

Abstract
Background
Phenotypic and molecular genetic data often provide conflicting patterns of intraspecific relationships confounding phylogenetic inference, particularly among birds where a variety of environmental factors may influence plumage characters. Among diurnal raptors, the taxonomic relationship of Buteo jamaicensis harlani to other B. jamaicensis subspecies has been long debated because of the polytypic nature of the plumage characteristics used in subspecies or species designations.
Results
To address the evolutionary relationships within this group, we used data from 17 nuclear microsatellite loci, 430 base pairs of the mitochondrial control region, and 829 base pairs of the melanocortin 1 receptor (Mc1r) to investigate molecular genetic differentiation among three B. jamaicensis subspecies (B. j. borealis, B. j. calurus, B. j. harlani). Bayesian clustering analyses of nuclear microsatellite loci showed no significant differences between B. j. harlani and B. j. borealis. Differences observed between B. j. harlani and B. j. borealis in mitochondrial and microsatellite data were equivalent to those found between morphologically similar subspecies, B. j. borealis and B. j. calurus, and estimates of migration rates among all three subspecies were high. No consistent differences were observed in Mc1r data between B. j. harlani and other B. jamaicensis subspecies or between light and dark color morphs within B. j. calurus, suggesting that Mc1r does not play a significant role in B. jamaicensis melanism.
Conclusions
These data suggest recent interbreeding and gene flow between B. j. harlani and the other B. jamaicensis subspecies examined, providing no support for the historical designation of B. j. harlani as a distinct species.
Background
The criteria necessary to recognize and define distinct species have been frequently debated [1-3]. One of the primary difficulties in species designation is that the process of speciation requires an extended period of time during which various attributes of species-level distinction are attained [2]. Depending on the length of time since divergence, only some fraction of phenotypic and molecular characters may have become fixed between incipient species. Therefore, different characters and associated species concepts may provide conflicting inference in the determination of species status [e.g.,[4,5]]. For example, the relative influence of genetic drift versus natural selection, relative sizes of populations sampled, and age of barriers to reproduction (if any) of the species involved will affect the degree to which characters become diagnostic, thus influencing phylogenetic inference. One manifestation of this phenomenon occurs during the comparison of phenotypic and molecular genetic data where conflicting patterns of divergence are detected [see [4,6]]. Attempts to accurately define evolutionary relationships are further confounded if expression of the phenotypic trait examined is influenced by environmental factors apart from any genomic control and therefore does not faithfully reflect evolutionary relationships.
Understanding the relationship between phenotypic and molecular genetic data sets may be particularly informative in studies of avian taxonomy where plumage characteristics have been historically used to designate subspecies as well as species [7]. Many species and subspecies designations based upon phenotype alone have not been supported by molecular genetic phylogenies [e.g.,[8,9]]. In fact, among avian researchers, the value of designating subspecies rank is subject to ongoing debate often as vigorous as the debate over species concepts [6,10]. Inconsistencies at both hierarchical levels are associated with the difficulty in understanding the underlying genetic and/or environmental basis of phenotypic characters, and consequently, interpreting phenotypic patterns alone may mislead evolutionary inference [6,10]. Phenotypic characters associated with melanin-based plumage may be particularly susceptible to misinterpretation in an evolutionary context due to environmental and temporal influences. Expression of melanin-based characters can vary with body condition [11,12], oxidative stress induced by environmental factors [13], and temporally within individuals [14].
Among diurnal raptors, phenotypic differences have fueled a long-standing controversy concerning intraspecific relationships (i.e., subspecies designations) within the North American raptor Buteo jamaicensis (red-tailed hawks). This controversy has been motivated primarily by the presence of substantial variation in plumage characteristics of B. j. harlani, of which almost all individuals display melanistic plumage. By contrast, the majority of B. j. calurus are not melanistic, and melanistic plumage has not been documented in either B. j. alascensis or B. j. borealis [15,16]. Other melanin-based plumage characters, such as tail pattern and coloration and throat coloration, show extreme variation within B. j. harlani but overlap to some degree with plumage characters observed in other B. jamaicensis subspecies [17]. The phenotypic variation within B. j. harlani has been cited as evidence to consider it as either a distinct species, B. harlani [18], or as a subspecies within B. jamaicensis [19-21]. Whether observed plumage differences within B. jamaicensis reflect long-standing evolutionarily distinct lineages or are a consequence of environmental, temporal, or physiological factors, particularly within the range of B. j. harlani, remains undetermined.
