

Abstract
Tissue factor (TF) antagonists targeting the factor VII (FVII) binding domain have been shown to interrupt acute vascular thrombus formation without impairing hemostasis in non-human primates. In this study, we evaluate whether a human/mouse chimeric monoclonal antibody (ALT-836, formerly known as Sunol-cH36) blocking the factor X/factor IX (FX/FIX) binding site of tissue factor could achieve similar clinical benefits in an arterial thrombosis model induced by surgical endarterectomy in chimpanzees. In this model, sequential surgical endarterectomies on right and left superficial femoral arteries were performed 30 days apart in five chimpanzees. A bolus (1 mg/kg) of ALT-836 was injected intravenously immediately preceding the restoration of flow in the endarterectomized femoral artery. Pre-surgical labeling of autologous platelets using 111In-Oxine and post-surgical gamma camera imaging of 111In-platelet deposition at endarterectomy sites was performed. The manipulated arterial segments were harvested for patency analysis 30 days following surgery. The results indicate that ALT-836 was highly effective at reducing acute vascular thrombosis, with no significant variations in surgical blood loss and template-bleeding time in the treated group compared to the control animals. These data suggest that ALT-836 is an effective and safe antithrombotic agent in preventing TF-initiated vascular thrombogenesis without compromising hemostasis.
Introduction
Thrombotic occlusion that is resistant to currently available antithrombotic therapy complicates interventional mechanical therapies for symptomatic atherosclerotic vascular disease (1–4). Consequently, there is a need for more effective prevention and interruption of platelet-dependent occlusive thrombi. Mechanically damaged vascular tissues initiate TF-dependent thrombin generation that converts fibrinogen to fibrin and mediates platelet recruitment by cleaving protease-activated receptors (PARs) leading to fibrin-stabilized vascular thrombosis. In this process, factor VII/VIIa (FVII/FVIIa) avidly binds with TF exposed on cellular membranes at sites of vascular disruption leading to the proteolytic activation of factor X (FX), and subsequent factor Xa-factor Va (FXa-FVa) complex cleavage of prothrombin to produce thrombin on platelet phospholipid surfaces (5, 6). The TF-FVlla complex also activates factor IX (FIX), which amplifies the formation of FXa by complexing with thrombin-activated factor VIIIa (FVIIIa), thereby greatly enhancing the rate of thrombin activation. Inactivation of thrombin, inhibition of thrombin activation of PARs, and interruption of thrombin generation have important effects on thrombogenesis, hemostasis, inflammation, and neointimal vascular responses to injury, with corresponding therapeutic opportunities (7).
Strategies to block thrombus formation have employed pharmacological agents that act at various points in the coagulation cascade, ranging from use of nonspecific inhibitors to specific inhibitors of coagulation factors or direct acting thrombin inhibitors (8). While inhibition of the coagulation cascade at the final stages can lead to bleeding complications, studies in various animal models have shown that inhibition of the TF-FVIIa complex can block or prevent thrombosis with little or no effect on bleeding parameters. Compounds including anti-TF antibodies to the FVIIa binding domain, active-site inactivated FVIIa (FVIIai), and small molecule TF-FVIIa inhibitors have each been shown to provide effective antithrombotic responses with less impact on hemostasis than activity-equivalent doses of FXa or thrombin inhibitors (9–11). However, due to the picomolar affinity of FVIIa for membrane-bound TF (12), there may be limitations in the ability of some of these inhibitors to effectively block TF-FVIIa complex formation in vivo. In addition, TF/VIIa-targeted antagonists may inhibit a variety of non-coagulant cellular processes related to cytokine responses, cell migration and wound healing that are mediated by TF-FVIIa complex formation or signaling (13). Since these coagulation-independent actions of TF-FVIIa may play a role in a number of physiological processes, the overall impact of using TF/VIIa targeted antagonists, such the inactivated form of factor VIIa (FVIIai), as anti-thrombotic therapy is unknown. Therefore, it is of interest to investigate approaches to prevent thrombosis without specifically blocking TF-FVIIa complex activity (14).
In this report, we describe the generation and identification of a high-affinity anti-human TF monoclonal antibody (mAb) that blocks the binding of FX and FIX to the TF-FVIIa complex, thereby preventing FX and FIX activation. However this antibody does not inhibit the formation of the TF-FVIIa complex. We further describe the evaluation of the recombinant chimeric form of H36 (ALT-836) as a safe and potent inhibitor of TF-initiated acute arterial thrombosis using a surgical endarterectomy model in chimpanzees.
Materials and methods
Generation of anti-TF mAbs
Female BALB/c mice were immunized four times over a 7-month period with 10–20 μg lipidated recombinant human TF (rhTF) in accordance with Institutional Animal Care and Use Committee (IACUC)-approved procedures. Hybridoma cell lines were generated using standard procedures and screened for production of anti-TF mAb with an rhTF-specific enzyme-linked immunosorbent assay (ELISA). Monoclonal antibodies were purified from cell culture media by protein A affinity chromatography.
Construction and expression of recombinant chimeric anti-TF antibody
Total RNA, isolated from the H36 hybridoma cell line using the RNeasy kit (QIAGEN, Valencia, CA), was used to generate cDNA for the H36 heavy chain (HC) and light chain (LC) variable regions by reverse transcriptase polymerase chain reaction (RT-PCR) using degenerate IgG-specific primers described previously (15). The HC and LC variable gene fragments were cloned into vectors containing HC leader/human IgG4 constant domain and LC leader/human kappa domain sequences, respectively. The chimeric HC H36 IgG4 and LC H36 kappa genes were subsequently cloned into a mammalian expression vector, pJAIgG4TF.A8. CHO-K1 cells were transfected with pJAIgG4TF.A8 and cells were selected based on the production of anti-TF antibody measured by ELISA. The transfected CHO cell line H9G12 was used for production of ALT-836 in a hollow fiber bioreactor system. ALT-836 was purified from the cell culture harvest using protein A and ion exchange chromatography.
Chromogenic and prothrombin time (PT) assays
Truncated rhTF (243 amino acids) was expressed in E. coli and purified by immunoaffinity on an anti-TF mAb-Sepharose column. TF preparations from non-human primates, canine, bovine, pig, rabbit, and mouse brains were extracted from acetone powders as described previously (16). All assays were conducted with rhTF, relipidated as previously described (17). Chromogenic assays were performed using purified human factors VII, VIIa, and X (Enzyme Research Laboratories, South Bend, IN) and chromogenic substrates S-2222 and S-2288 (Chromogenix, Milan, Italy) as previously described (18, 19).
