

Abstract
An efficient, convergent synthesis of totarol by a diastereoselective epoxide/alkene/arene bicyclization is described. The reported synthesis enables the preparation of related diterpenes totaradiol and totarolone as well as previously unavailable derivatives that exhibit comparable inhibition of the bacterial cell division protein FtsZ.
Bacterial cell division is a novel target for the development of new antibiotics to fight infections that are resistant to current therapies.1 FtsZ is the central protein of bacterial cell division that forms the Z-ring at mid-cell and enables septation.2 This GTPase is structurally related to eukaryotic tubulin, which has been successfully targeted as a treatment for cancer. Although tubulin is targeted by many small molecules, and several are in clinical use as drugs, inhibitors of FtsZ are significantly lower in number and in vitro potency. More significantly, three separate allosteric sites of inhibition on tubulin have been identified for taxol, colchicine, and the Vinca alkaloids. Similar knowledge regarding FtsZ is lacking, and no direct evidence for inactivation outside the GTP binding site has been reported.3 Our group is interested in developing efficient syntheses of natural products that target FtsZ with the long-term goal of using synthesis to elucidate the mechanism by which these compounds act on this protein.4,5
Totarol is a diterpene produced in the sap of Podocarpus totara, a conifer native to New Zealand. The wood from the tree is prized for its resistance to rot and the antimicrobial properties of the secondary metabolites in the sap are well-established.6 Totarol is approved for use as an antimicrobial additive in several consumer products, including toothpaste and acne treatments.7 Although several previous studies have probed the origin of totarol’s antimicrobial activity,8 FtsZ was only recently identified as a discrete molecular target.9
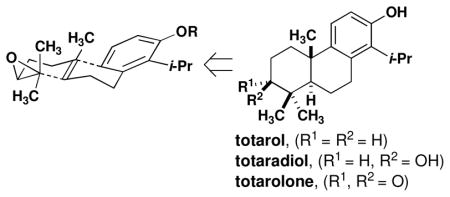
We have undertaken the synthesis of totarol and related diterpenes as part of a broader research program aimed at discerning the mechanism by which FtsZ can be inactivated by small molecules. Previous syntheses of totarol include routes to racemic material10 and semi-syntheses11 from chiral terpenoid precursors. Recent routes to related tricyclic systems have focused on electrophile-induced cyclization of polyene-derived precursors. Efficient syntheses of analogous ticyclic compounds have been reported using enantioselective protonation12 and halogenation13 of alkenes to effect polycyclization reactions. None of these efforts to date has resulted in the synthesis of the related diterpenoids totaradiol and totarolone. As a result, medicinal studies of totarol rely on preparing derivatives of the natural material, largely limiting these studies to modifications of the B and C rings.14 We recognized that an epoxide-initiated polycyclization would provide access to all three natural products and enable the synthesis of previously inaccessible A-ring derivatives for biochemical studies.
Retrosynthetic analysis reveals that a suitable precursor (4, Scheme 1) is produced by benzylic attachment of epoxygeraniol to a substituted arene. Cyclizations of related substrates have been described,15 and these substrates are produced by either copper- or palladium-catalyzed cross coupling reactions to allylic acetates or halides, respectively.16,17 Alternatively, direct coupling of Grignard reagents to allylic phosphonates has also been employed.18 A consistent synthetic challenge is the installation of the epoxide at one of the two tri-substituted alkenes. The established lack of regiocontrol in the Sharpless asymmetric dihydroxylation (SAD) reaction19 would necessitate installation of the epoxide before the coupling reaction (Scheme 1, route A). Given the liability posed by the use of benzylic Grignard reagents, we also considered an alternate coupling using dithiane 9 (Scheme 1, route B). The latter route would rely on steric control of the dihydroxylation.20 This synthetic scheme provides protected totaradiol directly from the polycyclization and totarol or totarolone by subsequent deoxygenation or oxidation, respectively.
Scheme 1.

Retrosynthesis of totarol, totaradiol and totarolone.
We initially explored route A by preparing the precursor to 4 in seven steps (Scheme 2). Acid 10 was converted to mixed anhydride 10b and then treated with 2-amino-2-methyl-1-propanol to amide 11.21 The resultant amide was cyclized to oxazoline 12 using triethylamine and methanesulfonyl chloride22 in 96% overall yield from 10. Regioselective displacement of the ortho methoxy group was achieved in high yield using conditions reported by Myers.23 Although many conditions have been reported for the solvolysis of dimethyloxazolines related to 13, this substrate proved problematic.21,24 We found that acidic hydrolysis in ethanol was more consistently high-yielding for conversion to ester 14.25 Reduction to alcohol 15 and conversion to either bromide 16 or chloride 17 were straightforward.