Previous research has examined the phylogeographic relationships of B. j. calurus and B. j. borealis across nine breeding populations and ten migration sites [22]. This earlier analysis of microsatellite data in B. jamaicensis indicated low levels of genetic differentiation among breeding sites within recognized subspecies. Fine-scale population substructure was identified between central California and Intermountain West populations of B. j. calurus (FST = 0.011 - 0.018) [22]; no other significant within-subspecies differentiation was identified. Significant differentiation was also identified between B. j. borealis and B. j. calurus, supporting historical subspecies designations (FST = 0.036 - 0.039) [22,23].
Building upon this previous work, we used molecular genetic data from the mitochondrial and nuclear genomes to investigate the taxonomic relationship of B. j. harlani to two common and widespread subspecies geographically adjacent to B. j. harlani; B. j. borealis and B. j. calurus. Specifically, we investigated whether B. j. harlani is distinct at the species level by comparing our results with the levels of differentiation previously documented among subspecies of B. jamaicensis. If B. j. harlani represents a distinct species, it should at a minimum conform to criteria proposed for assessing status as a subspecies, or an "evolutionary significant unit," [24-26]. These include reciprocal monophyly at mitochondrial DNA (mtDNA) and significant partitions at nuclear loci, relative to the other named subspecies. The results of our analyses help to resolve a long-standing taxonomic challenge and provide insight into the role of plumage variation in species designations.
Methods
Sample collection
We collected whole blood and feathers during the breeding season (May through June) from young in nests and adults on breeding territories of three putative B. jamaicensis subspecies; 178 B. j. calurus, 69 B. j. borealis, and 24 B. j. harlani (Table 1). Additionally, we collected samples from 21 melanistic individuals, presumably B. j. calurus, during autumn migration. We drew approximately 0.2 mL of blood from the medial metatarsal vein and plucked two feathers from the breast. We received approval for our methodology from the U.C. Davis Institutional Animal Care and Use Committee (protocol number 12260). We stored blood samples in 1.5 mL of Longmire's lysis buffer (100 mM Tris pH 8.0, 100 mM EDTA, 10 mM NaCl, 0.5% SDS) at ambient temperature in the field and at -80°C once delivered to the laboratory and we stored feathers in envelopes in cool and dry conditions. We extracted genomic DNA from 25 μL of blood/buffer solution or a single feather calamus using Qiagen DNeasy kits (Qiagen Inc.); we stored DNA at -80°C following extraction.
Table 1.
Sampling locations and the number of individuals for B. j. calurus, B. j. borealis, B. j. harlani, and B. jamaicensis spp (melanistic).
| Subspecies | Location | N |
|---|---|---|
| B. j. calurus | central California | 68 |
| Idaho | 35 | |
| Nevada/Utah | 39 | |
| southern California | 36 | |
| B. j. borealis | Alberta | 2 |
| Colorado | 11 | |
| Kentucky | 9 | |
| Michigan | 5 | |
| north Texas | 9 | |
| Wisconsin | 33 | |
| B. j. harlani | Anchorage, Alaska | 6 |
| Fairbanks, Alaska | 3 | |
| Kuskokwim, Alaska | 10 | |
| Palmer, Alaska | 4 | |
| Kluane, Yukon Territory | 1 | |
| B. jamaicensis spp. (melanistic) | central California | 21 |
Microsatellite data collection and analysis
We genotyped each individual at 17 microsatellite loci (BswA110w, BswA204w, BswA302w, BswA303w, BswA312w, BswA317w, BswB111aw, BswB220w, BswB221w, BswD122w, BswD127w, BswD210w, BswD220w, BswD234w, BswD310w, BswD313w, BswD327w) in six multiplex reactions following previously developed methods [27]. We separated PCR products using a 3730 DNA Analyzer (Applied Biosystems Inc.) and scored products using STRAND version 2.3.89 [28].
