

Abstract
Hepatic stellate cells (HSCs) are responsible for type I collagen deposition in liver fibrosis that leads to cirrhosis. The purpose of this study was to examine potential molecular signals that lead to increased α2(I) collagen gene expression by acetaldehyde, the primary metabolite of alcohol and malondialdehyde (MDA), a lipid peroxidation product known to be associated with chronic liver injury. MDA and the combination of MDA and acetaldehyde were employed to determine the effect on α2(I) collagen gene expression as assessed by transient transfection analysis and reverse transcriptase polymerase chain reaction (RT-PCR). Immunoblot and subsequent immunoprecipitation analysis examined stress-activated protein kinase (SAPK) activity. Cotransfection with a dominant negative mutant for c-jun nuclear kinase (dnJNK1) was also employed with the α2(I) collagen promoter. MDA increased α2(I) collagen gene expression nearly 2.5- to 3-fold, however there was no synergistic effect of the combination of acetaldehyde and MDA on α2(I) collagen gene activation and expression. Acetaldehyde, MDA, or both significantly increased JNK activity when compared to untreated stellate cells. The dnJNK1 expression vector abrogated α2(I) collagen transgene activity. In conclusion, JNK activation appears to be critical in the signaling cascade of oxidative metabolites of chronic alcohol-related liver injury and collagen gene activation.
Keywords: Acetaldehyde, Malondialdehyde, Stellate cells, Collagen, Fibrosis, Signal transduction, Oxidative stress, JNK, Free radicals
INTRODUCTION
Chronic liver disease is a leading cause of morbidity and mortality worldwide; and, alcoholic liver disease remains the leading cause of cirrhosis in Western countries. Over the past twelve years, investigators have examined whether or not the oxidative metabolite of alcohol, acetaldehyde, plays a role in augmenting the transcriptional activation of type I collagen genes. In 1987, Brenner and Chojkier were the first to report that acetaldehyde increased collagen gene transcription in cultured fibroblasts [1]. Lieber and colleagues shortly thereafter made an important observation that acetaldehyde stimulates collagen gene activation and protein synthesis in hepatic stellate cells, but not hepatocytes [2]. Indeed, the hepatic stellate cell is widely accepted as the principal collagen-producing cell once it becomes activated. Mezey and his colleagues confirmed that acetaldehyde can activate the α2(I) collagen promoter in NIH-3T3 fibroblasts [3]. In previous work in conjunction with this group we found that acetaldehyde could increase α2(I) collagen promoter gene activation, which was critically dependent on the nuclear factor I (NF-I) binding region in the gene’s promoter [4,5]. This region had been shown by deletion mutation analysis to be required for the transcriptional activation of the mouse α2(I) collagen promoter [6].
It is also evident, however, that acetaldehyde is not believed to be a major factor in the activation of hepatic stellate cells during alcoholic liver injury [7]. Furthermore, Maher and colleagues reported, after an exhaustive investigation, that acetaldehyde did not have a significant effect on collagen gene transcription, and concluded that further study was not warranted [8]. Nevertheless, the pathogenesis of alcoholic liver injury raises important questions regarding potential signaling mechanisms employed by highly reactive aldehydes. Furthermore, alcoholic hepatitis is not necessarily required for progression to cirrhosis, since periportal sclerosis and cirrhosis have been demonstrated in the absence of hepatocellular inflammation and necrosis [9]. Hence, the absence of conclusive evidence that necroinflammation is an absolute prerequisite for the development of fibrosis has led to a search for alternative mediators of liver fibrosis. These not only include acetaldehyde, but also products of lipid peroxidation, in particular malondialdehyde (MDA) and 4-hydroxynonenal.
Lipid peroxidation is a particular type of oxidation reaction resulting from attack by reactive free radicals on the polyunsaturated fatty-acid side chains of membrane phospholipids or lipoproteins. This reaction can lead to complete decomposition of the phospholipids, resulting in membrane disruption and the generation of aldehyde end products, including MDA [10]. There is sufficient evidence suggesting that lipid peroxidation can occur in both acute and chronic liver injury due to alcohol as reviewed by Cederbaum [11]. Lipid peroxidation products such as MDA may also play a role in other chronic liver diseases, including hepatitis C [12].
Because similar concentrations of acetaldehyde and MDA can coexist in the liver during ethanol metabolism it is not surprising that both acetaldehyde and MDA adducts have been detected in livers of ethanol-fed animals [13]. Tuma and colleagues recently demonstrated that acetaldehyde and MDA can react together in a synergistic manner and generate hybrid MDA-acetaldehyde protein adducts (MAA-adducts) and suggest that MAA adducts may represent a major species of adducts formed in the liver during ethanol metabolism in vivo [14]. A major aim of the present study was to determine whether a link between extracellular signaling by putative profibrogenic aldehydes can potentiate the activation of one or more members of the mitogen-activated (MAPK) or stress-activated (SAPK) protein kinase superfamily, which are known to be activated by extracellular stress [15,16]. Also, since MAA adducts have been immunologically detected in animals chronically fed alcohol [17], we examined whether the presence of both acetaldehyde and MDA would act in a synergistic manner to increase collagen gene expression. The work presented here demonstrates that c-jun N-terminal kinase (JNK), one of the major SAPK members, is critical to α2(I) collagen promoter activation in HSCs.
MATERIALS AND METHODS
Materials
Dulbecco’s minimal essential medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin, fungizone, trypsin, EDTA, dithiothreitol, cesium chloride, acrylamide, molecular weight markers, Lipofectamine, and agarose were purchased from Gibco-BRL (Rockville, MD, USA). Plastic 10 cm2 culture dishes and 75 cm2 culture flasks were from Falcon Division, Becton Dickinson Co. (Mountain View, CA, USA). 4-Methylpyrazole, cyanamide, pronase E, collagenase type IV, phenylmethylsulfonyl fluoride (PMSF), β-glycerophosphate, sodium orthovanadate, antipain, leupeptin, aprotinin, cymostatin, and pepstatin were all purchased from Sigma Chemical Co. (St. Louis, MO, USA). Acetaldehyde and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (Milwaukee, WI, USA). Malondialdehyde was purchased as malonaldehyde diethylacetate from Aldrich Chemical Co. (Milwaukee, WI, USA).