PT tests were conducted using relipidated rhTF and human plasma (Ci-Trol Control, Dade Behring, Deerfield, IL) using an automated coagulation timer (MLA Electra 800 or 900C, Medical Laboratory Automation, Pleasantville, N.Y) according to the manufacturer’s instructions. PT assays were initiated by injecting 0.2 mL of various concentrations of lipidated rhTF into plastic twin-well cuvettes containing 0.1 mL of plasma that had been preincubated with either 0.01 mL of buffer or antibody for 1–2 minutes at room temperature. The inhibition of TF procoagulant activity by anti-TF mAb was calculated using an rhTF standard curve in which the log rhTF concentration was plotted against log clot time.
Cellular TF assays
Factor X activation by TF expressed on cell surfaces was performed with the human bladder carcinoma cell line J82 (American Type Culture Collection (ATCC), Manassas, VA) in the presence of FVII as described by Fair and MacDonald (20). J82 cells (2 × 105) in 1 mL were preincubated with FVII (50 ng) for 2 hours at 37 °C in the absence or presence of various concentrations of H36, followed by addition of 0.3 mL of FX (50 μg/mL). FXa activity generated by J82 cells was determined using chromogenic assays described above.
MDA-MB231 breast cancer cells (ATCC) expressing TF were incubated at room temperature for one hour with anti-TF antibody mAb, human FVII (6.5 μg) or FX (10 μg). The cells were stained with fluorescein isothiocyanate (FITC)-labeled goat anti-mouse IgG (Jackson ImmunoResearch Laboratory, West Grove, PA) for 30 minutes at room temperature and analyzed on a FACScan (BD Biosciences, San Jose, CA).
Chimpanzee endarterectomy model
Animals
Five tuberculosis-negative, disease-free chimpanzees were selected for this study from the general population of the Yerkes National Primate Research Center. All procedures were approved by the Emory IACUC and the Interagency Animal Model Committee of the National Institutes of Health and were conducted in accordance with U. S. federal guidelines.
Femoral endarterectomy procedures
Femoral endarterectomy was performed through medial thigh incision as previously described (21). To prevent intravascular thrombosis during vessel crossclamp, a bolus of 100 U/kg of unfractionated heparin was administered prior to clamping the vessel. The superficial femoral artery was dissected free of surrounding tissues, cross-clamped at each end of the exposed vessel and cut proximal to the distal clamp. The artery was then everted and endarterectomy was performed by removing the normal intima and the upper half of the media for a distance of 1 cm in the central portion of the vessel, using forceps under 2.5 times optical magnification. Following endarterectomy the vessel was returned to its normal configuration and an end-to-end anastomosis was performed. Heparin anticoagulation was reversed by protamine sulfate (1 mg/kg) prior to unclamping and restoration of flow. ALT-836 (1 mg/kg) or placebo (PBS) was injected intravenously as a bolus immediately preceding the restoration of flow in the endarterectomized artery. Identification of the endarterectomized portion of the vessel for later gamma camera imaging was accomplished by affixing a plastic-sealed ~5 μCi 57Co radioisotope source to the perivascular adventitia prior to wound closure.
Measurements of vascular thrombosis
Autologous chimpanzee platelets were labeled with 1 mCi 111Indium (111In)-Oxine and reinjected at least 30 minutes prior to restoration of blood contact with the freshly endarterectomized segment (21, 22). Gamma camera images were acquired continuously for 30 minutes in 5 minutes intervals with a GE 400T scintillation camera (General Electric Milwaukee, WI). The data were stored and analyzed on a Medical Data System A Computer (Medasys Inc., Ann Arbor, MI). A medium-energy collimator was placed close to the animal. The total platelet deposition, including both labeled and unlabeled platelets, was calculated as described previously (23). The results are expressed as platelets deposited per 1 cm of vascular graft. The vessel injury site/blood ratio was determined as described previously (24).
Other assays
Platelet counts and white cell counts were performed as described previously (23). Plasma ALT-836 activity was determined using PT assays as described above. Bleeding time measurements were performed in duplicate on the shaved volar surface of the forearm using the standard template method described previously for studies in baboons (25). Measurement of surgical blood loss was quantitated as described previously (26).
Statistics
The Wilcoxon signed rank test and the Mann Whitney two-sample test were used to test for significance of differences. The analysis of variance (ANOVA) test for multivariate repeated measure analysis was applied for multiple comparisons. Mean ± standard error of the mean is given unless otherwise noted. P-values < 0.05 were considered significant.
Results
Screening of anti-TF monoclonal antibodies
A panel of mAbs was generated against purified, relipidated rhTF and tested for the ability to block TF cofactor function. The effect of each mAb on TF-FVIIa amidolytic activity toward synthetic peptide substrate S-2288 was examined. Since FVIIa alone exhibits very little catalytic activity in the absence of TF (27, 28), all assays contained equal molar amounts of rhTF and FVIIa. Antibody activity was determined both when each mAb was preincubated with rhTF prior to addition of FVIIa and when each mAb was added after formation of the TF-FVIIa complex. The results shown in Figure 1A indicate that five of the sixteen mAbs showed significant inhibition (38–68%) of TF-FVIIa amidolytic activity when each mAb was preincubated with rhTF for 30 minutes at 37°C prior to addition of FVIIa. However, inhibition of TF-FVIIa amidolytic activity by these mAbs was decreased when rhTF and FVIIa were allowed to complex prior to mAb addition, indicating that these five mAbs interfere with the binding of FVIIa to TF and formation of the catalytically active TF-FVIIa complex.
Figure 1. Inhibition of TF-FVIIa activity by anti-TF monoclonal antibodies.

(A) Lipidated recombinant human TF (5 nM) was preincubated with anti-TF antibody (50 nM) for 30 minutes at 37°C, followed by addition of FVIIa (5 nM) (black bars). Alternatively, lipidated recombinant human TF (5 nM) was preincubated with human FVIIa (5 nM) for 30 minutes at 37°C, followed by addition of individual mAb (50 nM) (white bars). The chromogenic substrate S-2288 was added and TF-FVIIa activity measured as described in Materials and Methods. The average percent inhibition of TF-FVIIa activity (± tandard deviation (SD), n = 3) relative to a control reaction without antibody is plotted. (B) Pre-formed TF-FVIIa complex (containing 0.08 nM TF and 0.08 nM FVIIa) was incubated with 0.8 nM of each individual mAb for 30 minutes at 37°C. Factor X was then added to start the activation. Newly formed FXa was then measured using the chromogenic substrate S-2222 as described in Materials and Methods. The average percent inhibition of TF-FVIIa-dependent FX activity (± D, n = 3) relative to a control reaction without antibody is plotted.
To examine the effect of anti-TF mAbs on the TF-FVIIa-dependent FXa activation, each mAb was added to preformed TF-FVIIa complex and human plasma FX was used as a substrate with the subsequent formation of the FXa product measured based on FXa enzymatic activity. The concentration of TF-FVIIa complex (containing 0.08 nM rhTF and 0.08 nM FVIIa) was much lower in this assay than that used in the FVIIa amidolytic assay (containing 5 nM of rhTF and FVIIa, respectively). In this assay only those mAbs that interfere with TF-FVIIa complex activity, rather than TF-FVIIa complex formation, displayed significant inhibition. Three mAbs (H36, M5 and M107) showed significant inhibition against TF-FVIIa-dependent FX activation when a 10 fold molar excess of mAb over TF was tested (Figure 1B). At this concentration, H36 inhibited 95% of TF-FVIIa-dependent FX activation.
