

Abstract
Chronic pancreatitis increases by 16 fold the risk of developing pancreatic ductal adenocarcinoma (PDAC), one of the deadliest human cancers. It also appears to accelerate cancer progression in genetically engineered mouse models. We now report that in a mouse model where oncogenic Kras is activated in all pancreatic cell types, two brief episodes of acute pancreatitis caused rapid PanIN progression and accelerated pancreatic cancer development. Thus, a brief inflammatory insult to the pancreas, when occurring in the context of oncogenic KrasG12D, can initiate a cascade of events that dramatically enhances the risk for pancreatic malignant transformation.
Keywords: KrasG12D, PDAC, acute pancreatitis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the United States, with a median survival of 6 months [1]. PDAC is generally believed to arise predominantly through progression of pancreatic intraepithelial neoplasia (PanIN), ranging from low grade PanINs (termed PanIN1A, -1B) to high grade PanINs (termed PanIN-2, -3), to ductal adenocarcinoma [2]. PDAC is characterized by a high frequency of KRAS mutations at early stages and the accumulation over time of multiple additional genetic abnormalities [3]. Relevant mouse models of PDAC have been generated by targeting a conditionally mutated Kras allele (KrasG12D) to early pancreatic progenitors, using Pdx1 (PK model) and Ptf1a promoters, and subsequently to all pancreatic cell types, underscoring the fact that oncogenic Kras is sufficient for PDAC initiation [4]. In these models, mouse PanIN (mPanIN) formation and progression faithfully recapitulated what is observed in human PDAC, and later in life (8–12 months old mice) these mouse models developed pancreatic cancer.
More recently, it was shown that activation of oncogenic Kras in pancreatic acinar (and in one model centroacinar as well) cells during embryogenesis was sufficient for PDAC initiation [5] [6] [7], and that subjecting the mice to chronic pancreatitis accelerated PDAC development [5]. In two mouse models, Kras oncogenic activation in adult acinar cells was sufficient for initiating PanIN formation but no pancreatic cancer was observed after one year [6; 7]. However, in another model, oncogenic Kras activation in adult acinar/centroacinar cells yielded PDAC, but only when the mice were administered high doses of caerulein over many weeks resulting in a chronic pancreatitis like state with intense fibrosis and inflammatory cell infiltrates [5]. These results in mice are consistent with epidemiologic studies showing that patients suffering from chronic pancreatitis have a 16-fold increased risk of developing pancreatic cancer [8].
We now show that the PK mouse model, when subjected to two brief episodes of acute pancreatitis which do not induce chronic pancreatitis like changes, rapidly develops high grade PanINs and exhibits accelerated PDAC formation.
Material and methods
Mouse colony generation
The LSL-KrasG12D (01XJ6 B6;129-Kras2tm4Tyj) mice were generated by D.A. Tuveson and T. Jacks [9] and obtained from MMHCC, NCI. The Pdx1-Cre mice were a gift from G. Gu [10]. All genotyping were done by PCR following the conditions of the providers.
Acute pancreatitis induction
Mice were subjected to a series of seven hourly intraperitoneal injections of caerulein that was repeated 48 hours later, based on preliminary experiments that revealed the presence of edematous pancreatitis following each series of injections [11]. Caerulein (Sigma, St. Louis MO) was diluted in 6% dextran 70, 0.9% NaCl and injected at a dose of 50 μg/kg of body weight. At least 5 compound mutant animals per experiment were injected with caerulein in parallel with five single mutant (LSL-KrasG12D only or Pdx1-Cre only) and wild type mice. A second group of compound mutant and control animals received injections of carrier buffer only. All animals were fasted for 12 hours before the experiment.
Histology and immunohistochemistry
Mice were perfused with PBS then 10% formalin/PBS. The pancreata were dissected and fixed overnight. For histology and immunostaining, pancreata were processed for paraffin embedding. Routine Hematoxylin and Eosin (H&E) staining was performed using standard procedures. For immunostaining, sections were deparaffinized, rehydrated and antigens were retrieved if required using a 2100-Retriever and antigen unmasking solution (Vector Laboratories). For cytokeratin 19 (CK19) immunostaining, Proteinase K antigen retrieval procedure was used. Immunostaining procedures were as described previously [12]. The antibodies and dilution used were: TromaIII (CK19 antibody developed by Rolf Kemler and obtained from Developmental Studies Hybridoma Bank, 1:10), Muc5a (Novocastra, 1:30), Ki67 (Novocastra, 1:200), Hes1 (a gift from Dr. T. Sudo, Toray Inc., Kamakura, Japan; 1:400), Pdx1 (Upstate, 1:5000), and SMA (Abcam, 1:100).