Luciferase reporter vectors and pCMVβ were purchased from Promega (Madison, WI, USA). The mouse α2(I) collagen cDNA was provided by Dr. Benoit de Crombrugghe from M. D. Anderson Hospital and Tumor Institute (Houston, TX, USA). Dr. T. H. Tan, Baylor College of Medicine (Houston, TX, USA), provided the dominant negative mutant expression vector construct dnJNK1. RNAzol B was from Tel-Test Inc. (Friendswood, TX, USA). Goat antirabbit IgG horseradish peroxidase (HRP) conjugate, protein A-agarose beads, antibodies to extracellular regulated kinase (ERK), c-jun N-terminal kinase (JNK), p38/mitogen-activated protein kinase (MAPK), and antiphosphospecific JNK, ERK, and p38 antibodies, as well as myelin basic protein substrate, ATF-2, SB202190 were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Stellate cell isolation and culture
Stellate cells were isolated from male Sprague-Dawley rats (500 g) obtained from Hilltop Lab Animals (Scottdale, PA, USA) or Charles River (Charles River, MA, USA). The animals received humane care in compliance with the guidelines of the Animal Care and Use Committee of the Johns Hopkins University and the University of Maryland. The method for isolation of hepatic stellate cells is described in detail in previous publications [5,18] and utilizes a Nycodenz gradient [19]. After harvest, stellate cells were cultured in DMEM containing penicillin (100 units/ml), streptomycin (100 μg/ml), fungizone (2.5 μg/ml), and 10% FBS. Cells were grown at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The medium was changed every 48 h. Cells were passaged as activated stellate cells and frozen. Cells used in these experiments were passaged up to 6 times after thawing.
Cell culture with acetaldehyde and malondialdehyde
After several passages, activated stellate cells were seeded into 75 cm2 sealable flasks until the monolayers were 75–80% confluent. The cell cultures were then incubated with 200 μM of acetaldehyde, or 200 μM of MDA, or both. MDA was generated from malondialdehyde bis (diethyl acetal) (Aldrich Chemical Co.) by incubation with sulfuric acid and neutralized with sodium bicarbonate. The generation of MDA is described in more detail by Esterbauer and colleagues [20]. The concentration of 200 μM MDA was based on the previous work by Maher and seminal work by Chojkier demonstrating that collagen synthesis in culture-activated hepatic stellate cells or fibroblasts was enhanced by 200 μM MDA [21,22]. The concentration of 200 μM acetaldehyde was determined based on previous determinations of acetaldehyde in culture for 24 h following initial incubation, which we described previously [4]. Cell viability was monitored under these culture conditions by trypan blue exclusion. All cell-culture additives were placed in serum-free (SF) medium containing 1mM 4-methylpyrazole, and 100 μM cyanamide. These agents inhibit alcohol and aldehyde dehydrogenase, respectively.
Identical studies were performed without either MDA or acetaldehyde, but with SF media alone. To account for artificial increases in acetaldehyde by the presence of aldehyde dehydrogenase inhibitors, studies were performed in the absence of 4-methylpyrazole or cyanamide. MDA is not metabolized by either alcohol or aldehyde dehydrogenase, and none of the experiments designed contained glutathione-S-transferase inhibitors. The data presented here represent the results of studies with and without 4-methylpyrazole and cyanamide. Typically, five supplemental growth factors were added to SF media in the absence of serum. These factors include transferrin (0.5 mg/l), selenous acid (5 μg/l), fetuin (0.5 mg/l), bovine serum albumin (0.5 mg/l), and linoleic acid (0.5 mg/l). Serum-free media were used to prevent formation of adducts between acetaldehyde and serum proteins.
RNA isolation and reverse transcriptase-polymerase chain reaction (RT-PCR) analysis for α2(I) collagen messages
Total RNA was isolated from cultured stellate cells by the method described by Chomczynski and Sacchi [23]. For each PCR, 1 μg of total RNA was reverse-transcribed to complementary DNA (cDNA) using RETRO-script First Strand Synthesis Kit for RT-PCR (Ambion Inc., Austin, TX, USA) as recommended by the manufacturer. The resulting cDNA was then subjected to 30 cycles of PCR. The set of primers for the α2(I) collagen gene, which give an amplified fragment of 183 bp, consisted of a forward primer 5′TCGACTAAGTTGGAGG-GAACGGTC3′ and a reverse primer 5′TTGGCATGT-TGCTAGGCACGAC3′ from rat [accession #X66209]. PCR products were quantified by Quantum RNA 18S Internal Standards (Ambion Inc.). After amplification, each sample was then applied to 1.2% agarose/ethidium bromide gel and electrophoresed. The resulting gel was photographed under ultraviolet illumination. Scanning laser densitometry was used to quantitate PCR products. Previously we confirmed PCR product identity by sequencing collagen gene products. Data are expressed as relative density units of PCR products.
Transient transfection analysis
To determine the effect of the additives in vitro, transient transfection with the α2(I) collagen promoter was performed. The original promoter (−2000 to +54 bp) was subcloned into the pGL3-enhancer luciferase reporter vector (Promega) by the Mezey laboratory. This wild-type α2(I) collagen vector is termed pGL3-1009. Transfections were conducted by the calcium phosphate precipitation method [24] or with the use of Lipofectamine (Life Technologies, Inc.; Gaithersburg, MD, USA). Two to 10 μg of pGL3-1009 were added to 80% confluent cell cultures in sealable flasks. After 4 h incubation, the cells were washed twice with PBS and shocked with 10% DMSO in DMEM for 3 min. The media were removed, and the cells were provided with fresh DMEM containing 10% FBS for 16 h for recovery.
To downregulate JNK, the expression vector dnJNK 1 was cotransfected with the α2(I) collagen promoter using Lipofectamine. One day before transfection, HSCs were seeded in T-25 plug-sealed flasks at a density of 20 × 104 cells per flask in DMEM containing 10% FBS. The transfection mixture contained 2μg of pGL3-1009, 2μg of dnJNK1, 0.5μg of the internal control plasmid pSV-βgal, and the transfection reagents. After 3 h incubation, 4 ml of DMEM containing 15% FBS was added to the transfection mixture and the cells were incubated at 37°C in 5% CO2 for 12 h. Following this incubation, cells were treated with SF-DMEM for 12 h, after which HSCs were treated with nothing, acetaldehyde (200 μM), MDA (200 μM), or both. After 24 h, HSCs were fed with fresh SF medium containing the additives. Total additive exposure time to HSCs was 40 h. The basic pGL3 vector, which lacks either a promoter or enhancer, was used as one negative control; whereas the pGL3 control vector, which contains both the SV40 promoter and enhancer, was used as a positive control. Luciferase activity was determined using a Monolight 3010 Luminometer (Analytical Luminescence Laboratory; San Diego, CA, USA). Results are expressed in relative light units per microgram of protein quantitated by the method of Lowry [25].