Effect of H36 on FX activation
To characterize the inhibitory mechanism of H36, the mAb was titrated at a fixed TF-FVIIa complex concentration (0.08 nM TF, 0.08 nM FVIIa) in the TF-FVIIa-dependent FX activation assay. When H36 was preincubated with TF-FVIIa complex for 30 minutes at 37°C prior to FX addition, the inhibition of FX activation by H36 was very potent, with an IC50 value of 0.02 nM (IC50 value refers to the antibody concentration required for inhibiting 50% TF-FVIIa-dependent FX activation). At an H36/rhTF molar ratio of one or greater, the inhibition of FX activation reached saturation at about 95% inhibition (Figure 2A). Under the same assay conditions, H36 Fab also inhibited TF-FVIIa-dependent FX activation, with an IC50 value of 0.031 nM. WhenTF-FVIIa, FX and H36 were mixed simultaneously to start FX activation, the IC50 value increased to 0.96 nM, indicating less inhibition of FX activation by H36. These data indicate that the order by which H36 and FX were added to TF-FVIIa complex had a dramatic effect on the inhibitory ability of H36, suggesting that anti-TF antibody H36 and FX can compete or sterically block each other’s binding to TF. A similar titration study demonstrated that FIX activation by TF-FVIIa was also inhibited by H36 (data not shown).
Figure 2. Inhibition of coagulation by anti-TF antibodies H36 and ALT-836.

(A) Inhibition of TF-FVIIa-mediated FX activation by anti-TF monoclonal antibodies murine H36 (▲) and human-mouse chimeric H36 (ALT-836) (□). Preformed TF-FVIIa complex (containing 0.08 nM TF and 0.08 nM FVIIa) was preincubated in a microfuge tube at 37°C for 30 minutes with buffer, H36 or ALT-836 (0.02 nM, 0.04 nM, 0.08 nM, and 0.16 nM) separately. Factor X was then added to the above mixture to a final concentration of 30 nM and allowed to incubate for 10 minutes at 37°C. Factor X activation was quenched with 50 mM EDTA and the newly generated FXa concentration was determined with chromogenic substrate S-2222. (B) Human bladder carcinoma cells (J82, 2 ×105) in 1 mL were preincubated with FVII (50 ng) for 2 hours at 37 °C in the absence or presence of the indicated concentrations of H36, followed by addition of 0.3 mL of FX (50 μg/mL). Newly formed FXa levels were determined as described in Materials and Methods. (C) Effect of anti-TF monoclonal antibodies murine H36 (▲) and ALT-836 (□) on PT clotting times. PT assays were performed using Innovin and Ci-Trol Control Level I in the absence and presence of the anti-TF antibody. Data represent the means ± SD for triplicates.
Binding of H36 to cell-surface TF
Flow cytometry analysis on human breast MB231 cells was used to assess the binding of H36 to TF expressed on the cell surface. As shown in Figure 3A and 3B, H36 can stain MB231 cells and addition of FX, but not FVII, inhibits this staining. In contrast, staining of MB231 cells by the anti-TF antibody I43, which is known to bind to the FVII binding site on TF (data not shown), is strongly affected by the addition of FVII (Figure 3C).
Figure 3. TF-specific staining of MDA MB231 cells with anti-TF mAbs H36 and I43.
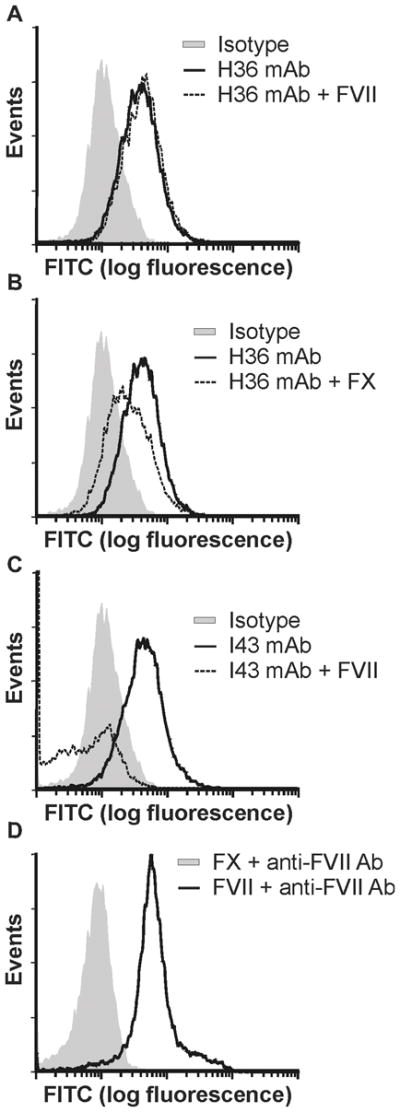
(A) Tumor cells (2.2 × 105 in 0.2 mL) were incubated with 0.5 μg of H36 in the absence (gray shading) or presence (dark line) of 6.5 μg human FVII then stained with FITC-labeled goat anti-mouse IgG. Staining was compared to cells treated with an IgG4 isotype control (dashed line). (B) Tumor cells were stained with H36 as in (A) in the absence (gray shading) or presence (dark line) of 10 μg of human FX. (C) Tumor cells were incubated with 0.7 μg of I43 in the absence (gray shading) or presence (dark line) of 6.5 μg human FVII then stained with FITC-labeled goat anti-mouse IgG. (D) Tumor cells were incubated with FVII (6.5 μg) (dark line) or FX (10 μg) (dashed line), and then with FVII antibody, followed by staining with FITC-labeled goat anti-mouse IgG. Positive staining of FVII treated cells verifies the ability of FVII to bind cell surface TF.
J82 cells were used to further determine the effect of H36 on cellular TF activity. When J82 cells were preincubated with FVII and H36 for 2 hours at 37°C prior to addition of FX, H36 exhibited inhibition of cell surface TF-FVIIa complex-mediated FX activation (Figure 2B). When H36, J82 cells, FVII, and FX were added simultaneously to start FX activation, less inhibition of FX activation was observed (data not shown). These findings are consistent with the data obtained in the cell-free assay. Collectively, these data indicate that H36 is able to bind to cell surface-associated human TF and blocks FX activation; and H36 and FX compete for binding to TF-FVIIa complex displayed on cell surfaces.