Scheme 2.

Synthesis of cyclization precursor 4
Synthesis of cyclization precursor 4 was attempted using a copper-catalyzed coupling (eq 1).15 Although conversion of chloride 16b in to the requisite Grignard reagent 6b has been reported,26 we observed sluggish consumption of magnesium turnings under standard conditions. Next, bromide 16a was used to form Grignard reagent 6a. Benzylic bromides often provide significant quantities of Würtz coupling products when allowed to react with magnesium metal. In this case, we had hoped that the bulky ortho substituent would suppress this side reaction. Transfer of 6a into a solution of epoxygeranyl acetate (5) and Li2CuCl4 or CuCN provided 4 in highly variable yields along with Würtz product 18.27 The long sequence to prepare 16 paired with the variable yields in the coupling reaction prompted us to consider an alternate route to 4.
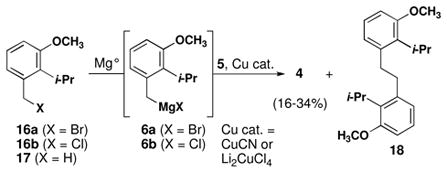 |
(1) |
We began route B by preparing the dithiane precursor of 9. Although a sequence analogous to Scheme 2 would be suitable, an alternative route from the benzonitrile would avoid excessive changes in oxidation state. Direct displacement of methoxy groups ortho to aryl cyano groups is possible.28 Myers noted that this reaction is often lower yielding than the analogous reactions of oxazolines.29 That said, the introduction of an additional methoxy group seems to increase the yield of displacement. Nitrile 19 was treated with i-PrMgCl to produce the displacement product 20 in high yield (eq 2). Reduction and dithioacetal formation proceeded smoothly to produce 21 in three steps from commercially available benzonitrile 19.
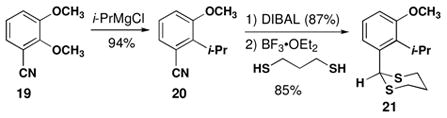 |
(2) |
The cyclization precursor was prepared by regio- and enantioselective dihydroxylation (Scheme 2). Lithiation of 21 under standard conditions followed by alkylation with geranyl bromide produced alkene 7 in high yield. Although previous attempts at regioselective dihydroxylation of related substrates have only been modestly successful, treatment of 22 with ADmix-β produced diol 22 as a single regioisomer in 90–95% ee.30 Desulfurization31 and dehydration provided epoxide 4 in seven steps from commercially available materials.
Benzyl geraniol derivative 4 was converted into diterpenes 1–3 by a short sequence. Cyclization of 4 proceeded smoothly in the presence of indium tribromide (2.0 equiv) to give tricyclic alcohol 24 in 58% yield as a single detectable diastereomer (Scheme 3).14 This result is in marked contrast to the acid-mediated cyclization of an alkene substrate in a recent syntheses of totarol in which 25% of the cis diastereomer is formed and the overall yield is 50%.9
Scheme 3.

Synthesis of totaradiol and totarolone
Two combinations of oxidation and demethylation of 24 were investigated. Deprotection of 24 produced totaradiol in good yield.32 Subsequent Oppenauer oxidation to totarolone (2) was achieved in a maximum yield of 33%.33 Given the limitations placed on oxidation conditions with the free phenol present, the order of operation was reversed. Jones oxidation of 24 to 25 was proceeded in 74% yield.34 Demethylation under conditions used for 24 were complicated by the formation of vinyl sulfide 26, which was isolated in 72% yield. This material could be hydrolyzed to 2 in high yield.35 Other attempts at direct conversion of 25 to 2 were unsuccessful, seemingly due to side reaction of the ketone. Synthetic totaradiol (3) and totarolone (2) each exhibited 13C and 1H NMR spectra and optical rotations in good agreement with reported values.
Final conversion of totarolone to totarol was achieved using the Wolff-Kischner reduction in modest yield (Scheme 4).36 The synthetic sample of (+)-totarol (1) produced by this exhibited identical chromatographic properties (TLC, GC-MS) when compared to an authentic sample and the 1H NMR spectrum was also identical. The longest linear sequence of reactions was 11 steps from commercially available nitrile 19.