Because of the limited genetic differentiation previously documented among breeding populations within B. j. calurus and B. j. borealis [23] and the focus of our study on the taxonomic status of B. j. harlani in relation to other subspecies, we treated subspecies as our unit of analysis for the majority of the population genetic analyses. We tested for deviations from Hardy-Weinberg equilibrium in all loci using GENEPOP version 3.4 [29] and we tested each pair of loci in each putative subspecies (B. j. calurus, B. j. borealis, and B. j. harlani) for genotypic disequilibrium in GENEPOP. After we assessed significance of pairwise tests using a sequential Bonferroni correction for multiple comparisons [30], we tested for the presence of null alleles and scoring error in MICROCHECKER [31]. Previous research has indicated that the rate of scoring error for this set of microsatellites is 0.8% [23].
We used the program CONVERT version 1.31 [32] to determine the number of private alleles in each presumed subspecies. We used MICROSATELLITE TOOLKIT [33] to calculate observed and expected heterozygosity of the total sample and within each subspecies as well as the average number of alleles per locus. Allelic richness, weighted by sample size, was estimated for each subspecies using FSTAT version 2.9.3.2 [34].
We determined pairwise estimates of variance in allele frequency (θST) for all subspecies pairs in ARLEQUIN version 3.11 [35] and we used a sequential Bonferroni correction for multiple tests to determine significance of θST values [30]. We analyzed the relationships between sampling localities with greater than 20 individuals by averaging allele frequency differences among sampling sites [36]. We used the resulting divergence matrices with both neighbor-joining and UPGMA algorithms to make a clustering tree in the program PHYLIP [37] and assessed node support using 10,000 bootstrap replicates.
In order to further test for a distinction between subspecies and investigate the possibility of cryptic population differentiation beyond subspecies groupings, we implemented a Bayesian clustering analysis in STRUCTURE version 2.1 [38]. The unit of analysis was the individual and the algorithm implemented in Structure probabilistically grouped individuals together into the most likely number of populations without regard to presumed subspecies. This approach allowed us to determine the most likely number of population groups (K) without a priori information about subspecies or region of origin. We used the population admixture model with a flat prior and assumed allele frequencies were correlated. We allowed the program to run with a 500,000 iteration burn-in and a run length of 1,000,000 iterations and used this parameter set to explore K = 1 through K = 5 and averaged log Pr(X|K) estimates across 10 runs for each value of K. We determined the most likely number of populations by selecting the set of K values where the log Pr(X|K) estimation was maximized and then selecting the minimum value for K that did not sacrifice explanatory power [39,40]. We grouped individuals into clusters based upon the highest proportion of ancestry to each inferred cluster.
Mitochondrial data collection
We sequenced a 430 base pair segment of domain I of the mitochondrial control region using primers 16065F [41] and H15454 [42]. We prepared PCR products for sequencing with 0.5 μL exonuclease I and 1 μL shrimp alkaline phosphatase per 25 μL PCR and delivered PCR products to the UC Davis Sequencing Facility using primers 14965F and H15414 [43]. We aligned sequences by eye using SEQUENCHER version 4.7 (Gene Codes Corporation).
Phylogeographic analyses of mtDNA sequence
We conducted phylogenetic analyses of control region sequences in PAUP* version 4.0b8 [44], using maximum parsimony (MP), maximum likelihood (ML) and distance (minimum evolution, ME). We used MODELTEST version 3.06 [45] to determine the minimum parameter nucleotide substitution model that best fit the mtDNA sequence data under the Akaike Information Criterion [46]. In each analysis, we conducted heuristic tree searches, with 20 and 100 random additions of taxa for maximum likelihood and parsimony analyses, respectively, each followed by tree bisection-reconnection topological rearrangements. We applied midpoint rooting, employing the close-neighbor interchange algorithm for finding the tree, beginning with an initial neighbor-joining tree. We assessed the robustness of nodes using tree reconstructions of bootstrap-resampled data sets for 1,000 replicates under distance and parsimony criteria, and 200 replicates for ML criteria.
We calculated the number of variable sites, number of haplotypes, haplotype diversity, and nucleotide diversity in DNAsp [47]. Due to the low levels of divergence within subspecies previously documented [22], we calculated pairwise estimates of variance in haplotype frequency (ΦST) between each presumed subspecies using haplotype frequencies in ARLEQUIN version 3.11 and we assessed significance using 10,000 permutations. We used a sequential Bonferroni correction for multiple tests to determine significance of ΦST values [30].