In-vitro specificity and potency
PT assays were conducted to determine the specificity and potency of H36 on the extrinsic coagulation pathway. H36 did not have any significant effect on TF-initiated clotting times in assays using plasma and lipidated TF from canine, bovine, pig, rabbit, and mouse, but significantly inhibited TF cofactor function in comparable assays using human, baboon, rhesus and cynomolgus monkey components (data not shown), demonstrating the specificity of H36 for TF of primates. Prolongation of clotting time by H36 was observed over a broad range of rhTF concentrations in PT assays using human plasma (Table 1). Moreover, an H36 concentration only 1.3 fold higher than the rhTF concentration (i.e. 16 nM H36 vs. 11.5 nM rhTF) provided >90% inhibition of TF cofactor activity. These results are consistent with the apparent association constant of H36 for rhTF of 3.1 × 1010 M−1 determined by surface plasmon resonance (data not shown). H36 did not prolong the clotting time in APTT assays using human plasma.
Table 1.
Inhibition of TF-initiated coagulation by H36 in human plasma
| Final [rhTF] in PT Assay, nM | Final [H36] in PT Assay, nM | % Inhibition of rhTF Function |
|---|---|---|
| 0.005 | 3.2 | 46 |
| 0.02 | 3.2 | 65 |
| 6.5 | 74 | |
| 0.09 | 3.2 | 65 |
| 6.5 | 90 | |
| 13 | 96 | |
| 0.5 | 13 | 97 |
| 32 | 99. | |
| 2.3 | 16 | 95 |
| 32 | 100 | |
| 11.5 | 16 | 93 |
| 32 | 99 | |
Construction and expression of ALT-836 (Human-mouse chimeric H36)
To minimize the human anti-mouse antibody (HAMA) immunological response that may occur when mouse antibodies are used as human therapeutics, the murine H36 mAb was converted to a human-mouse chimeric antibody for clinical development. This was accomplished by isolating the H36 variable regions from the hybridoma cell line and subsequently cloning the H36 HC variable region as a fusion to the human IgG4 HC constant domain and the H36 LC variable region as a fusion to the human kappa LC constant domain. This chimeric antibody was designated ALT-836 (previously Sunol-cH36) (29, 30). IgG4 was selected since this immunoglobulin isotype is known to be ineffective in eliciting antibody-dependent cellular cytotoxicity function and complement fixation (31). As shown in Figure 2A, the activity of H36 to inhibit TF-FVIIa-dependent FX activation was not significantly affected by the chimerization process. Data from PT assays using rhTF also indicated that ALT-836 showed comparable activity to H36 in inhibiting TF-dependent coagulation in human plasma (Figure 2C).
Effect of ALT-836 on thrombus formation at sites of femoral endarterectomy
Preliminary experiments with LPS-induced monocytes isolated from baboon, chimpanzee and human indicated that ALT-836 had less inhibitory activity against baboon TF than human or chimpanzee TF (data not shown). Thus, chimpanzees were chosen to test the antithrombotic effect of ALT-836 in a femoral endarterectomy model with a randomized, placebo-controlled crossover study design. In this model, TF exposed at the sites of vascular injury initiates blood coagulation resulting in vascular thrombosis as measured by platelet deposition (11). Sequential surgical endarterectomies on right and left superficial femoral arteries were performed 30 days apart in five chimpanzees, with pre-surgical labeling of autologous platelets using 111In-Oxine and post-surgical gamma camera imaging of 111In-platelet deposition at endarterectomy sites. The ALT-836 antibody (1 mg/kg) or placebo was injected intravenously as a bolus immediately preceding the restoration of flow in the endarterectomized femoral artery. The sequence for ALT-836 or placebo and right or left femoral artery endarterectomy were each randomly determined. Using this design, each animal served as its own control for the treatment comparison regarding 111In-platelet deposition at sites of surgical femoral endarterectomy.
Treatment with a bolus of ALT-836 (1 mg/kg) at the time of the endarterectomy significantly reduced acute vascular thrombosis. Platelet deposition at sites of endarterectomy was significantly decreased at 5 minutes (Figure 3A), from 1.29 ± 0.42 × 109 platelets in controls to 0.36 ± 0.19 × 109 platelets (P = 0.001) in the treated group. Similarly, at 30 minutes following endarterectomy, platelet deposition was decreased from 1.53 ± 0.93 × 109 platelets in controls to 0.37 ± 0.03 × 109 platelets (P = 0.02) in the treated group (Figure 4A).
Figure 4. ALT-836 effects on platelets and vessel injury/blood ratio in chimpanzees.

(A) Effect of ALT-836 (1 mg/kg) (white bars) compared to PBS control (black bars) on platelet deposition at injury sites induced by surgical endarterectomies in chimpanzees. Platelet deposition was measured by scintillation camera imaging of autologous 111In-platelets. (B) Effect of ALT-836 (1 mg/kg) (white bars) compared to PBS control (black bars) on vessel injury/blood ratios in chimpanzees following surgical endarterectomies. The vessel injury/blood ratios were calculated from 111In-platelet activity at the endarterectomy site and 111In-platelet activity in the blood. Data represent the means ± SEM (n = 5). * P < 0.02; ALT-836 vs. control.
Due to the continuous clearance of circulating 111In platelet activity, platelet accumulation after the acute postoperative period was expressed as the ratio of the 111In platelet activity at the endarterectomy site to the 111In platelet activity in the blood. This measurement, the vessel injury site/blood ratio, is independent of the size of the animal, the amount of isotope injected, or the extent to which the isotope may have decayed and represents a reproducible measure of thrombus over time (24). The vessel injury site/blood ratio was shown to be affected by ALT-836 treatment (Figure 4B). Five minutes postoperatively, the vessel injury site/blood ratio decreased over 3-fold in ALT-836 treated animals compared to controls (P = 0.002) and at 30 minutes the ratio decreased over 5-fold with treatment (P = 0.02). At 24 hours postoperatively, the ratio was not significantly different between the two groups. To assess longer-term effects of ALT-836, the endarterectomized vessel segments were harvested 30 days following surgery, and the patency of these vessels was examined. Twenty percent of the control vessels from the controls were patent, whereas 80% of the vessels treated with ALT-836 were patent (not significant).
Effect of ALT-836 on hemostatic function and surgical blood loss
To evaluate the hemostatic risk profile of ALT-836, bleeding times and surgical blood loss were measured (Table 2). No significant change in mean bleeding time was observed in control versus ALT-836-treated chimpanzees at baseline or at 60 minutes post infusion. Additionally, surgical blood loss was measured by hemoglobin determination from sponges collected during surgery and was not found to be significantly different for the five ALT-836 treatment procedures compared to the control procedures. However, the prothrombin time was prolonged from baseline 11.2 ± 0.7 seconds to 177 ± 27 seconds (P<0.0001) at 60 minutes following ALT-836 injection (Table 2), implying that there is sufficient anti-TF mAb available to inhibit TF-mediated coagulation in vivo. No rebleeding events occurred in the control or treated animals. No increase in the hematoma formation was observed at the surgical site in the ALT-836 treated animals. Thus, hemostatic function was unaffected by the anti-TF antibody therapy at a dose that is potently antithrombotic, despite the dramatic prolongation of the PT clotting time in vitro. There were no significant changes in the platelet count or WBC count between the groups although there was an increase in the WBC count in both groups in response to the surgery (Table 2).
