBlanc H, Lawrence D, Varfolomeev E, et al. Tumor-cell resistance to death receptor-induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat Med. 2002;8:274–81. doi: 10.1038/nm0302-274. [DOI] [PubMed] [Google Scholar]
- 23.Theodorakis P, Lomonosova E, Chinnadurai G. Critical requirement of BAX for manifestation of apoptosis induced by multiple stimuli in human epithelial cancer cells. Cancer Res. 2002;62:3373–6. [PubMed] [Google Scholar]
- 24.Bingle CD, Craig RW, Swales BM, Singleton V, Zhou P, Whyte MK. Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem. 2000;275:22136–46. doi: 10.1074/jbc.M909572199. [DOI] [PubMed] [Google Scholar]
- 25.Bae J, Leo CP, Hsu SY, Hsueh AJ. MCL-1S, a splicing variant of the anti-apoptotic BCL-2family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem. 2000;275:25255–61. doi: 10.1074/jbc.M909826199. [DOI] [PubMed] [Google Scholar]
- 26.Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006;13:994–1002. doi: 10.1038/sj.cdd.4401908. [DOI] [PubMed] [Google Scholar]
- 27.Mathai JP, Germain M, Marcellus RC, Shore GC. Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene. 2002;21:2534–44. doi: 10.1038/sj.onc.1205340. [DOI] [PubMed] [Google Scholar]
- 28.Hershko T, Ginsberg D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J Biol Chem. 2004;279:8627–34. doi: 10.1074/jbc.M312866200. [DOI] [PubMed] [Google Scholar]
- 29.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams JM, Cory S. The Bcl-2apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson DE. Noncaspase proteases in apoptosis. Leukemia. 2000;14:1695–703. doi: 10.1038/sj.leu.2401879. [DOI] [PubMed] [Google Scholar]
- 32.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 33.Scorrano L, Ashiya M, Buttle K, et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 34.Sugiyama T, Shimizu S, Matsuoka Y, Yoneda Y, Tsujimoto Y. Activation of mitochondrial voltage-dependent anion channel by a pro-apoptotic BH3-only protein Bim. Oncogene. 2002;21:4944–56. doi: 10.1038/sj.onc.1205621. [DOI] [PubMed] [Google Scholar]
- 35.Sun MG, Williams J, Munoz-Pinedo C, et al. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9:1057–65. doi: 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- 36.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–6. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russo SM, Tepper JE, Baldwin AS, Jr, et al. Enhancement of radiosensitivity by proteasome inhibition: implications for a role of NF-κB. Int J Radiat Oncol Biol Phys. 2001;50:183–93. doi: 10.1016/s0360-3016(01)01446-8. [DOI] [PubMed] [Google Scholar]
- 38.Adams J. Preclinical and clinical evaluation of proteasome inhibitor PS-341 for the treatment of cancer. Curr Opin Chem Biol. 2002;6:493–500. doi: 10.1016/s1367-5931(02)00343-5. [DOI] [PubMed] [Google Scholar]
- 39.Hideshima T, Chauhan D, Richardson P, et al. NF-κB as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–47. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 40.Mitsiades N, Mitsiades CS, Poulaki V, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A. 2002;99:14374–9. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding WX, Ni HM, Yin XM. Absence of Bax switched MG132-induced apoptosis to non-apoptotic cell death that could be suppressed by transcriptional or translational inhibition. Apoptosis. 2007;12:2233–44. doi: 10.1007/s10495-007-0142-0. [DOI] [PubMed] [Google Scholar]
- 42.Wolff S, Erster S, Palacios G, Moll UM. p53′s mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res. 2008;18:733–44. doi: 10.1038/cr.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pietsch EC, Perchiniak E, Canutescu AA, Wang G, Dunbrack RL, Murphy ME. Oligomerization of BAK by p53 utilizes conserved residues of the p53 DNA binding domain. J Biol Chem. 2008;283:21294–304. doi: 10.1074/jbc.M710539200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frank S, Gaume B, Bergmann-Leitner ES, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–25. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 45.Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem. 2005;280:35742–50. doi: 10.1074/jbc.M505970200. [DOI] [PubMed] [Google Scholar]
- 46.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–60. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 47.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–56. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–73. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 49.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 50.Kim H, Rafiuddin-Shah M, Tu HC, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2subfamilies. Nat Cell Biol. 2006;8:1348–58. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 51.Weber A, Paschen SA, Heger K, et al. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2proteins. J Cell Biol. 2007;177:625–36. doi: 10.1083/jcb.200610148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lomonosova E, Subramanian T, Chinnadurai G. Mitochondrial localization of p53 during adenovirus infection and regulation of its activity by E1-19K. Oncogene. 2005;24:6796–808. doi: 10.1038/sj.onc.1208836. [DOI] [PubMed] [Google Scholar]
- 53.Liu H, Ma Y, Pagliari LJ, et al. TNF-α-induced apoptosis of macrophages following inhibition of NF-κB: a central role for disruption of mitochondria. J Immunol. 2004;172:1907–15. doi: 10.4049/jimmunol.172.3.1907. [DOI] [PubMed] [Google Scholar]
- 54.Colell A, Ricci JE, Tait S, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–97. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.