Western blot of whole stellate cell lysates
To determine whether the oxidant additives resulted in phosphorylation of p38, JNK, and ERK in stellate cell culture, in vitro, cells were harvested and lysates subjected to SDS-PAGE and immunoblot as described previously. Antibodies to phosphorylated p38, JNK, and ERK were employed at 1:500 dilution. Stellate cell lysates from sealed-cell culture flasks under one of four conditions (no additives, acetaldehyde, acetaldehyde and MDA, or MDA alone) were grown as described and collected following 40 h incubation with additives in serum-free media. Cells were washed with PBS and collected by rubber policemen in 50 mM Tris-HCl (pH 7.5), 2 mM EDTA, 10 mM MgCl2, 5 mM DTT, and 5 mM PMSF. On ice, the harvested cells were homogenized with a Dounce homogenizer. Homogenates were resuspended in 250 mM Tris-HCl (pH 7.5), 0.25 M KCl, 10 mM MgCl2, 5 mM DTT, 5 mM PMSF, 2 mM EDTA, and 50% glycerol. Cell extracts were centrifuged at 15,000 × g. Sixty μg of extract was then subject to 10% SDS-PAGE at 50 V for 16 h [26]. Equal protein loading was confirmed by immunoblot for β-actin. Following electrophoresis, the gels were equilibrated in 50 mM Tris-HCl, 40 mM glycine, 130 μM SDS, and 5 M methanol. Gel transfer was completed to Bio-Rad Trans-blot 0.45 micron nitrocellulose membrane. The membrane was blocked with TBS-Tween20 containing 5% milk for 1 h. The membranes were subsequently washed with Tris-buffered saline and Triton X. Antiphospho JNK and p38 were applied at 1:1000 dilution. After subsequent washes, the blots were exposed to 1:5000 dilution of antirat IgG peroxidase conjugate for 1 h. Membrane-bound antigen-antibody complexes were detected with an enhanced chemiluminescence system from Amersham (Braunschweig, Germany). Protein loading was standardized according to the method of Lowry [25].
To confirm the presence of ERK activity in HSCs, cells were pretreated with 10 ng/ml of EGF and treated and untreated lysates were subjected to SDS-PAGE, transblotting, and immunodetection of phosphorylated ERK.
Immunoprecipitation and kinase assays for MAPK pathways
Stellate cell culture and the four experimental conditions described previously were established exactly as conducted in transfection and RT-PCR analysis. Cells were washed twice with ice-cold PBS. Protein was extracted on ice with a lysis buffer by cell scraper and included 20 mM Tris-HCl, pH 7.4, 1% Triton X-100, 10% glycerol, 137 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 1 mM Na orthovanadate, and 1 mM PMSF. Protease inhibitor buffers included 1 mg/ml antipain, 1 mg/ml leupeptin, 10 mg/ml aprotinin, 1 mg/ml chymostatin, and 1 mg/ml pepstatin. Stellate cell extracts were passed through a 20-gauge sterile syringe and centrifuged at 6000 × g for 10 min. The resultant supernatant was transferred to a fresh tube and the lysate was precleared with 20 μl of protein A-agarose beads. Fifty microliters were stored for protein assay. After subsequent centrifugation at 1000 × g for 5 min, the second supernatant was transferred to a new microfuge tube. All experiments were done in triplicate on ice at 4°C. One microgram of polyclonal antibodies to ERK, JNK, and p38 was added to the cell supernatant protein A-agarose extracts for 2 h at 4°C.
The antibody-supernatant mixture was subsequently incubated for 16 h with 20 μl of protein A-agarose beads on a laboratory rocker. The beads were collected following centrifugation at 1000 × g for 5 min. Each experimental tube included 18 μl of kinase buffer including 20 mM Hepes, pH 7.4, 20 mM β-glyercophosphate, 20 mM MgCl2, 2 mM DTT, and 100 μM sodium orthovanadate. The kinase assay was initiated by addition of 1 μl of myelin basic protein for ERK activity, or 5 μl of ATF-2 protein substrate for either JNK or p38 activity. Five microcuries of 32P[γ]ATP were added last at room temperature. Assays were incubated for 30 min in a 30°C water bath and terminated with Laemmli loading buffer. Resultant assays were subjected to 10% SDS-PAGE at 50 V for 16 h [26]. Dried gels were exposed to Kodak X-Omat film at −80°C for autoradiography.
Statistical analysis
Data comparison was analyzed between means ± standard error (SE) of each of the three treatment groups (acetaldehyde, MDA, or both) compared to results from untreated stellate cells. Results were considered significant if p < .05, by the Student’s t-test.
RESULTS
Since MDA may play a role in the pathogenesis of liver diseases other than alcoholic liver disease, we initially examined whether MDA alone would increase corresponding RNA production, by RT-PCR with the α2(I) collagen primers. The time of exposure to the additives was 48 h. Figure 1 demonstrates a representative agarose gel image for RT-PCR analysis of the α2(I) collagen message following HSCs exposure to 200 μM MDA, 200 μM acetaldehyde, or equimolar concentrations of both for 48 h. These RT-PCR data have been quantitated by scanning laser densitometry as indicated. The α2(I) collagen products were expressed relative to 18S RNA product. These data indicate that 200 μM MDA, 200 μM acetaldehyde, or equimolar concentrations of both, significantly increase the α2(I) collagen message (p < .03 Student’s t-test) compared to untreated cells. We have previously demonstrated that 200 μM acetaldehyde increases α2(I) collagen message in prior publications by Northern analysis [5] and RT-PCR [27]. These data, while indicating a significant increase in α2(I) collagen gene expression compared to untreated HSCs by acetaldehyde or MDA, show no significant difference in resultant gene expression between treatment conditions. Also, no synergy appeared to exist from the presence of HSC exposure to both aldehydes.
Fig. 1.

RT-PCR analysis of α2(I) collagen message after treatment of hepatic stellate cell cultures (HSC) with 200 μM MDA, 200 μM acetaldehyde, or the combination of both MDA and acetaldehyde. Total RNA from HSCs grown in SF media containing 200 μM MDA, 200 μM acetaldehyde (AC), or both (MDA/AC) was reverse transcribed with primer pairs for α2(I) collagen gene and 18S RNA. Control RNA was from untreated HSCs grown in SF media in the absence of additives. Results are expressed as relative units quantitated to 18S RNA. This experiment was performed three times in triplicate. Data analysis of resolved agarose gel electrophoresis products by Student’s t-test. All treatment data compared to controls, *p < .03.
Similarly to determine whether MDA alone or the combination of MDA and acetaldehyde activate the collagen reporter gene more than either aldehyde alone, transient transfection analysis was performed with the α2(I) collagen promoter. These experiments, repeated three times in triplicate, reveal in Fig. 2 that 200 μM MDA had a significant effect (p < .04) at 48 and 72 h (p < .02) on α2(I) collagen transgene activity compared to control activity. Note that data for each time point was compared to untreated HSCs lysates for identical lengths of time. These time points were chosen based on prior findings in Fig. 1, that at 48 h MDA was found to increase α2(I) collagen gene transcription. While transcriptional activation did appear to begin increasing at 24 h, this was not statistically significant compared to basal luciferase transgene activity at 24 h. Note that these experiments did not contain inhibitors of glutathione-S-transferase, the key metabolizing enzyme for MDA. Similar transfection experiments with the combination of 200 μM MDA and varying concentrations of acetaldehyde 50 and 100 μM with and without inhibitors of alcohol and aldehyde dehydrogenase did not act synergistically to increase α2(I) collagen transgene activity. In fact, the combination of both aldehydes was inhibitory until 200 μM concentrations of both MDA and acetaldehyde were employed (data not shown). Even with both aldehydes present (Fig. 3) there was no statistical difference between luciferase activity from MDA and acetaldehyde-treated HSCs when compared to 200 μM acetaldehyde or 200 μM MDA alone.