Table 2.
Effect of ALT-836 on hematological parameters in endarterectomized chimpanzees
| Parameters* | Control (mean ± SD, n = 5) | ALT-836 (mean ± SD, n = 5) | P |
|---|---|---|---|
| Bleeding Time (min) | 2.0 ± 0.2 | 2.5 ± 0.6 | ns |
| Surgical Blood Loss (mL) | 5.9 ± 2.2 | 4.7 ± 3.0 | ns |
| PT Time (sec): | |||
| Pre-Surgery Baseline | nd | 11.2 ± 0.7 | nd |
| 5 minutes post RF | nd | 180 ± 40 | 0.0001 |
| 15 minutes post RF | nd | 157 ± 52 | 0.0001 |
| 30 minutes post RF | nd | 161 ± 60 | 0.0001 |
| 60 minutes post RF | nd | 177 ± 27 | 0.0001 |
| 90 minutes post RF | nd | 202 ± 7.7 | 0.0001 |
| 24 hours post RF | nd | 18.1 ± 5.1 | 0.036 |
| Platelet Count (×103/μL): | |||
| Pre-Surgery Baseline | 222 ± 73 | 233 ± 52 | ns |
| 24 hours Post Surgery | 273 ± 61 | 247 ± 90 | ns |
| WBC (×103/μL): | |||
| Pre-Surgery Baseline | 7.8 ± 1.5 | 9.8 ± 1.8 | ns |
| 24 hours Post Surgery | 18.1 ± 4.5 | 19.7 ± 6.5 | ns |
ns, not significant; P < 0.05 considered statistically significant nd, not determined
post RF, post restoration of flow following conclusion of endarterectomy
Activity of plasma ALT-836 following bolus injections in chimpanzees
The activity levels on ALT-836 in the plasma following bolus intravenous injections in chimpanzees were estimated by FX activation and PT assays (Table 3). The ALT-836 levels 5 minutes after injection averaged 26.5 ± 5.3 μg/mL (~180 nM) using the PT assay, and 18.8 ± 2.8 μg/mL (~125 nM) using the FX activation assay. Twenty-four hours post-injection the mean antibody levels fell to one third of the initial blood levels, approximating a 16-hour half-life assuming first-order pharmacokinetics.
Table 3.
ALT-836 plasma levels following intravenous injection
| Time After ALT-836 Intravenous Injection | Plasma Concentrations of ALT-836 (μg/mL) Determined by Prothrombin Time Assay | Plasma Concentrations of ALT-836 (μg/mL) Determined by Factor X Activation Assay |
|---|---|---|
| 5 min | 26.5 ± 5.3 | 18.8 ± 2.8 |
| 90 min | 27.0 ± 2.1 | 16.5 ± 1.4 |
| 24 hour | 8.9 ± 3.7 | 6.3 ± 0.5 |
Discussion
Arterial thrombosis is the major cause of morbidity and mortality in patients with acute coronary artery syndromes. There is substantial evidence that this acute thrombotic process is triggered by TF-dependent activation of the coagulation cascade, thrombin generation, fibrin deposition, and platelet activation following the rupture of an atherosclerotic plaque (32). Additionally, interventional mechanical procedures such as endarterectomy, balloon catheter angioplasty, and thrombolytic therapy may trigger TF-dependent thrombosis. Therapies such as aspirin, heparins, and platelet inhibitors are proven to reduce the risks associated with acute coronary syndromes but are ineffective at preventing coronary thrombosis unless administered at doses that significantly compromise hemostatic function. In contrast, studies with FVIIa-specific antagonists support the notion that inhibition of TF-FVIIa activity provides a novel opportunity to selectively intervene in mechanical or pathological activation of the coagulation pathway without major untoward effects on hemostasis (9–11). However, it is not clear whether blocking of FX activation by TF-FVIIa complex is capable of achieving similar effects.
To directly address this, we generated an anti-TF monoclonal antibody, H36, which potently inhibits human TF-mediated blood coagulation by blocking FX/FIX binding to the by TF-FVIIa complex. This antibody is highly species-specific and recognizes an epitope of TF displayed on the cell surface. Consistent with a previous report (33), H36-specific blockade of the FX/FIX binding site on TF was found to be more effective at inhibiting TF-dependent coagulation than antibody-specific blockade of the FVII binding site. These results are likely due to the inefficiency of anti-TF antibodies to affect the high affinity (Kd<50 pM) stable interactions between lipidated TF and FVII (12, 34). In contrast, FX initially binds the anionic phospholipid surface (Kd=10–100 nM) and then forms a transient ternary complex with TF-FVIIa (35). In the absence of phospholipid interactions, the binding affinity of FX for the TF-FVIIa complex has been reported to be in the micromolar range (36). Thus, H36 with a high binding affinity for TF (Kd=32 pM) is able to effectively block formation of the TF-FVIIa-FX complex resulting in potent inhibition of FX activation and clot formation in vitro and TF-initiated acute arterial thrombosis in vivo.
Several groups have published data characterizing the interactions of various TF antibodies with TF and TF-VIIa (33, 37–40). These studies suggest that the location of antibody binding is an important determinant of the potency of the inhibition of TF function. Antibodies which, like H36 and ALT-836, bind to TF and inhibit TF-FVIIa substrate binding (D3, 5G9, 5G6) display a much higher anticoagulant potency than those antibodies that inhibit the TF association with FVIIa. Of particular interest in these studies is the apparent difference in the D3 and 5G9 antibodies when compared to the 5G6 antibody (33, 37–39). These studies determined that although all three of the antibodies were potent anticoagulants, the D3 and 5G9 antibodies differed in that they could also weakly inhibit the amidolytic activity of the TF-FVIIa complex suggesting subtle differences in epitope binding. While the exact epitope of H36 and ALT-836 has not been determined, it would appear that the function of these antibodies is similar to that of 5G6 in that there is no detectable inhibition of TF-VIIa amidolytic activity (Fig. 1a).
Also of interest are more recent studies that suggest that antibodies targeting different epitopes on TF can selectively inhibit diverse functions. Versteeg et al. (14) recently reported a unique pair of isotype-matched antibodies targeting separate epitopes on TF, one of which could inhibit coagulation and the other which could inhibit activation of PAR2 and disrupt the interaction of TF with integrins. These findings suggest that the independent actions of TF may allow for the development of approaches that can inhibit specific functions of the protein while not affecting the activity of the other protein functions. These findings also underline the difficulties in predicting the activity of different TF antagonists as potential anti-thrombotic drugs when the molecules bind to different regions of TF or inhibit different TF functions. The clinical outcomes of FVII and FX blockage, using for example FVIIai or ALT-836, could be vastly different.