Scheme 4.

Synthesis of totarol from totarolone
In summary, we have reported a concise, enantioselective route to (+)-totarol and the first syntheses of the related diterpenes (+)-totaradiol and (+)-totarolone. This route sets the stage for a detailed study of the mechanism by which FtsZ is inactivated by this natural product.
Displayed equations should be assigned “Normal Text” on the Styles toolbar. Displayed equations can only be one column wide. If artwork needs to be two columns wide, it must be relabeled as a Figure or Scheme.
Supplementary Material
Scheme 1.

Synthesis of Grignard reagents 6a and 6b.
Acknowledgments
This research was supported by startup funds from UC Davis, a grant from the UC Davis Academic Senate Committee on Research, and by the NIH/NIAID (R56AI80931-01). MBK acknowledges the United States Department of Education for a GAANN fellowship. Gorkem Gunbas (Mascal Research Group, UC Davis) is acknoweldged for helpful discussions.
Footnotes
Supporting Information Available Experimental procedures for the preparation of all new compounds. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.a) Vollmer W. Appl Microbiol Biotechnol. 2006;73:37–47. doi: 10.1007/s00253-006-0586-0. [DOI] [PubMed] [Google Scholar]; b) Lock RL, Harry EJ. Nat Rev Drug Discov. 2008;7:324–338. doi: 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- 2.a) Romberg L, Levin PA. Annual Review of Microbiology. 2003;57:125–154. doi: 10.1146/annurev.micro.57.012903.074300. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Margolin W. Nat Rev Mol Cell Biol. 2005;6:862–871. doi: 10.1038/nrm1745. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dajkovic A, Lutkenhaus J. J Mol Microbiol Biotechnol. 2006;11:140–151. doi: 10.1159/000094050. [DOI] [PubMed] [Google Scholar]; d) Vollmer W. Chem Biol. 2008;15:93–94. doi: 10.1016/j.chembiol.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Indirect techniques, including site-directed mutagenesis and mapping of mutations to resistant strains, have provided evidence for the mode of inactivation of one FtsZ inhibitor: Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG. Science. 2008;321:1673–1675. doi: 10.1126/science.1159961.
- 4.a) Wang J, Galgoci A, Kodali S, Herath KB, Jayasuriya H, Dorso K, Vicente F, Gonzalez A, Cully D, Bramhill D, Singh S. J Biol Chem. 2003;278:44424–44428. doi: 10.1074/jbc.M307625200. [DOI] [PubMed] [Google Scholar]; b) Urgaonkar S, La Pierre HS, Meir I, Lund H, RayChaudhuri D, Shaw JT. Org Lett. 2005;7:5609–5612. doi: 10.1021/ol052269z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Domadia PN, Bhunia A, Sivaraman J, Swarup S, Dasgupta D. Biochemistry. 2008;47:3225–3234. doi: 10.1021/bi7018546. [DOI] [PubMed] [Google Scholar]; d) Kanoh K, Adachi K, Matsuda S, Shizuri Y, Yasumoto K, Kusumi T, Okumura K, Kirikae T. J Antibiot. 2008;61:192–194. doi: 10.1038/ja.2008.29. [DOI] [PubMed] [Google Scholar]
- 5.Curcumin has been reported to target many proteins, including both FtsZ and tubulin: Rai D, Singh JK, Roy N, Panda D. Biochem J. 2008;410:147–155. doi: 10.1042/BJ20070891.Gupta KK, Bharne SS, Rathinasamy K, Naik NR, Panda D. FEBS J. 2006;273:5320–5332. doi: 10.1111/j.1742-4658.2006.05525.x.
- 6.Much of the history of totarol has been reviewed. See: Bendall JG, Cambie RC. Aust J Chem. 1995;48:883–917.
- 7.a) Nixon D, Hobbs D. NZ Family Phys. 2006;33:253–255. [Google Scholar]; b) [accessed on August 16, 2009]; http://www.cosmeticsdesign.com/Formulation-Science/Totarol-goes-global.