Fig. 2.

Transient transfection of primary passaged stellate cells with the α2(I) collagen collagen promoter exposed to 200 μM MDA for increasing time intervals as compared to untreated cells. Ten μg of the wild-type (pGL3-1009) promoter was transfected by the calcium phosphate precipitation method [24]. Exposure time to MDA is as indicated and pGL3-1009 luciferase transgene activity was measured at these time points. Experiments shown were performed in triplicate on three separate occasions and transfection efficiency was assayed by cotransfection with 0.5 μg β-galactosidase. The data shown represent the average of these studies ± SE. Data analysis was by Student’s t-test. Results shown were standardized to protein assayed (RLU/μg protein). Forty-eight hour and 72 h transgene results were significant compared to control (untreated) cells. Protein sample analysis by the method of Lowry [25]. Transfection efficiency for all transfection assays was between 75–85%.
Fig. 3.

Transient transfection of the α2(I) collagen promoter in hepatic stellate cells exposed to 200 μM acetaldehyde, MDA, or both. Two to ten μg of the wild-type (pGL3-1009) promoter was transfected by the calcium phosphate precipitation method [24] or by Lipofectamine. Exposure time to all culture additives was 48 h. Experiments shown were performed in triplicate on three separate occasions and transfection efficiency was assayed by cotransfection with 0.5 μg β-galactosidase. The data shown represent the average of these studies ± SE. Data analysis was by Student’s t-test. Results shown were standardized to protein assayed (RLU/μg protein). Protein sample analysis by the method of Lowry [25]. No significant difference exists between treatment groups, but a significant difference (p < .05) did exist between each treatment group and untreated, or control lysates.
Since Mendelson and colleagues have implicated that oxidative stress in the liver results in the activation of JNK [28], we set out to examine whether acetaldehyde and/or MDA would result in downstream phosphorylation of the transcription factor ATF-2 or MBP. These 2 molecules serve as in vitro phosphorylation substrates for JNK, p38, and ERK, respectively. To examine these potential MAPK pathways, we first performed immunoblot analysis under the treatment conditions outlined in Figs. 1–3 and probed HSC lysates for phosphorylated JNK (p-JNK), p38 (p-p38), and ERK (p-ERK). Such information would implicate MEK activation or mitogen-activated protein kinase kinase activation by oxidant byproducts. Figure 4 is a representative immunoblot demonstrating that phosphorylated ERK, or p-ERK, was detectable under the three experimental conditions defined in experiments outlined, but there did not appear to be an increase in phosphorylated ERK from treatment. Phosphorylated p38 (p-p38) was detectable under all conditions, including the control HSC extracts, but again, no significant difference between control HSC extracts or treatment groups was observed. By contrast, however, a characteristic doublet pattern of phosphorylated JNK (p-JNK) was demonstrated and was repeatedly detectable from HSCs extracts exposed to MDA, acetaldehyde, or both. In these studies, there was a significant difference between p-JNK in the control cell extracts and that of the treated cells. Note that the antibody used did not cross-react with other MAPKs and reflects the dually phosphorylated JNK isoforms p46 and p54.
Fig. 4.
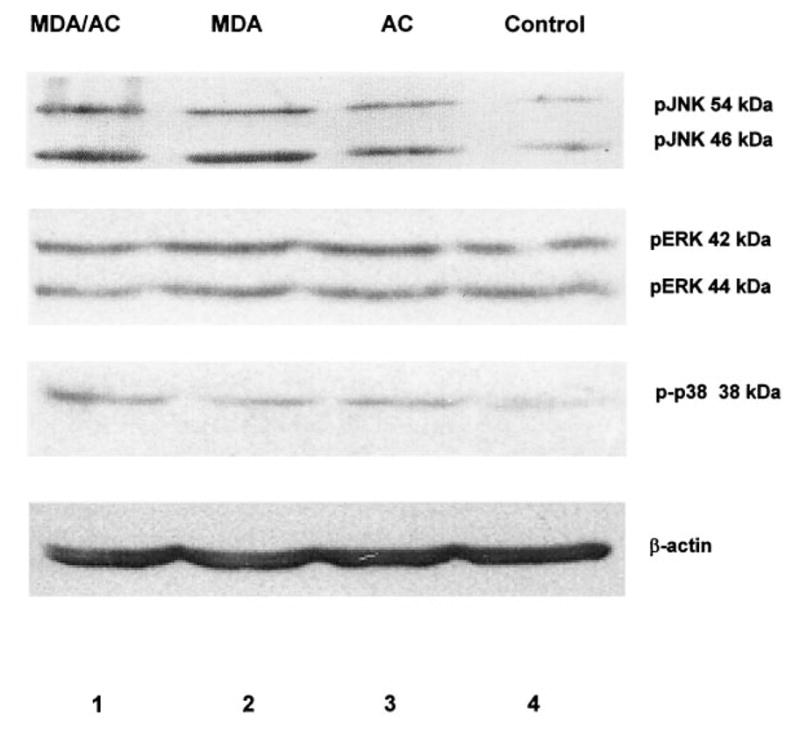
Immunoblot analysis to detect phosphorylated MAPK proteins (p-JNK, p-p38, or p-ERK) in rat hepatic stellate cells exposed to MDA and/or acetaldehyde. Fifty μg of cell lysate were subjected to 10% SDS-PAGE and transblotted [26]. Equal protein loading was based on Lowry protein assay and immunodetection of β-actin [25]. Primary antibodies to phosphorylated kinases were diluted at 1:1000; secondary conjugate antibodies for enhanced chemiluminescence were diluted at 1:5000. The results shown represent of five separate studies. Lane 1: Cell exposure to both 200 μM MDA and 200 μM acetaldehyde (AC). Lane 2: Exposure to 200 μM MDA alone. Lane 3: Exposure to 200 μM AC alone. Lane 4: SF media only, control (C). Phosphorylated proteins were detected for all MAPKs examined. Only pJNK was significantly different under the experimental conditions compared to untreated, or control experiments. No significant difference appeared present between treatment groups. Time of exposure to aldehydes was 48 h.
Based on the immunoblot analysis we determined, by immunoprecipitation, whether JNK, p38, or ERK activation resulted in phosphorylation of the downstream substrate, ATF-2. Similarly, we examined whether experimental treatment outlined resulted in ERK phosphorylation of its substrate myelin basic protein (MBP). Figure 5 is a representative immunoprecipitation analysis obtained following the experimental rationale outlined. ERK phosphorylation of MBP was barely detectable under either control or experimental conditions. P38 activity was present under all conditions but did not differ significantly from basal (C) activity in lane 4 of Fig. 5. JNK activity, however, was significantly increased under each of the three experimental conditions to which the cells were exposed as compared to untreated HSCs extract activity. However, no difference in JNK phosphorylation activity was observed between treatment groups (lanes 1–3 of Fig. 5). Taken together, Figs. 4 and 5 indicate that JNK activation and its subsequent phosphorylation of downstream elements is significantly affected by the presence of either MDA, acetaldehyde, or the combination of both. Again, there did not appear to be more ATF-2 phosphorylation by the immunoprecipitation of activated JNK activity from stellate cell lysates exposed to both MDA and acetaldehyde compared to lysates from exposure to either aldehyde alone.