Since our data show that a narrow concentration range (6–16 nM, 2.5 fold) of H36 inhibits >90% of TF cofactor activity at a very broad range of TF concentrations (0.1–12 nM, 120 fold), this antibody may have potential for use in the management of various diseases, such as acute coronary syndrome and coagulopathy. TF levels and activity are known to vary significantly depending on the type of disease and/or its progression. For example, elevated circulating TF activity has been observed in patients with cardiovascular risk factors including diabetes, hypercholesterolemia, smoking and hypertension (41, 42). Moreover, patients with unstable angina show higher plasma TF levels than those with stable angina (238 pg/mL vs. 189 pg/mL), and elevated plasma TF levels may predict future cardiovascular events in these patients (43). There are higher levels of functionally active TF in the plaque of patients with unstable angina or acute myocardial infarction than in those with stable angina (44, 45). In one study, a median TF concentration of 66 pg/mg-plaque (2 nM TF assuming a homogeneous distribution) was detected in samples from patients with unstable angina patients and 32 pg/mg-plaque (1 nM) in those from stable angina patients (44). In addition, histologically defined thrombus was only detected in atherosclerotic lesions where TF was present (45), supporting the view that the presence and amount of functional TF in coronary atherosclerotic plaques may be critical for thrombosis associated with acute coronary events (32). A number of recent reports have also established the presence of low levels of circulating TF in the form of microparticles which support coagulation and inflammation (46). Several disorders of the vasculature and hematopoetic system such as thrombotic stroke, coronary artery disease, anti-phospholipid antibody syndrome, sickle cell disease, type 2 diabetes, and cancer are associated with increased levels of TF-bearing microparticles (47–52). The levels of tissue factor in microparticles from the plasma of normal individuals have generally been reported to be in the low picomolar range (<10 pM) while TF levels from the plasma of patients with CAD have been reported to be 10–15 times higher (15–132 pM) (53). These blood borne TF microparticles are thought to play a crucial role in the rapid thrombus formation after plaque rupture leading to myocardial infraction (54). Given that pico- to nanomolar levels of TF are found in circulation and in atherosclerotic plaques associated with unstable disease in patients, administration of H36 (or the chimeric version, ALT-836) at a dosage sufficient to provide nanomolar serum Ab concentrations could provide effective control of TF-induced vascular thrombosis in these patients.
ALT-836 has been used in a Phase I clinical trial for a cardiovascular indication (29). In this study, a total of 26 patients, on a daily dose of aspirin (81 – 325 mg), received a single intravenous bolus of ALT-836 at levels ranging from 0.03 to 0.3 mg/kg body weight. In the 0.08 mg/kg dose cohort, the maximal concentration of ALT-836 in the serum was 13 nM with a terminal half-life of about 72 hours. This level is several fold higher than that estimated above to inhibit procoagulant TF activity following atherosclerotic plaque rupture. In addition, equivalent concentrations of ALT-836 effectively inhibited clot formation and thrombin generation in whole blood and this activity was more pronounced with prior administration of aspirin (29). No major bleeding events were observed for any ALT-836 dose level in the Phase I study; though dose-related spontaneous minor bleeding was reported that was consistent with platelet-mediated bleeding without thrombocytopenia (29). In a separate clinical trial, no spontaneous bleeding episodes were observed in patients treated with ALT-836 at the 0.1 mg/kg level while undergoing peripheral vascular grafts (unpublished data). Collectively, these data indicate that ALT-836 can be safely administered to patients resulting in serum Ab levels with significant antithrombotic activity.
The potential of ALT-836 is further illustrated in the non-human primate arterial thrombosis model reported here. ALT-836 demonstrated a high degree of effectiveness in decreasing TF-dependent vascular thrombogenesis induced by surgical endarterectomy in chimpanzees. Following a bolus injection of ALT-836 (1 mg/kg) platelet accumulation was significantly decreased at femoral endarterectomy sites when compared to non-treated controls. Furthermore, the long-term patency rate for ALT-836 treated vessels was greater than for the control vessels. The lack of an effect on bleeding times and surgical blood loss verified that hemostasis was preserved when ALT-836 was administered at this dose. The results observed for ALT-836 are comparable to those reported for active-site inactivated FVIIai treatment (1 mg/kg), which provided a >10 fold decrease in 111In-platelet deposition at the site of carotid endarterectomy without increasing bleeding time or surgical blood loss in a similar surgical endarterectomy model in baboons (11). FVIIai was also shown to inhibit vascular lesion formation following surgical carotid endarterectomy and femoral artery balloon catheter angioplasty in this model (11). In contrast, administration of either antithrombins or inhibitors of platelet aggregation (anti-GPIIb/IIIa mAb) at doses effective at eliciting a comparable antithrombotic effect caused >8 fold prolongation in bleeding time and a >50 fold increase in surgical bleeding, indicating a striking inhibition of hemostatic function (11). Thus, it is clear from the results presented here and these previous studies that direct inhibition of the TF-dependent coagulation pathway at either the TF FX/FIX or FVIIa binding site can achieve potent antithrombotic effects without compromising hemostasis.
In addition to its high binding affinity and specificity to TF, ALT-836 has a number of features desirable for a therapeutic TF antagonist. Ease of manufacture via standard industrial methods and a long-term stability in solution at 2–8°C (>4 years) distinguish ALT-836 from FVIIai. ALT-836 has also been tested in cynomolgus monkeys at doses as high as 50 mg/kg administered as a single bolus and four doses of up to 25 mg/kg over a 4-week period with no apparent systemic or overt toxicity and no abnormal bleeding events reported (unpublished data). In clinical studies, a single bolus infusion of ALT-836 exhibited a serum half-life of about 3 days in subjects with stable coronary artery disease compared to a 3.8 to 5.8 hour half-life reported for FVIIai in healthy human subjects (29, 55). Based on these findings, potent activity in preventing vascular thrombogenesis and an acceptable safety profile in the chimpanzee endarterectomy model reported here, ALT-836 may be an attractive clinical candidate as an antithrombotic agent in patients with unstable angina or as a transient inhibitor of TF-induced thrombotic complications associated with a variety of interventional procedures.
Acknowledgments
The authors gratefully thank Jeffrey Stinson for providing expression vectors, Lawrence Luepschen for technical assistance in establishing chromogenic assay conditions and Dr. Laurence A. Harker for his thoughtful advice on the conduct of the chimpanzee studies and his critical reading of the manuscript.