Results
Acute pancreatitis causes lesion progression
The mouse model used in this study is the PK model where the Pdx1-driven Cre recombinase starts being expressed in early pancreatic progenitors [10]. Following Cre mediated removal of a transcriptional stop region surrounded by LoxP sites (LoxP-Stop-LoxP or LSL), a mutated Kras allele is activated in these progenitors and subsequently in all pancreatic cells types [4; 13]. By 2 months of age, the PK mice develop low-grade mPanINs in limited numbers, as well as very limited areas of acinar to ductal metaplasia (ADM). An increase in lesions number but not in grade is observed at 6 months of age in conjunction with the appearance of extensive ADM lesions. After a lag phase of 8 to 12 months, the animals occasionally develop pancreatic cancer [4].
To determine whether acute pancreatitis could accelerate mPanIN progression and PDAC formation, mutant mice were subjected to 2 episodes of acute pancreatitis induced by caerulein, a cholecystokinin (CCK) analog that binds and activates the CCK receptor [11; 14]. Despite the fact that the dosage of caerulein (50 ng/g of body weight/injection, administered by 7 intra-peritoneal injections and repeated 48 hours later) that we used in our experimental settings are relatively low compared to the dosage used in other publications (from 100–400 ng/g of body weight and twice 8 injections) [15] [16], we observed acute pancreatitis, as demonstrated by the presence of marked edema a few hours following the last caerulein injection. H&E staining of pancreas sections collected at different time points shows that at 24 hours, an important inflammatory infiltrate is present and acinar cells are rapidly dying, whereas one week later complete regeneration of the pancreas is observed (not shown) as described [15]. Three cohorts of 5 to 8 PK mutant animals at 2–3 months of age were subjected to two episodes of acute pancreatitis and sacrificed 30, 60 and 80 days later. As early as 1 month following the two acute pancreatitis episodes, all caerulein-treated PK pancreata presented numerous high grade mPanINs (Fig. 1C–D) as well as extensive ADM (Fig. 1B) while untreated PK pancreata showed only isolated and low-grade lesions and limited ADM (Fig. 1A). In all caerulein-treated control animals, carrying one or no mutation, the pancreata appeared to be normal with occasional inflammatory cells present at 1-month post treatment (not shown). There was no evidence for residual inflammation by 60 days. These data allowed us to conclude that two episodes of acute inflammation can cause rapid neoplastic progression in the pancreas even in the absence of chronic pancreatitis, but only in the context of Kras oncogenic activation.
Figure 1.

PanIN progression 2 months following acute pancreatitis episodes. H&E staining of untreated (A) and caerulein treated NK pancreata (B–D). While untreated pancreata display rare lesions (A), treated pancreata show extensive ADM and high number of mPanIN-1 (B), as well as high-grade lesions mPanIN-2 (C) and -3 (D). Islets (dotted outlines), normal ducts (d), mPanIN-1 (empty arrowheads), and mPanIN-2 and -3 (solid arrowheads) are readily seen. Scale bar: 100μm for A–B, 20μm for C–D.
As observed in human PDAC, while mPanIN-1 lesions exhibited a low index of proliferation, there was increased proliferation in mPanIN-2 and -3 as evidenced by increased Ki67 staining (Fig. 2A). mPanINs also expressed cytokeratin19 (CK19), underscoring their epithelial nature (Fig. 2B), and displayed mucus accumulation as shown by Muc5a expression (Fig. 2C). High-grade PanINs also exhibited the Notch signaling marker Hes1 (Fig. 2D) and the early pancreatic progenitor marker Pdx1 (Fig. 2E), both known to be reactivated in early stages of PDAC. ADM was also observed, as shown by the co-expression in the same cells of acinar (amylase) and ductal (CK19) markers (Fig. 2F), as observed by others [17].
Figure 2.

High grade PanIN characterization following acute pancreatitis. Ki67 staining shows a clear increase in proliferation in high-grade lesions (A) when compared to mPanIN-1 (*). High-grade PanINs express CK19 (B) and Muc5a (C). Markers of early progenitors Hes1, panel D) and Pdx1 (panel E) are reactivated; as expected, Pdx1 is also seen in the islet cells (dotted outline). ADM is observed as shown by coexpression of CK19 (green) and Amylase (red) in same cells (see inset). Scale bar: 20μm.
Among the three groups of mice that were subjected to AP, two PK animals from the 80 days post caerulein group developed PDAC. The carcinomas were locally invasive, the cancer cells exhibited a ductal like morphology (Fig. 3A) and displayed increased proliferation (Fig. 3B). In addition, there were foci of cancer cells that were surrounded by a reactive stroma characterized by smooth muscle actin (SMA) expression (Fig. 3C), indicating the presence of activated pancreatic stellate cells.
Figure 3.