Fig. 5.
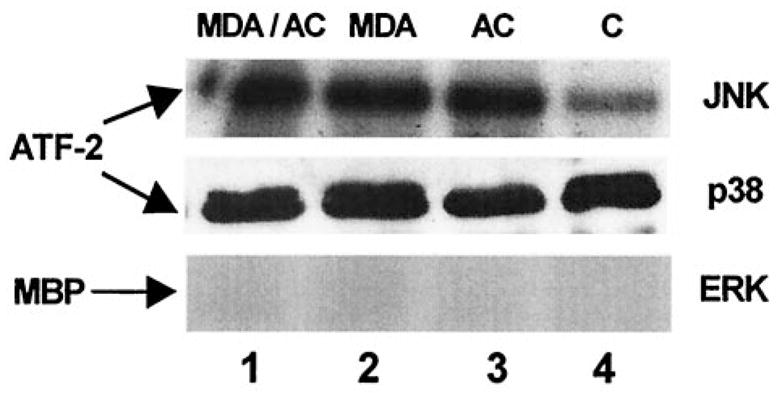
Immunoprecipitation of MAPK activity in cultured rat hepatic stellate cells exposed to MDA and/or acetaldehyde. One microgram of polyclonal antibodies to ERK, JNK, or p38 were added to cell lysate bound to protein A-agarose beads. The kinase assay was initiated by addition of 1 μl myelin basic protein (MBP) for ERK activity and 5 μl of ATF-2 for either JNK or p38 activity. These studies were performed three times in triplicate for each MAPK activity. Equal protein loading was based on Lowry protein assay [25]. Assays were subjected to 10% SDS-PAGE and autoradiography [26]. Lane 1: Cell exposure to both 200 μM MDA and 200 μM acetaldehyde (AC). Lane 2: Exposure to 200 μM MDA alone. Lane 3: Exposure to 200 μM AC alone. Lane 4: SF media only, control (C). JNK activity was significantly higher in the treatment groups (lanes 1–3) compared to control (lane 4). P38 activity was not significantly different between treatment groups (lanes 1–3) compared to control activity. ERK phosphorylation activity, as measured here by MBP phosphorylation, was barely detectable. Phosphorylation activity between treatment groups was not significantly different.
We gratefully obtained a dominant negative mutant for JNK1 (dnJNK1), and cotransfected it with the wild-type α2(I) collagen promoter. We hypothesized that if JNK activity, and the presence of phosphorylated JNK, was increased by the presence of either acetaldehyde or MDA, and that if both metabolites have the potential to increase collagen gene expression, then the absence of JNK activity should decrease collagen transgene expression. Figure 6 represents the summary of triplicate transfection analysis designed exactly as in Figs. 4 and 5 employing equimolar (200 μM) of acetaldehyde, MDA, or both. The dnJNK1 mutant significantly decreased the luciferase transgene activity under all conditions (p < .041, Student’s t-test). The empty vector pCMVβ, in which the mutant was inserted, did not significantly affect α2(I) collagen transgene activity. Acetaldehyde alone, or the combination of MDA and acetaldehyde, did increase α2(I) collagen transgene activity, but no significant difference between treatment groups was observed; and, as in the all the previous studies, no synergy between MDA and acetaldehyde to increase transgene activity was apparent. Transfection efficiency in all studies was measured by β-galactosidase activity and was calculated to be between 75 and 85%.
Fig. 6.

Effect of MDA and/or acetaldehyde on the α2(I) collagen promoter cotransfected with a dominant negative expression vector for JNK (dnJNK1). Transient transfection experiments with HSCs were identical to those described previously, except that either 2 μg of dnJNK1, or 2 μg pCMVβ, the empty vector in which dnJNK1 was inserted were cotransfected with 2 μg of the α2(I) collagen promoter. Assays were performed three times in triplicate. The symbol * indicates p values (Student’s t-test), comparing collagen transgene expression between cotransfection experiments with pCMVβ and the collagen promoter, and the same experiments with the collagen promoter and dnJNK1. DnJNK1 did reduce collagen transgene activity in the absence of additives, however this was not significant. By contrast, the presence of dnJNK1 significantly decreased collagen transgene activity that was enhanced by MDA, acetaldehyde, or both. Transfection efficiency was assayed by employing β-galactosidase at 0.5 μg/flask. U = untreated; MDA = malondialdehyde; AC = acetaldehyde; MDA/AC = both; DnJNK1 = dominant negative mutant for JNK. X-axis summarizes each experimental condition. Aldehyde concentrations were 200 μM.
Figure 7 is the correlating RT-PCR analysis corresponding to the series of transfection studies displayed in Fig. 6, and demonstrates a statistically significant reduction in α2(I) collagen PCR products, following stellate cell transfection with the dnJNK1, when compared to basal collagen gene expression. These data again confirm that JNK activity appears to be central in mediating the effect of either or both aldehydes; on the other hand, we failed to demonstrate synergy in collagen mRNA expression from transgene studies with both MDA and acetaldehyde.
Fig. 7.

RT-PCR analysis of α2(I) collagen message from stellate cells transfected with dnJNK1 and treated with 200 μM MDA, acetaldehyde, or both. To determine whether the transfection data in Fig. 6 corresponds to JNK-mediated collagen gene transcription, total RNA from stellate cells grown in SF media containing 200 μM MDA, 200 μM acetaldehyde, or both and previously transfected with 2 μg of dnJNK1, was reverse transcribed with primer pairs for α2(I) collagen and 18S RNA. Control RNA was from stellate cells grown in SF media in the absence of additives alone, and in the absence of additives and dnJNK1. Results are expressed as relative units quantitated to the 18S RNA product. These experiments were performed three times in triplicate. Data analysis by Student’s t-test. P values were measured against PCR product for each treatment in the absence of dnJNK1. To control for the insertion of the expression vector, the pCMVβ empty vector was used as a control. U = untreated; MDA = malondialdehyde; AC = acetaldehyde; MDA/AC = both; DnJNK1 = dominant negative mutant for JNK. X-axis summarizes each experimental condition. Ethidium bromide stained agarose gel (top) corresponds to densitometric histogram for each experimental condition.
DISCUSSION
Since the discovery that HSCs are the principal collagen-producing cells in the liver, intensive investigation has been focused on oxidant stress and liver fibrosis. Several investigators, including our group, have reported that acetaldehyde, the oxidative metabolite of alcohol, may in part be responsible for increased collagen gene activation and transcription in the activated stellate cell phenotype. This is significant in alcoholic liver disease, since inflammation is not necessarily a requirement for collagen deposition in the natural history.