References
- 1.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Harker LA, Maraganore JM, Hirsch J. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. J.P. Lippincott; 1994. Novel antithrombotic agents; pp. 1638–60. [Google Scholar]
- 3.Braunwald E. Shattuck lecture--cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med. 1997;337:1360–9. doi: 10.1056/NEJM199711063371906. [DOI] [PubMed] [Google Scholar]
- 4.Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–71. doi: 10.1161/01.cir.92.3.657. [DOI] [PubMed] [Google Scholar]
- 5.Krishnaswamy S, Field KA, Edgington TS, et al. Role of the membrane surface in the activation of human coagulation factor X. J Biol Chem. 1992;267:26110–20. [PubMed] [Google Scholar]
- 6.Furie B, Furie BC. Molecular and cellular biology of blood coagulation. N Engl J Med. 1992;326:800–6. doi: 10.1056/NEJM199203193261205. [DOI] [PubMed] [Google Scholar]
- 7.Harker LA, Hanson SR, Kelly AB. Antithrombotic strategies targeting thrombin activities, thrombin receptors and thrombin generation. Thromb Haemost. 1997;78:736–41. [PubMed] [Google Scholar]
- 8.Weitz JI, Hirsh J, Samama MM. New antithrombotic drugs: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition) Chest. 2008;133:234S–56S. doi: 10.1378/chest.08-0673. [DOI] [PubMed] [Google Scholar]
- 9.Shirk RA, Vlasuk GP. Inhibitors of factor VIIa/tissue factor. Arterioscler Thromb Vasc Biol. 2007;27:1895–900. doi: 10.1161/ATVBAHA.107.148304. [DOI] [PubMed] [Google Scholar]
- 10.Himber J, Kirchhofer D, Riederer M, et al. Dissociation of antithrombotic effect and bleeding time prolongation in rabbits by inhibiting tissue factor function. Thromb Haemost. 1997;78:1142–9. [PubMed] [Google Scholar]
- 11.Harker LA, Hanson SR, Wilcox JN, et al. Antithrombotic and antilesion benefits without hemorrhagic risks by inhibiting tissue factor pathway. Haemostasis. 1996;26:76–82. doi: 10.1159/000217245. [DOI] [PubMed] [Google Scholar]
- 12.Neuenschwander PF, Morrissey JH. Roles of the membrane-interactive regions of factor VIIa and tissue factor. The factor VIIa Gla domain is dispensable for binding to tissue factor but important for activation of factor X. J Biol Chem. 1994;269:8007–13. [PubMed] [Google Scholar]
- 13.Rao LV, Pendurthi UR. Tissue factor-factor VIIa signaling. Arterioscler Thromb Vasc Biol. 2005;25:47–56. doi: 10.1161/01.ATV.0000151624.45775.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Versteeg HH, Schaffner F, Kerver M, et al. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111:190–9. doi: 10.1182/blood-2007-07-101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stinson JR, Wittman V, Wong HC. PCR Strategies. Academic Press; San Diego: 1995. Generation of single-chain antibody fragments by PCR; pp. 300–12. [Google Scholar]
- 16.Broze GJ, Jr, Leykam JE, Schwartz BD, et al. Purification of human brain tissue factor. J Biol Chem. 1985;260:10917–20. [PubMed] [Google Scholar]
- 17.Barenholz Y, Gibbes D, Litman BJ, et al. A simple method for the preparation of homogeneous phospholipid vesicles. Biochemistry. 1977;16:2806–10. doi: 10.1021/bi00631a035. [DOI] [PubMed] [Google Scholar]
- 18.Neuenschwander PF, Branam DE, Morrissey JH. Importance of substrate composition, pH and other variables on tissue factor enhancement of factor VIIa activity. Thromb Haemost. 1993;70:970–7. [PubMed] [Google Scholar]
- 19.Broze GJ, Jr, Warren LA, Novotny WF, et al. The lipoprotein-associated coagulation inhibitor that inhibits the factor VII-tissue factor complex also inhibits factor Xa: insight into its possible mechanism of action. Blood. 1988;71:335–43. [PubMed] [Google Scholar]
- 20.Fair DS, MacDonald MJ. Cooperative interaction between factor VII and cell surface-expressed tissue factor. J Biol Chem. 1987;262:11692–8. [PubMed] [Google Scholar]
- 21.Lumsden AB, Kelly AB, Schneider PA, et al. Lasting safe interruption of endarterectomy thrombosis by transiently infused antithrombin peptide D-Phe-Pro-ArgCH2Cl in baboons. Blood. 1993;81:1762–70. [PubMed] [Google Scholar]
- 22.Kelly AB, Marzec UM, Krupski W, et al. Hirudin interruption of heparin-resistant arterial thrombus formation in baboons. Blood. 1991;77:1006–12. [PubMed] [Google Scholar]
- 23.Harker LA, Hunt P, Marzec UM, et al. Regulation of platelet production and function by megakaryocyte growth and development factor in nonhuman primates. Blood. 1996;87:1833–44. [PubMed] [Google Scholar]
- 24.Kotze HF, Lotter MG, Badenhorst PN, et al. Kinetics of In-111-platelets in the baboon: I. Isolation and labelling of a viable and representative platelet population. Thromb Haemost. 1985;53:404–7. [PubMed] [Google Scholar]
- 25.Hanson SR, Harker LA. Interruption of acute platelet-dependent thrombosis by the synthetic antithrombin D-phenylalanyl-L-prolyl-L-arginyl chloromethyl ketone. Proc Natl Acad Sci U S A. 1988;85:3184–8. doi: 10.1073/pnas.85.9.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suleymanov OD, Szalony JA, Salyers AK, et al. Pharmacological interruption of acute thrombus formation with minimal hemorrhagic complications by a small molecule tissue factor/factor VIIa inhibitor: Comparison to factor Xa and thrombin inhibition in a nonhuman primate thrombosis model. J Pharmacol Exp Ther. 2003;306:1115–21. doi: 10.1124/jpet.103.052779. [DOI] [PubMed] [Google Scholar]
- 27.Bom VJ, Bertina RM. The contributions of Ca2+, phospholipids and tissue-factor apoprotein to the activation of human blood-coagulation factor X by activated factor VII. Biochem J. 1990;265:327–36. doi: 10.1042/bj2650327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruf W, Rehemtulla A, Morrissey JH, et al. Phospholipid-independent and-dependent interactions required for tissue factor receptor and cofactor function. J Biol Chem. 1991;266:16256. [PubMed] [Google Scholar]
- 29.Morrow DA, Murphy SA, McCabe CH, et al. Potent inhibition of thrombin with a monoclonal antibody against tissue factor (Sunol-cH36): results of the PROXIMATE-TIMI 27 trial. Eur Heart J. 2005;26:682–8. doi: 10.1093/eurheartj/ehi094. [DOI] [PubMed] [Google Scholar]