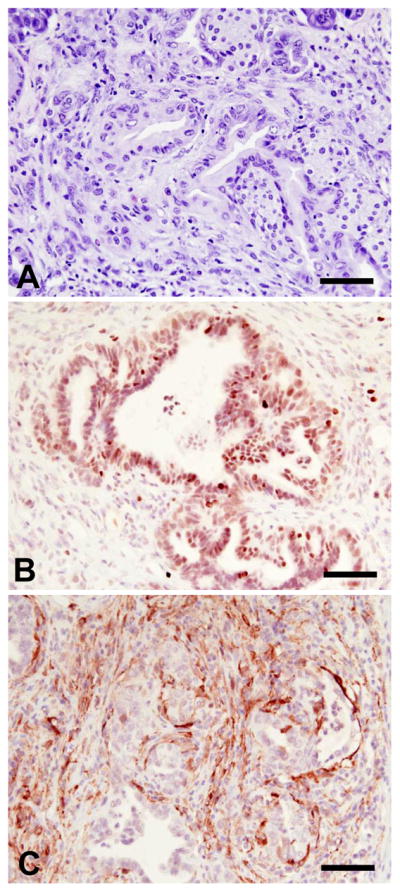
PK mice develop PDAC following acute pancreatitis. H&E staining of ductal adenocarcinoma (A) reveals the presence of many nuclei that are enlarged, pleomorphic and crowded, with the cancer cells exhibiting a ductal-like morphology. Adenocarcinoma cells are highly proliferative as shown by Ki67 expression (B). They are surrounded by fibroblasts characterized by SMA expression (C). Scale bar: 20μm.
Discussion
Caerulein-induced acute pancreatitis is a well-studied animal model in which caerulein normally causes a mild, edematous pancreatitis, which is self-limiting and highly reproducible. Initiation of acute pancreatitis in this model is mediated through premature intracellular activation of zymogens in the acinar cells, leading to acinar cell death and an inflammatory response associated with mild pancreatic edema [18]. While many acinar cells are lost in this process, full pancreatic function is restored approximately a week following caerulein treatment as the result of a robust acinar regenerative response.
In the present study we determined that two episodes of acute pancreatitis in the PK mouse model lead to a remarkable acceleration in the formation of high-grade mPanINs, reactivation of Notch signaling, and progression to PDAC. Extensive characterization of the events following caerulein-induced acute pancreatitis showed that during the 24 hours following an episode of acute pancreatitis, large numbers of acinar cells undergo apoptosis and that one week later, the acinar compartment is fully regenerated [15]. By contrast, ductal, centroacinar or endocrine cells do not exhibit an increase in apoptosis or proliferation [15]. Acute pancreatitis also causes a rapid activation of signaling pathways such as Notch and Hedgehog, which are known to be upregulated in PDAC early lesions [15; 19]. Taken together with the present findings, these observations indicate that acute pancreatitis-induced acinar cell regeneration and associated alterations in key signaling pathways, in conjunction with oncogenic Kras activation, combine to markedly increase the propensity for pancreatic malignant transformation. Our findings thus suggest that PanINs and PDAC may arise from the acinar cell compartment of the exocrine pancreas.
Several recent reports support the concept that acinar cells could give rise to PDAC. First, restricted activation of a mutated Kras allele in acinar cells (and centroacinar cells in some cases), using the elastase or the Mist1 promoters for Cre expression, leads to mPanIN development in adult mice [6; 7] and infrequent invasive PDAC [5]. Second, activation of KrasG12D in the Nestin lineage, constituted essentially of acinar cells, leads to the development of mPanINs in similar number and grade that was seen in the PK model [20]. Third, chronic pancreatitis induced by repeated injections of caerulein over many weeks causes PDAC progression in mice carrying an activated KrasG12V allele in adult acinar/centroacinar cells [5]. Fourth, ductal cells, ADM lesions and islet cells display cilia; by contrast, pancreatic acinar cells, all grades of PanINs, and pancreatic cancer cells are characterized by the absence of cilia in both humans and mouse models [21], suggesting that PanINs, PDAC, and acinar cells have a common origin. Moreover, lineage tracing experiments suggest that new acinar cells originate from preexisting acinar cells [16; 22; 23]. Taken together, these observations support the hypothesis that induction of acinar cell proliferation by pancreatic injury such as an episode of acute pancreatitis, when occurring in the context of an activated mutated Kras allele, can lead to deregulated growth and progression to PDAC.