Since available animal models do not reliably reproduce alcoholic cirrhosis, exacting the signaling pathogenesis of collagen gene activation is difficult to demonstrate in vivo. Also the limitations of studying oxidative metabolites in vitro are equally challenging because of the inability to maintain steady state concentrations of acetaldehyde. However, several important in vivo studies in which rats were chronically fed ethanol, do implicate the role of other aldehydes—including MDA in the pathogenesis of chronic alcoholic liver disease [29,30]. Moreover, recent studies implicate the probable formation of malondialdehyde-acetaldehyde protein adducts with albumin [31], and more importantly, a role for such adducts in atherosclerotic-induced vascular disease [32] as well as in animals chronically fed ethanol [14]. Therefore, the purpose of the current studies was dual. First, to determine, in an in vitro model for liver fibrosis, whether the combination of MDA and acetaldehyde could synergistically increase collagen gene expression and transgene activation. Second, to determine which of the stress-activated protein kinases is responsible for aldehydes enhancing HSC-associated collagen gene expression.
From the work presented here we did not find that the combination of acetaldehyde and malondialdehyde together acted synergistically to increase collagen gene transcription or promoter activation. Similarly, we did not, in the immunoprecipitation experiments (Fig. 5) demonstrate a quantitative increase in the phosphorylation of ATF by JNK from HSC lysates exposed to both aldehydes when compared to either aldehyde alone. We did examine whether altering the concentration (50, 100, or 150 μM) of either MDA or acetaldehyde together would change these outcomes, but in fact 200 μM concentrations of both additives together was optimal; lesser concentrations of both aldehydes together resulted in less transgene activity or mRNA expression than either alone. Furthermore, using confocal microscopy with anti-MAA antibodies [17] we failed to detect specific MAA epitopes in cultured HSC, since similar staining patterns were obtained with cells exposed to MDA alone. We also did not detect protein adducts by immunoblot analysis with these antibodies.
It may not be surprising that the combination of acetaldehyde and MDA failed to result in a synergistic response in vitro. Acetaldehyde is much more highly reactive than MDA and may have effectively removed both aldehydes from potentially altering collagen gene expression in the culture model we employed. The immunoblot probing for p-JNK (Fig. 4) and the immunoprecipitation analysis (Fig. 5) also corroborate the findings that a lack of synergy exists between acetaldehyde and MDA in activation of JNK and phosphorylation of its downstream transcription factors. Recently Parola and colleagues suggested that 4-hydroxy-2, 3-nonenal (HNE), another aldehydic end product of lipid peroxidation, interacts directly with JNK isoforms to alter gene transcriptional regulation [33]. This observation is related to the lack of crucial HNE-metabolizing enzymatic activities in HSCs and may explain that aldehyde altering adducts affecting transcription may not require cell-surface signaling to alter collagen gene expression. Indeed while we demonstrate that JNK activation is critical in aldehyde-associated HSC collagen gene activation, the phosphorylation activity was from whole cell lysates. This work clearly shows, nonetheless, that JNK is critical to stress-induced collagen gene expression, however.
Whalen and colleagues have recently reported that activated HSC lose most forms of glutathione-S-transferase [34]. Thus, lipid peroxidation products such as MDA do not require inhibitors to prevent elimination of the culture additive in order to study the potential long-term effect of aldehydes in vitro. MDA, or in the future HNE, would serve as more stable, less volatile aldehydes in vitro, and will be useful in studying signal transduction mechanisms that are activated not only in alcohol-related liver disease but other chronic liver diseases in which lipid peroxidation products have been implicated [35–37].
We anticipated that ERK activity would not be increased by the presence of MDA, acetaldehyde, or both, because of the role of ERK in mitogenic control of cell proliferation [38] (Figs. 4 and 5). None of these oxidative metabolites is known to activate HSC proliferation. Conversely, we did expect that either JNK or p38 MAPK activities, or both, might be increased by the presence of these metabolites. We found that JNK activity, and the presence of phosphorylated JNK (p-JNK), was increased under the three experimental conditions to which stellate cells were subjected when compared to untreated stellate cells. Phosphorylated JNK, however, was found to be increased in the presence of MDA alone or in combination with acetaldehyde as well as with acetaldehyde alone. A significant increase in JNK activity, as measured by phosphorylation of ATF-2, and p-JNK, was present in the MDA-treated HSCs, as well as activity from the combination of acetaldehyde and MDA or acetaldehyde alone (Fig. 5).
The link between JNK activation and collagen gene expression is substantiated by the results shown in Fig. 6. The dnJNK1 mutant significantly reduced α2(I) collagen luciferase transgene activity under all experimental conditions while the pCMVβ vector, an expression vector, lacking the dnJNK1 construct, did not. These transfection data clearly corroborate the finding that increased JNK phosphorylation activity is required for stress-induced collagen gene activation in HSCs by these aldehydes. More convincingly, neither significant phosphorylation of ATF-2, detection of p-JNK, or abrogation of collagen transgene activity by dnJNK1, was found in untreated HSC collagen gene expression.
The data presented here relating acetaldehyde and MDA to increased JNK phosphorylation and collagen gene expression, raise important questions for future investigation and provide a framework for potential signaling of aldehydes in liver fibrosis. These data strongly suggest that JNK activation is a requisite for oxidant stress-induced collagen gene expression, and as such, SAPK activation is not required in basal collagen gene activity. It is worthwhile noting that Chen and Davis also very recently reported that acetaldehyde-induced collagen gene expression is JNK-dependent [39]. Our report strongly implicates JNK in lipid peroxidation-associated collagen gene activation. While circulating antibodies to MAA adducts, and MAA adducts themselves, have been detected in rats chronically fed ethanol, it does not appear that the in vitro model used here would best serve to determine the pathologic significance of such adducts. Perhaps an immune-mediated phenomenon does take place in vivo that has potential for enhancing liver fibrosis. Such a hypothesis should be further examined in vitro to elucidate a putative mechanism relating anti-MAA antibodies and HSC-associated collagen production.
Whether oxidant stress-related collagen gene signaling in activated HSCs results in signal “cross-talk” will be worthwhile to pursue. In prior work, we, and others, have shown that acetaldehyde increases protein kinase C (PKC) activity [27,40], and that PKC activation also increases collagen gene transcription. Our data implicating aldehyde-associated SAPK activation, parallels recent data in which stretch and phorbol ester (PMA) increase nuclear protein binding to AP-1 in kidney mesangial cells, which are known to be responsible for glomerulosclerosis. The mechanism of enhanced AP-1 binding activity, a putative binding site in collagen gene regulation [41,42], appears to be mediated by PKC and subsequent activation of SAPK/JNK [43]. Similar PKC-JNK activation mechanisms were also recently reported in an in vivo ischemic injury model in rabbits [44]. There are emerging data suggesting that the perpetuation of nascent collagen gene expression in hepatic stellate cells may not require the TGFβ1 signaling mechanism [45]. Hence, current investigations are ongoing to examine the critical role of oxidant stress in cross-talk signaling between two potentially important pathways in fibrosis—the SAPK pathway and the TGFβ pathway. Establishing a molecular link in a culture model between lipid peroxidation products, including MDA, and HNE, and specific signal transduction pathways augmenting collagen gene activation will provide critical answers to key questions regarding perpetuation of liver fibrosis. Furthermore, such information will lay the groundwork for novel pharmacologic and molecular agents that inhibit liver fibrosis.