- 30.Welty-Wolf KE, Carraway MS, Ortel TL, et al. Blockade of tissue factor-factor X binding attenuates sepsis-induced respiratory and renal failure. Am J Physiol Lung Cell Mol Physiol. 2006;290:L21–31. doi: 10.1152/ajplung.00155.2005. [DOI] [PubMed] [Google Scholar]
- 31.Bruggemann M, Williams GT, Bindon CI, et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. 1987;166:1351–61. doi: 10.1084/jem.166.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113:722–31. doi: 10.1161/CIRCULATIONAHA.105.567297. [DOI] [PubMed] [Google Scholar]
- 33.Kirchhofer D, Moran P, Chiang N, et al. Epitope location on tissue factor determines the anticoagulant potency of monoclonal anti-tissue factor antibodies. Thromb Haemost. 2000;84:1072–81. [PubMed] [Google Scholar]
- 34.Shaw AW, Pureza VS, Sligar SG, et al. The local phospholipid environment modulates the activation of blood clotting. J Biol Chem. 2007;282:6556–63. doi: 10.1074/jbc.M607973200. [DOI] [PubMed] [Google Scholar]
- 35.Monroe DM, Key NS. The tissue factor-factor VIIa complex: procoagulant activity, regulation, and multitasking. J Thromb Haemost. 2007;5:1097–105. doi: 10.1111/j.1538-7836.2007.02435.x. [DOI] [PubMed] [Google Scholar]
- 36.Shobe J, Dickinson CD, Edgington TS, et al. Macromolecular substrate affinity for the tissue factor-factor VIIa complex is independent of scissile bond docking. J Biol Chem. 1999;274:24171–5. doi: 10.1074/jbc.274.34.24171. [DOI] [PubMed] [Google Scholar]
- 37.Ruf W, Edgington TS. An anti-tissue factor monoclonal antibody which inhibits TF:VIIa complex is a potent anticoagulant in plasma. Thrombosis and Haemostasis. 1991;66:529–33. [PubMed] [Google Scholar]
- 38.Faelber K, Kirchhofer D, Presta L, et al. The 1.85 A resolution crystal structures of tissue factor in complex with humanized Fab D3h44 and of free humanized Fab D3h44: revisiting the solvation of antigen combining sites. J Mol Biol. 2001;313:83–97. doi: 10.1006/jmbi.2001.5036. [DOI] [PubMed] [Google Scholar]
- 39.Huang M, Syed R, Stura EA, et al. The mechanism of an inhibitory antibody on TF-initiated blood coagulation revealed by the crystal structures of human tissue factor, Fab 5G9 and TF. G9 complex. J Mol Biol. 1998;275:873–94. doi: 10.1006/jmbi.1997.1512. [DOI] [PubMed] [Google Scholar]
- 40.Ohto U, Mizutani R, Nakamura M, et al. Crystal structure of a humanized Fab fragment of anti-tissue-factor antibody in complex with tissue factor. J Synchrotron Radiat. 2004;11:105–8. doi: 10.1107/s0909049503023513. [DOI] [PubMed] [Google Scholar]
- 41.Sambola A, Osende J, Hathcock J, et al. Role of risk factors in the modulation of tissue factor activity and blood thrombogenicity. Circulation. 2003;107:973–7. doi: 10.1161/01.cir.0000050621.67499.7d. [DOI] [PubMed] [Google Scholar]
- 42.Felmeden DC, Spencer CG, Chung NA, et al. Relation of thrombogenesis in systemic hypertension to angiogenesis and endothelial damage/dysfunction (a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial [ASCOT]) Am J Cardiol. 2003;92:400–5. doi: 10.1016/s0002-9149(03)00657-x. [DOI] [PubMed] [Google Scholar]
- 43.Soejima H, Ogawa H, Yasue H, et al. Heightened tissue factor associated with tissue factor pathway inhibitor and prognosis in patients with unstable angina. Circulation. 1999;99:2908–13. doi: 10.1161/01.cir.99.22.2908. [DOI] [PubMed] [Google Scholar]
- 44.Ardissino D, Merlini PA, Ariens R, et al. Tissue-factor antigen and activity in human coronary atherosclerotic plaques. Lancet. 1997;349:769–71. doi: 10.1016/S0140-6736(96)11189-2. [DOI] [PubMed] [Google Scholar]
- 45.Marmur JD, Thiruvikraman SV, Fyfe BS, et al. Identification of active tissue factor in human coronary atheroma. Circulation. 1996;94:1226–32. doi: 10.1161/01.cir.94.6.1226. [DOI] [PubMed] [Google Scholar]
- 46.Giesen PL, Nemerson Y. Tissue factor on the loose. Semin Thromb Hemost. 2000;26:379–84. doi: 10.1055/s-2000-8456. [DOI] [PubMed] [Google Scholar]
- 47.Simak J, Gelderman MP, Yu H, et al. Circulating endothelial microparticles in acute ischemic stroke: a link to severity, lesion volume and outcome. J Thromb Haemost. 2006;4:1296–302. doi: 10.1111/j.1538-7836.2006.01911.x. [DOI] [PubMed] [Google Scholar]
- 48.Chironi GN, Boulanger CM, Simon A, et al. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335:143–51. doi: 10.1007/s00441-008-0710-9. [DOI] [PubMed] [Google Scholar]
- 49.Dignat-George F, Camoin-Jau L, Sabatier F, et al. Endothelial microparticles: a potential contribution to the thrombotic complications of the antiphospholipid syndrome. Thromb Haemost. 2004;91:667–73. doi: 10.1160/TH03-07-0487. [DOI] [PubMed] [Google Scholar]
- 50.Shet AS, Aras O, Gupta K, et al. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–83. doi: 10.1182/blood-2003-03-0693. [DOI] [PubMed] [Google Scholar]
- 51.Diamant M, Nieuwland R, Pablo RF, et al. Elevated numbers of tissue-factor exposing microparticles correlate with components of the metabolic syndrome in uncomplicated type 2 diabetes mellitus. Circulation. 2002;106:2442–7. doi: 10.1161/01.cir.0000036596.59665.c6. [DOI] [PubMed] [Google Scholar]
- 52.De Cicco M. The prothrombotic state in cancer: pathogenic mechanisms. Crit Rev Oncol Hematol. 2004;50:187–96. doi: 10.1016/j.critrevonc.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 53.Butenas S, Bouchard BA, Brummel-Ziedins KE, et al. Tissue factor activity in whole blood. Blood. 2005;105:2764–70. doi: 10.1182/blood-2004-09-3567. [DOI] [PubMed] [Google Scholar]
- 54.Bogdanov VY, Balasubramanian V, Hathcock J, et al. Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nat Med. 2003;9:458–62. doi: 10.1038/nm841. [DOI] [PubMed] [Google Scholar]
- 55.Erhardtsen E, Nilsson P, Johannessen M, et al. Pharmacokinetics and safety of FFR-rFVIIa after single doses in healthy subjects. J Clin Pharmacol. 2001;41:880–5. doi: 10.1177/00912700122010780. [DOI] [PubMed] [Google Scholar]