Estimation of polarity in gene flow
To provide a coarse estimate of gene exchange among subspecies, we calculated the number of migrants per generation (Nem) for nuclear microsatellite and number of female migrants per generation (Nfm) for mtDNA among populations using MIGRATE version 2.0.6 [48-50]. To test for asymmetry in gene flow, we estimated full models, θ (4Neμ or Nfμ, composite measure of effective population size and mutation rate), and all pairwise migration parameters individually from the data and compared them to a restricted island model for which θ was averaged and pairwise migration parameters were symmetrical between populations.
We ran MIGRATE using maximum likelihood search parameters; ten short chains (1,000 used trees out of 20,000 sampled), five long chains (10,000 used trees out 200,000 sampled), and five adaptively heated chains (start temperatures: 1, 1.5, 3, 6, and 12; swapping interval = 1). We ran full models three times to ensure the convergence of parameter estimates; we ran restricted models once. We evaluated the alternative model for goodness-of-fit given the data using a log-likelihood ratio test. The resulting statistic from the log-likelihood ratio test is equivalent to a χ2 distribution with the degrees of freedom equal to the difference in the number of parameters estimated in the two models [50].
Melanocortin 1 receptor sequencing
To determine if differences in the coding region of melanocortin 1 receptor (Mc1r) gene are associated with subspecies or color variation in B. jamaicensis, we sequenced Mc1r from 23 individuals (17 B. j. calurus; 6 B. j. harlani) using primers MSHR9 and MSHR72 [51]. We amplified an 829 bp fragment from the 945 bp avian Mc1r coding region, excluding 56 bases from the 5' end and 54 bases from the 3' end of exon 1. We used the following polymerase chain reaction (PCR) conditions for a 15 μL total reaction: 1.5 μl 10× reaction buffer, 0.3 μL of dNTPs (10 μM), 0.5 μL each primer (10 μM), 0.1 μL Taq polymerase, and 20 ng DNA. We used the following PCR cycling parameters: 94°C for 3:00, (94°C for 0:30, 61°C for 0:45, 72°C for 1:00) × 40 cycles, 72°C for 10:00, 15°C forever. We performed cycle sequencing for both strands with Big Dye v. 2 using the amplification primers and two additional internal primers (intF 5'-AGCAACCTGGCAGAGACACT-3' and intR 5'-TGTAGAGCACCAGCATGAGG-3'; 4 μM) following standard sequencing protocols. We sequenced all PCR products on an ABI PRISM® 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA) and edited the sequences in the program Sequencher version 4.7 (Gene Codes Corporation). We identified all heterozygous sites by visual inspection of chromatogram peaks and confirmed each site using sequences from both strands.
Results
Microsatellite data
None of the 17 microsatellite loci significantly differed from Hardy-Weinberg equilibrium expectations and we detected no evidence of linkage disequilibrium. We found no evidence of null alleles. Observed heterozygosity, average number of alleles, private alleles, and allelic richness for each sampling site are summarized in Table 2.
Table 2.
Genetic diversity indices for microsatellite and mitochondrial sequence data.
| Nμsat | HO ± sd | Aa ± sd | Ap | Ar | Nmt | HN | HP | π | Hd | |
|---|---|---|---|---|---|---|---|---|---|---|