- 8.Muroi H, Kubo I. Biosci, Biotechnol, Biochem. 1994;58:1925–1926. [Google Scholar]; b) Muroi H, Kubo I. J Appl Bacteriol. 1996;80:387–394. doi: 10.1111/j.1365-2672.1996.tb03233.x. [DOI] [PubMed] [Google Scholar]; c) Kubo I, Muroi H, Himejima M. J Nat Prod. 1992;55:1436–1440. doi: 10.1021/np50088a008. [DOI] [PubMed] [Google Scholar]; d) Haraguchi H, Oike S, Muroi H, Kubo I. Planta Med. 1996;62:122–125. doi: 10.1055/s-2006-957832. [DOI] [PubMed] [Google Scholar]; e) Constantine GH, Karchesy JJ, Franzblau SG, LaFleur LE. Fitoterapia. 2001;72:572–574. doi: 10.1016/s0367-326x(01)00272-6. [DOI] [PubMed] [Google Scholar]
- 9.Jaiswal R, Beuria TK, Mohan R, Mahajan SK, Panda D. Biochemistry. 2007;46:4211–4220. doi: 10.1021/bi602573e. [DOI] [PubMed] [Google Scholar]
- 10.a) Barltrop JA, Rogers NAJ. J Chem Soc. 1958:2566–2572. [Google Scholar]; b) Tada M, Kurabe J, Yasue H, Ikuta T. Chem Pharm Bull. 2008;56:287–291. doi: 10.1248/cpb.56.287. Additional synthetic work is described in reference 6. [DOI] [PubMed] [Google Scholar]
- 11.a) Matsumoto T, Suetsugu A. Bull Chem Soc Jpn. 1979;52:1450–1453. [Google Scholar]; b) Marcos IS, Cubillo MA, Moro RF, Diez D, Basabe P, Sanz F, Urones JG. Tetrahedron Lett. 2003;44:8831–8835. [Google Scholar]; c) Marcos IS, Cubillo MA, Moro RF, Carballares S, Diez D, Basabe P, Llamazares CF, Beneitez A, Sanz F, Broughton HB, Urones JG. Tetrahedron. 2005;61:977–1003. [Google Scholar]
- 12.a) Ishihara K, Ishibashi H, Yamamoto H. J Am Chem Soc. 2001;123:1505–1506. [Google Scholar]; b) Ishibashi H, Ishihara K, Yamamoto H. J Am Chem Soc. 2004;126:11122–11123. doi: 10.1021/ja0472026. [DOI] [PubMed] [Google Scholar]; c) Youn SW, Pastine SJ, Sames D. Org Lett. 2004;6:581–584. doi: 10.1021/ol036385i. [DOI] [PubMed] [Google Scholar]
- 13.Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]
- 14.a) Evans GB, Furneaux RH. Bioorg Med Chem. 2000;8:1653–1662. doi: 10.1016/s0968-0896(00)00095-x. [DOI] [PubMed] [Google Scholar]; b) Evans GB, Furneaux RH, Gainsford GJ, Murphy MP. Bioorg Med Chem. 2000;8:1663–1675. doi: 10.1016/s0968-0896(00)00096-1. [DOI] [PubMed] [Google Scholar]; c) Evans GB, Furneaux RH, Gravestock MB, Lynch GP, Scott GK. Bioorg Med Chem. 1999;7:1953–1964. doi: 10.1016/s0968-0896(99)00162-5. [DOI] [PubMed] [Google Scholar]
- 15.Zhao J-F, Zhao Y-J, Loh T-P. Chem Commun. 2008:1353–1355. doi: 10.1039/b718337b. [DOI] [PubMed] [Google Scholar]
- 16.a) Baeckvall JE, Sellen M. Chem Commun. 1987:827–829. [Google Scholar]; b) Baeckvall JE, Sellen M, Grant B. J Am Chem Soc. 1990;112:6615–6621. [Google Scholar]; c) Gansaeuer A, Justicia J, Rosales A, Rinker B. Synlett. 2005:1954–1956. [Google Scholar]
- 17.Rosales V, Zambrano JL, Demuth M. J Org Chem. 2002;67:1167–1170. doi: 10.1021/jo010786z. [DOI] [PubMed] [Google Scholar]
- 18.Araki S, Sato T, Butsugan Y. Chem Commun. 1982:285–286. [Google Scholar]
- 19.a) Crispino GA, Sharpless KB. Tetrahedron Lett. 1992;33:4273–4274. [Google Scholar]; b) Corey EJ, Noe MC, Lin S. Tetrahedron Lett. 1995;36:8741–8744. [Google Scholar]; c) Corey EJ, Zhang J. Org Lett. 2001;3:3211–3214. doi: 10.1021/ol016577i. [DOI] [PubMed] [Google Scholar]; d) Hoshino T, Yonemura Y, Abe T, Sugino Y. Org Biomol Chem. 2007;5:792–801. doi: 10.1039/b615897h. [DOI] [PubMed] [Google Scholar]
- 20.A high level of steric control has been observed in a substrate related to 4 that is shorter by one carbon between the phenyl ring and the central alkene. See: Neighbors JD, Mente NR, Boss KD, Zehnder DW, Wiemer DF. Tetrahedron Lett. 2008;49:516–519.