Recovery from acute pancreatitis in humans does not necessarily lead to irreversible damage and is associated with pancreatic regeneration [24]. The most frequent causes for acute pancreatitis in Western countries are gallstone disease and alcohol consumption [25]. Heavy alcohol consumption, independently of chronic pancreatitis, has been associated with an increased incidence of PDAC [26]. Conceivably, alcohol may exert this effect by precipitating acute pancreatitis. It is not clear, however, whether such brief episodes of acute pancreatitis act by promoting a pro-inflammatory environment in the pancreas, by inducing bursts of acinar cell regeneration, or by upregulating signaling pathways and transcription factors involved in embryonic development of the pancreas and in PDAC [15]. Irrespective of the mechanisms, if confirmed in future studies, the present findings suggest that prevention of acute pancreatitis may be an important strategy for decreasing the incidence of PDAC.
Acknowledgments
This work was supported by grants from the National Cancer Institute: CA-101306 and CA-075059 to M. K. and CA-127095 to C. C, and by a Lustgarten Foundation award for Pancreatic Cancer Research to C.C.
References
- 1.Warshaw AL, Fernandez-del Castillo C. Pancreatic carcinoma. N Engl J Med. 1992;326:455–65. doi: 10.1056/NEJM199202133260706. [DOI] [PubMed] [Google Scholar]
- 2.Hruban RH, Wilentz RE, Maitra A. Identification and analysis of precursors to invasive pancreatic cancer. Methods Mol Med. 2005;103:1–13. doi: 10.1385/1-59259-780-7:001. [DOI] [PubMed] [Google Scholar]
- 3.Tuveson DA, Hingorani SR. Ductal pancreatic cancer in humans and mice. Cold Spring Harb Symp Quant Biol. 2005;70:65–72. doi: 10.1101/sqb.2005.70.040. [DOI] [PubMed] [Google Scholar]
- 4.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 5.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 6.De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–12. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, Maitra A. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–8. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20:197–209. doi: 10.1016/j.bpg.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 10.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–57. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 11.Neuschwander-Tetri BA, Burton FR, Presti ME, Britton RS, Janney CG, Garvin PR, Brunt EM, Galvin NJ, Poulos JE. Repetitive self-limited acute pancreatitis induces pancreatic fibrogenesis in the mouse. Dig Dis Sci. 2000;45:665–74. doi: 10.1023/a:1005423122127. [DOI] [PubMed] [Google Scholar]
- 12.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–26. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willemer S, Elsasser HP, Adler G. Hormone-induced pancreatitis. Eur Surg Res. 1992;24(Suppl 1):29–39. doi: 10.1159/000129237. [DOI] [PubMed] [Google Scholar]
- 15.Jensen JN, Cameron E, Garay MV, Starkey TW, Gianani R, Jensen J. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–41. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Desai BM, Oliver-Krasinski J, De Leon DD, Farzad C, Hong N, Leach SD, Stoffers DA. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest. 2007;117:971–7. doi: 10.1172/JCI29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol. 2007;171:263–73. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grady T, Mah'Moud M, Otani T, Rhee S, Lerch MM, Gorelick FS. Zymogen proteolysis within the pancreatic acinar cell is associated with cellular injury. Am J Physiol. 1998;275:G1010–7. doi: 10.1152/ajpgi.1998.275.5.G1010. [DOI] [PubMed] [Google Scholar]
- 19.Siveke JT, Lubeseder-Martellato C, Lee M, Mazur PK, Nakhai H, Radtke F, Schmid RM. Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology. 2008;134:544–55. doi: 10.1053/j.gastro.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Carriere C, Seeley ES, Goetze T, Longnecker DS, Korc M. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2007;104:4437–42. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009;69:422–30. doi: 10.1158/0008-5472.CAN-08-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strobel O, Dor Y, Alsina J, Stirman A, Lauwers G, Trainor A, Castillo CF, Warshaw AL, Thayer SP. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133:1999–2009. doi: 10.1053/j.gastro.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fendrich V, Esni F, Garay MV, Feldmann G, Habbe N, Jensen JN, Dor Y, Stoffers D, Jensen J, Leach SD, Maitra A. Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology. 2008;135:621–31. doi: 10.1053/j.gastro.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebert M, Yokoyama M, Ishiwata T, Friess H, Buchler MW, Malfertheiner P, Korc M. Alteration of fibroblast growth factor and receptor expression after acute pancreatitis in humans. Pancreas. 1999;18:240–6. doi: 10.1097/00006676-199904000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Steer ML. Classification and pathogenesis of pancreatitis. Surg Clin North Am. 1989;69:467–80. doi: 10.1016/s0039-6109(16)44831-0. [DOI] [PubMed] [Google Scholar]
- 26.Silverman DT, Brown LM, Hoover RN, Schiffman M, Lillemoe KD, Schoenberg JB, Swanson GM, Hayes RB, Greenberg RS, Benichou J, et al. Alcohol and pancreatic cancer in blacks and whites in the United States. Cancer Res. 1995;55:4899–905. [PubMed] [Google Scholar]