| B. j. calurus | 178 | 0.68 ± 0.01 | 14.2 ± 10.3 | 34 | 6.9 | 122 | 14 | 8 | 0.0019 | 0.57 ± 0.003 |
| central California | 68 | 0.68 ± .01 | 11.4 ± 7.6 | 9 | 6.3 | 29 | 6 | 2 | 0.0012 | 0.43 ± 0.11 |
| Idaho | 35 | 0.66 ± 0.02 | 10.1 ± 6.3 | 5 | 6.5 | 34 | 6 | 2 | 0.0012 | 0.37 ± 0.10 |
| Nevada/Utah | 39 | 0.71 ± 0.02 | 11.2 ± 7.2 | 9 | 6.8 | 29 | 8 | 1 | 0.0016 | 0.57 ± 0.11 |
| southern California | 36 | 0.65 ± 0.02 | 9.6 ± 5.1 | 3 | 6.5 | 30 | 4 | 0 | 0.0009 | 0.35 ± 0.10 |
| B. j. borealis | 69 | 0.73 ± 0.01 | 12.1 ± 7.9 | 11 | 7.1 | 52 | 13 | 8 | 0.0019 | 0.520 ± .007 |
| Alberta | 2 | 0.79 ± 0.07 | 2.9 ± 1.1 | 0 | --- | --- | --- | --- | --- | --- |
| Colorado | 11 | 0.73 ± 0.03 | 7.1 ± 3.4 | 1 | 6.6 | 6 | 1 | 0 | --- | --- |
| Kentucky | 9 | 0.72 ± 0.04 | 6.2 ± 2.5 | 0 | --- | 10 | 5 | 1 | 0.0022 | 0.67 ± 0.16 |
| Michigan | 5 | 0.76 ± 0.05 | 5.1 ± 2.0 | 1 | --- | 5 | 3 | 1 | 0.0023 | 0.70 ± 0.22 |
| north Texas | 9 | 0.73 ± 0.04 | 6.7 ± 3.4 | 1 | --- | 5 | 2 | 1 | 0.0008 | 0.33 ± 0.22 |
| Wisconsin | 33 | 0.73 ± 0.02 | 9.8 ± 5.3 | 5 | 6.6 | 26 | 8 | 5 | 0.0022 | 0.47 ± 0.12 |
| B. j. harlani | 24 | 0.69 ± 0.03 | 9.0 ± 5.3 | 5 | 6.9 | 24 | 5 | 0 | 0.016 | 0.59 ± 0.05 |
| B. jamaicensis spp. | ||||||||||
| melanistic migrants | 21 | 0.68 ± 0.03 | 9.0 ± 5.2 | 1 | 6.9 | --- | --- | --- | --- | --- |
Key: Nμsat = the number of samples for which microsatellite data was collected, HO = observed heterozygosity, Aa = average number of alleles per locus, Ap = number of private alleles, Ar = allelic richness averaged across 17 loci, Nmt = the number of samples for which sequence data was collected, HN = the number of haplotypes, HP = the number of private haplotypes, π = nucleotide diversity, Hd = haplotype diversity.
Strong bootstrap support (99.8%) in the UPGMA tree (Figure 1) indicated a distinction between all B. j. calurus samples and the combined sample of B. j. borealis and B. j. harlani. No other nodes were supported by bootstrap analysis. Neighbor joining tree topology (not shown) was identical to that observed in the UPGMA tree. This result is consistent with previous UPGMA clustering that supported a distinction between B. j. borealis and B. j. calurus for all breeding and migration sites [22].
Figure 1.
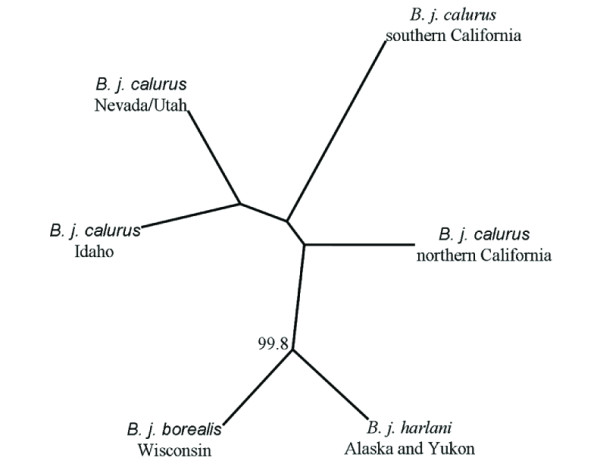
Unrooted UPGMA clustering tree based on microsatellite allele frequency differences. Only breeding sites with greater than 20 individuals were included; we assessed support using 10,000 bootstrapped data sets. Nodes with greater than 50% bootstrap support are indicated. The results shown here are consistent with previous research that found support in UPGMA clustering for a distinction between B. j. calurus and B. j. borealis breeding and migration sites [22].
Pairwise θST values for 17 microsatellite loci among subspecies are summarized in Table 3. We observed significant differentiation between B. j. calurus and both B. j. borealis and B. j. harlani. However, we did not detect significant differences in variance in allele frequency between B. j. borealis and B. j. harlani. A sample of 21 melanistic individuals trapped in migration in central California, presumably B. j. calurus, was significantly different than both B. j. harlani and B. j. borealis individuals, but genetically indistinct from B. j. calurus individuals.
Table 3.