Acknowledgments
We gratefully acknowledge Dr. Tsu-Than, Baylor College of Medicine, for the generous donation of the JNK1 dominant negative mutant. We also acknowledge Dr. Esteban Mezey and colleagues for the use of their pGL3-1009 wild-type α2(I) collagen promoter. This work was supported by a grant from the Alcohol Beverage Medical Research Foundation, NIH DK K11 02321, and the Department of Medicine of the University of Maryland.
ABBREVIATIONS
- AC
acetaldehyde
- ERK
extracellular-regulated kinase
- HSCs
rat hepatic stellate cells
- JNK
c-jun n-terminal kinase
- MAPK
mitogen-activated protein kinase
- MDA
malondialdehyde
- RLU
relative light units
- SAPK
stress-activated protein kinase
- SE
standard error
- TBS
Tris-buffered saline
References
- 1.Brenner DA, Chojkier M. Acetaldehyde increases collagen gene transcription in cultured human fibroblasts. J Biol Chem. 1987;262:17690–17695. [PubMed] [Google Scholar]
- 2.Moshage H, Casini A, Lieber CS. Acetaldehyde selectively stimulates collagen production in cultured rat liver fat-storing cells but not in hepatocytes. Hepatology. 1990;12:511–518. doi: 10.1002/hep.1840120311. [DOI] [PubMed] [Google Scholar]
- 3.Pares A, Potter JJ, Rennie L, Mezey E. Acetaldehyde activates the promoter of the mouse α2(I) collagen gene. Hepatology. 1994;19:498–503. doi: 10.1002/hep.1840190231. [DOI] [PubMed] [Google Scholar]
- 4.Anania FA, Potter JJ, Rennie-Tankersley LL, Mezey E. Effects of acetaldehyde on nuclear protein binding to the nuclear factor 1 consensus sequence in the α2(I) collagen promoter. Hepatology. 1995;21:1640–1648. [PubMed] [Google Scholar]
- 5.Anania FA, Potter JJ, Rennie-Tankersley L, Mezey E. Activation by acetaldehyde of the promoter of the mouse α2(I) collagen gene when transfected into rat-activated stellate cells. Arch Biochem Biophys. 1996;331:187–193. doi: 10.1006/abbi.1996.0297. [DOI] [PubMed] [Google Scholar]
- 6.Rossi P, Karsenty G, Roberts AB, Roche NS, Sporn MB, de Crombrugghe B. A nuclear factor 1 binding site mediates the transcriptional activation of a type I collagen promoter by transforming growth factor-β. Cell. 1988;52:405–414. doi: 10.1016/s0092-8674(88)80033-3. [DOI] [PubMed] [Google Scholar]
- 7.Friedman SL. Acetaldehyde and alcoholic fibrogenesis: fuel to the fire, but not the spark. Hepatology. 1990;12:609–612. doi: 10.1002/hep.1840120326. [DOI] [PubMed] [Google Scholar]
- 8.Maher JJ, Zia S, Tzagarakis C. Acetaldehyde-induced stimulation of collagen synthesis and gene expression is dependent on conditions of cell culture: studies with rat lipocytes and fibroblasts. Alcohol Clin Exp Res. 1994;18:403–409. doi: 10.1111/j.1530-0277.1994.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 9.Popper H, Lieber CS. Histogenesis of alcoholic fibrosis and cirrhosis in the baboon. Am J Pathol. 1980;98:695–716. [PMC free article] [PubMed] [Google Scholar]
- 10.Day CP. Is necroinflammation a prerequisite for fibrogenesis? Hepato-Gastroenterol. 1996;43:104–120. [PubMed] [Google Scholar]
- 11.Cederbaum AI. Role of lipid peroxidation and oxidative stress in alcohol toxicity. Free Radic Biol Med. 1989;7:537–539. doi: 10.1016/0891-5849(89)90029-4. [DOI] [PubMed] [Google Scholar]
- 12.De Maria N, Colantoni A, Fagiuoli S, Liu GJ, Rogers BK, Farinati F, Van Thiel DH, Floyd RA. Association between reactive oxygen species and disease activity in chronic hepatitis C. Free Radic Biol Med. 1996;21:291–295. doi: 10.1016/0891-5849(96)00044-5. [DOI] [PubMed] [Google Scholar]
- 13.Niemela O, Parkkila S, Yla-Herttuala S, Halstead C, Witztum JL, Lanca A, Israel Y. Covalent protein adducts in the liver as a result of ethanol metabolism and lipid peroxidation. Lab Invest. 1994;70:537–546. [PubMed] [Google Scholar]
- 14.Tuma DJ, Thiele GM, Xu DS, Klassen LW, Sorrell MF. Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology. 1996;23:872–880. doi: 10.1002/hep.510230431. [DOI] [PubMed] [Google Scholar]
- 15.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 16.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Proinflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 17.Rolla R, Vay D, Mottaran E, Parodi M, Traverso N, Arico S, Sartori M, Bellomo G, Klassen LW, Thiele GM, Tuma DJ, Albano E. Detection of circulating antibodies against malondialdehyde-acetaldehyde adducts in patients with alcohol-induced liver disease. Hepatology. 2000;31:878–884. doi: 10.1053/he.2000.5373. [DOI] [PubMed] [Google Scholar]
- 18.Friedman SL, Roll FJ. Isolation and culture of hepatic lipocytes, Kupffer cells, and sinusoidal endothelial cells by density gradient centrifugation with stractan. Anal Biochem. 1987;161:207–218. doi: 10.1016/0003-2697(87)90673-7. [DOI] [PubMed] [Google Scholar]
- 19.Schafer S, Zerbe O, Gressner AM. The synthesis of proteo-glycans in fat-storing cells of rat liver. Hepatology. 1987;7:680–687. doi: 10.1002/hep.1840070411. [DOI] [PubMed] [Google Scholar]
- 20.Esterbauer H, Lang J, Zadravec S, Slater TF. Detection of malondialdehyde by high-performance liquid chromatography. Meth Enzymol. 1984;105:319–328. doi: 10.1016/s0076-6879(84)05041-2. [DOI] [PubMed] [Google Scholar]
- 21.Maher JJ, Tzagarakis C, Gimenez A. Malondialdehyde stimulates collagen production by hepatic lipocytes only upon activation in primary culture. Alc Alcohol. 1994;29:605–610. [PubMed] [Google Scholar]
- 22.Chojkier M, Houglum K, Solis-Herruzo J, Brenner DA. Stimulation of collagen gene expression by ascorbic acid in cultured human fibroblasts. A role for lipid peroxidation. J Biol Chem. 1989;264:16957–16962. [PubMed] [Google Scholar]