- 21.Sakaguchi J, Higashi T, Azuma T, Suzuki T, Iwasaki N, Kondo N, Nagata O, Kato H, Hanaoka M. Chem Pharm Bull. 2001;49:788–790. doi: 10.1248/cpb.49.788. [DOI] [PubMed] [Google Scholar]
- 22.Ogasawara M, Yoshida K, Kamei H, Kato K, Uozumi Y, Hayashi T. Tetrahedron-Asymmetry. 1998;9:1779–1787. [Google Scholar]
- 23.Meyers AI, Willemsen JJ. Tetrahedron. 1998;54:10493–10511. [Google Scholar]
- 24.a) Meyers AI, Gabel R, Mihelich ED. J Org Chem. 1978;43:1372–1379. [Google Scholar]; b) Fukuda Y, Seto S, Furuta H, Ebisu H, Oomori Y, Terashima S. J Med Chem. 2001;44:1396–1406. doi: 10.1021/jm000107x. [DOI] [PubMed] [Google Scholar]
- 25.Kaplan LJ, Weisman GR, Cram DJ. J Org Chem. 1979;44:2226–2233. These conditions were optimized for execution in a microwave reactor. See supporting information. [Google Scholar]
- 26.Use of the Grignard reagent was reported by Nishizawa and coworkers. Although the preparation does not appear in the experimental section of this publication, the procedure was provided to us by Prof. nishizawa. See: Imagawa H, Iyenaga T, Nishizawa M. Synlett. 2005:703–705. doi: 10.1021/ol047472t.
- 27.In one run, 18 was isolated as an mixture with bromide 16a and 17 (from protonation of 6a) in 43% yield.
- 28.a) Richtzenhain H, Nippus P. Chem Ber. 1949;82:408–417. [Google Scholar]; b) Albert JS, Ohnmacht C, Bernstein PR, Rumsey WL, Aharony D, Alelyunas Y, Russell DJ, Potts W, Sherwood SA, Shen L, Dedinas RF, Palmer WE, Russell K. J Med Chem. 2004;47:519–529. doi: 10.1021/jm030197g. [DOI] [PubMed] [Google Scholar]; c) Parker JS, Smith NA, Welham MJ, Moss WO. Org Process Res Dev. 2004;8:45–50. [Google Scholar]
- 29.Meyers AI, Mihelich ED. J Am Chem Soc. 1975;97:7383–7385. [Google Scholar]
- 30.Enantiomeric excess was determined by chiral HPLC after cyclization to 24 (Scheme). See supporting information for details.
- 31.Gutierrez CG, Stringham RA, Nitasaka T, Glasscock KG. J Org Chem. 1980;45:3393–3395.Use of Raney nickel resulted in competitive alkene reduction, as observed by Bruggemann. See: Bruggemann M, Holst C, Hoppe D. Eur J Org Chem. 2001:647–654.
- 32.Node M, Nishide K, Fuji K, Fujita E. J Org Chem. 1980;45:4275–4277. [Google Scholar]
- 33.Edsall AB, Mohanakrishnan AK, Yang D, Fanwick PE, Hamel E, Hanson AD, Agoston GE, Cushman M. J Med Chem. 2004;47:5126–5139. doi: 10.1021/jm049647a. [DOI] [PubMed] [Google Scholar]
- 34.Martin R, Entchev EV, Dabritz F, Kurzchalia TV, Knolker HJ. Eur J Org Chem. 2009:3703–3714. [Google Scholar]
- 35.Imanishi T, Yamashita M, Matsui M, Tanaka T, Iwata C. Chem Pharm Bull. 1989;37:3114–3116. [Google Scholar]
- 36.Huang M. J Am Chem Soc. 1949;71:3301–3303. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.