Microsatellite pairwise θST comparisons between B. jamaicensis subspecies.
| B. jamaicensis spp. (melanistic) | B. j. calurus | B. j. borealis | B. j. harlani | |
|---|---|---|---|---|
| B. jamaicensis spp. (melanistic) | ---------- | |||
| B. j. calurus | 0.004 | ---------- | ||
| B. j. borealis | 0.023* | 0.012* | ---------- | |
| B. j. harlani | 0.023* | 0.020* | 0.004 | ---------- |
Key: *'s indicate significant differences following Bonferroni correction. Melanistic individuals were sampled during autumn migration within the range of B. j. calurus; however the sample may include individuals that originated outside the range of B. j. calurus.
Bayesian clustering analysis of breeding individuals without regard to subspecies or region of origin revealed a maximum log Pr(X|K) for K = 2 reflecting two distinct population clusters. One cluster corresponded to a group composed of B. j. calurus individuals while the second group was comprised of both B. j. borealis and B. j. harlani. As an additional test for the existence of a distinct B. j. harlani cluster we also examined population clustering for K = 3. We continued to find a distinct B. j. borealis/harlani cluster while the B. j. calurus cluster was further subdivided into a central California cluster and an intermountain desert cluster, consistent with the findings of Hull et al. 2008 [22]. Finally, we performed a separate run including only B. j. borealis and B. j. harlani samples to test whether any fine-scale structure could be identified. For this subset of the data, the maximum log Pr(X|K) occurred at K = 1, but was very similar to the value at K = 2. For K = 2, no pattern of cluster ancestry was apparent between B. j. borealis and B. j. harlani; a Yates corrected χ2 test also showed no difference in the proportion of ancestry (χ21 = 1.52, P = 0.22).
Mitochondrial data
We collected control region sequence data for 198 individuals from presumptive populations of B. j. borealis (n = 52), B. j. calurus (n = 122), and B. j. harlani (n = 24) and observed 29 haplotypes (GenBank accession numbers HM454161 - HM454189). Of these haplotypes, eight were unique to B. j. borealis and five unique to B. j. calurus; we did not identify any unique haplotypes in B. j. harlani. Haplotype and nucleotide diversity were generally lower in B. j. calurus than in either B. j. borealis or B. j. harlani which were relatively similar to each other (Table 2). Distribution and frequency of control region haplotypes among sampling sites is shown in Figure 2. Pairwise ΦST comparisons among subspecies revealed significant differentiation among all pairs of subspecies (Table 4).
Figure 2.
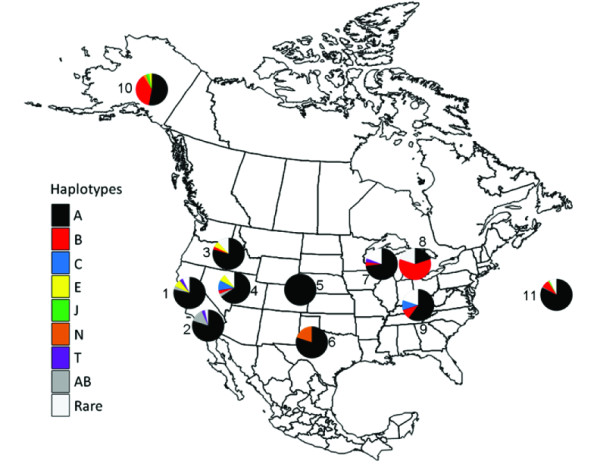
Map of North American B. jamaicensis sampling locations. We collected all samples from breeding sites during the breeding season from either young in nests or territorial adults. We collected samples from sites 1-4 within the breeding range of B. j. calurus; samples from sites 5-9 within the breeding range of B. j. borealis; samples from site 10 within the breeding range of B. j. harlani; and we collected the pooled samples from site 11 from migrants of unknown breeding origin, all samples from site 11 appeared phenotypically similar to typical or melanistic B. j. calurus. Sampling areas are shown with associated frequencies of mitochondrial control region haplotypes.
Table 4.
Mitochondrial pairwise ΦST comparisons between B. jamaicensis subspecies
| B. j. calurus | B. j. borealis | B. j. harlani | |
|---|---|---|---|
| B. j. calurus | ---------- | ||
| B. j. borealis | 0.032* | ---------- | |
| B. j. harlani | 0.203* | 0.059* | ---------- |
Key: *'s indicates significant differences following Bonferroni correction.