- 23.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1983;132:6–13. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 24.Graham FL, Van der Eb AJ. A new technique for the assay of infectivity of human adenovirus DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 25.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 26.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 27.Anania FA, Womack L, Potter JJ, Mezey E. Acetaldehyde enhances murine α2(I) collagen promoter activity by Ca2+-independent PKC activation in rat hepatic stellate cells. Alcohol Clin Exp Res. 1999;23:279–284. [PubMed] [Google Scholar]
- 28.Mendelson KG, Contois L-R, Tevosian SG, Davis RJ, Paulson KE. Independent regulation of JNK/p38 mitogen-activated protein kinases by metabolic oxidative stress in the liver. Proc Natl Acad Sci USA. 1996;93:12908–12913. doi: 10.1073/pnas.93.23.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moser J, Bagchi D, Akubue PI, Stohs SJ. Excretion of malondialdehyde, formaldehyde, acetaldehyde and acetone in the urine of rats following acute and chronic administration of ethanol. Alc Alcohol. 1993;28:287–295. [PubMed] [Google Scholar]
- 30.Niemela O, Parkkila S, Pasanen M, Iimur Y, Bradford B, Thurman RG. Early alcoholic liver injury: formation of protein adducts with acetaldehyde and lipid peroxidation products, and expression of CYP2E1 and CYP3A. Alcohol Clin Exp Res. 1998;22:2118–2124. doi: 10.1111/j.1530-0277.1998.tb05925.x. [DOI] [PubMed] [Google Scholar]
- 31.Kearley ML, Patel A, Chien J, Tuma DJ. Observation of a new nonfluorescent malondialdehyde-acetaldehyde-protein adduct by 13C NMR spectroscopy. Chem Res Toxicol. 1999;12:100–105. doi: 10.1021/tx980132u. [DOI] [PubMed] [Google Scholar]
- 32.Hill GE, Miller JA, Baxter BT, Klassen LW, Duryee MJ, Tuma DJ, Thiele GM. Association of malondialdehyde-acetaldehyde (MAA) adducted proteins with atherosclerotic-induced vascular inflammatory injury. Atherosclerosis. 1998;141:107–116. doi: 10.1016/s0021-9150(98)00153-1. [DOI] [PubMed] [Google Scholar]
- 33.Parola M, Robino G, Marra F, Pinzani M, Bellomo G, Leonarduzzi G, Chiarugi P, Camandola S, Poli G, Waeg G, Gentilini P, Dianzani MU. HNE interacts directly with JNK isoforms in human hepatic stellate cells. J Clin Invest. 1998;102:1942–1950. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whalen R, Rockey DC, Friedman SL, Boyer TD. Activation of rat hepatic stellate cells leads to loss of glutathione-S-transferases and their enzymatic activity against products of oxidative stress. Hepatology. 1999;30:927–933. doi: 10.1002/hep.510300404. [DOI] [PubMed] [Google Scholar]
- 35.Niemela O, Parkkila S, Britton RS, Brunt E, Janney C, Bacon B. Hepatic lipid peroxidation in hereditary hemochromatosis and alcoholic liver injury. J Lab Clin Med. 1999;133:451–460. doi: 10.1016/s0022-2143(99)90022-7. [DOI] [PubMed] [Google Scholar]
- 36.Parola M, Leonarduzzi G, Robino G, Albano E, Poli G, Dianzani MU. On the role of lipid peroxidation in the pathogenesis of liver damage induced by long-standing cholestasis. Free Radic Biol Med. 1996;20:351–359. doi: 10.1016/0891-5849(96)02055-2. [DOI] [PubMed] [Google Scholar]
- 37.Parola M, Pinzani M, Casini A, Albano E, Poli G, Gentilini A, Gentilini P, Dianzani MU. Stimulation of lipid peroxidation or 4-hydroxynonenal treatment increases procollagen alpha 1(I) gene expression in human liver fat-storing cells. Biochem Biophys Res Commun. 1993;161:1044–1050. doi: 10.1006/bbrc.1993.1927. [DOI] [PubMed] [Google Scholar]
- 38.Talarmin H, Rescan C, Cariou S, Glaise D, Zanninelli G, Bilodeau M, Loyer P, Guguen-Guillouzo C, Baffet G. The mitogen-activated protein kinase kinase/extracellular signal-regulated kinase cascade activation is a key signaling pathway involved in the regulation of G(1) phase progression in proliferating hepatocytes. Mol Cell Biol. 1999;19:6003–6011. doi: 10.1128/mcb.19.9.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen A, Davis BH. The DNA-binding protein BTEB mediates acetaldehyde-induced, jun N-terminal kinase-dependent alpha (I) collagen gene expression in rat hepatic stellate cells. Mol Cell Biol. 2000;20:2818–2826. doi: 10.1128/mcb.20.8.2818-2826.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casini A, Galli G, Salzano R, Ceni E, Franceschelli F, Rotella CM, Surrenti C. Acetaldehyde induces c-fos and c-jun proto-oncogenes in fat-storing cell cultures through protein kinase C activation. Alc Alcohol. 1994;29:303–314. [PubMed] [Google Scholar]
- 41.Sherwood AL, Bottenus RE, Martzen MR, Bornstein P. Structural and functional analysis of the first intron of the human alpha 2(I) collagen-encoding gene. Gene. 1990;89:239–244. doi: 10.1016/0378-1119(90)90011-f. [DOI] [PubMed] [Google Scholar]
- 42.Fabbro C, Braghetta P, Girotto D, Piccolo S, Volpin D, Bressan GM. Cell type-specific transcription of the alpha 1(VI) collagen gene. Role of the AP1 binding site and of the core promoter. J Biol Chem. 1999;274:1759–1766. doi: 10.1074/jbc.274.3.1759. [DOI] [PubMed] [Google Scholar]
- 43.Ingram AJ, James L, Ly H, Thai K, Scholey JW. Stretch activation of jun N-terminal kinase/stress-activated protein kinase in mesangial cells. Kidney Int. 2000;58:1431–1439. doi: 10.1046/j.1523-1755.2000.00305.x. [DOI] [PubMed] [Google Scholar]
- 44.Ping P, Zhang J, Huang S, Cao X, Tang XL, Li RC, Zheng YT, Qiu Y, Clerk A, Sugden P, Han J, Boll R. PKC-dependent activation of p46/p54 JNKs during ischemic preconditioning in conscious rabbits. Am J Physiol. 1999;277:H1771–1785. doi: 10.1152/ajpheart.1999.277.5.H1771. [DOI] [PubMed] [Google Scholar]
- 45.Dooley S, Delvoux B, Lahme B, Mangasser-Stephan K, Gressner AM. Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology. 2000;31:1094–1106. doi: 10.1053/he.2000.6126. [DOI] [PubMed] [Google Scholar]