Analysis of mtDNA sequence data suggested that the best model fit to the data under the hierarchical likelihood ratio criterion was the HKY+I(0.90) +G (Γ = 0.85) model [52]. Under the Akaike Information Criterion (AIC) criterion the best fit model was the K81uf+I(0.90) +G(Γ = 0.85) [53] model with an invariant site parameter (TrN+I). The log likelihood score for the HKY+I(0.90) +G (Γ = 0.85 model (-lnL = 769.17) was similar to that for the K81uf+I(0.90) +G(Γ = 0.85) model (-ln L 768.02, AIC = 1550.05). For the HKY+I model, we estimated the transition to transversion ratio at 14.42.
All gene tree reconstruction methods showed results similar to the among-haplotype relationships shown in the ME tree (Figure 3): haplotypes did not form monophyletic clades with respect to subspecies, and node support was weak. Both ME and ML analyses gave marginal bootstrap support in few cases with no support greater than 59% (data for ME analysis only given in Figure 3).
Figure 3.

Neighbor joining distance mid-point rooted minimum evolution tree of B. jamaicensis haplotypes. Branch length indicates degree of genetic differentiation. Bootstrap values providing > 50% support for nodes are indicated. Gray circles indicate haplotypes sampled from breeding sites of B. j. calurus, white triangles indicate haplotypes sampled from breeding sites of B. j. borealis, black squares indicate haplotypes sampled from breeding sites of B. j. harlani, and asterisks indicate haplotypes that were sampled from migrant hawks in California with plumage consistent with B. j. calurus. We applied midpoint rooting, employing the close-neighbor interchange algorithm for finding the tree, beginning with an initial neighbor-joining tree.
We observed asymmetrical gene flow among all subspecies and across both marker types. The full model (all parameters allowed to vary independently) had significantly higher likelihoods than the restricted island model (equal inter-group migration rate and equal θ across populations; microsatellite: LnLrestricted = -1078.32, LnLfull = -801.21, P < 0.0001; mtDNA: LnLrestricted = -527.48, LnLfull = 6.20, P < 0.0001). Nem and θ (4Neμ) values calculated in MIGRATE from microsatellite genotypes and mtDNA ranged from 2.56 -5.60 migrants per generation among subspecies, with θ ranging from 1.031 - 1.62 (Figure 4A), and <0.01 to 70.49 female migrants per generation among subspecies, with θ ranging from 0.004 - 0.007 (Figure 4B). The bias in the variances and the means indicate that, on average over generations, gene flow has been greater from B. j. borealis and B. j. harlani to B. j. calurus than B. j. calurus to the other two subspecies (Figure 4A, B). While gene flow estimates between B. j. harlani and B. j. borealis appear more symmetrical (overlapping confidence intervals) when based on microsatellite loci (Figure 4A), estimates based on mtDNA suggest significantly greater levels of gene flow from B. j. borealis to B. j. harlani than vice versa (Figure 4B). These estimates provide a rough estimation of the rate of gene exchange among subspecies and should not be viewed as precise measures. Rather, these results should be considered in the context of whether B. j. harlani is sufficiently isolated from other B. jamaicensis subspecies to be considered a full species.
Figure 4.

Results of full model migration matrix. We allowed all parameters to vary independently. We calculated migration direction and rates from: (A) 17 microsatellite loci and (B) mtDNA control region data. Nem and Nfm = estimate for number of migrants/females per generation based on nuclear microsatellites and mtDNA, respectively. Θ = population size (4Neμ; see text). 95% confidence intervals for parameter values are in parentheses. Arrow thickness is proportionate to estimated levels of gene flow (thicker arrows indicate higher relative gene flow).
Mc1r results
We sequenced 829 base pairs corresponding to 276 out of 314 amino acids in the avian Mc1r, excluding 19 amino acids in the N-terminus domain and 17 amino acids in the C-terminus domain (GenBank accession numbers HM454190 - HM454192). We found that six internal nucleotides were absent relative to the Gallus gallus Mc1r. We found three synonymous substitutions within conserved transmembrane regions I and III, but no non-synonymous substitutions in the 829 bp of Mc1r sequenced from the 23 B. jamaicensis individuals sampled (Table 5). None of the three synonymous substitutions were consistently associated with rufous, dark, light, partial albino, or B. j. harlani color morphs. These data suggest that the Mc1r locus in B. jamaicensis is not responsible for breast color variation or the variable melanin-based plumage patterns observed in B. j. harlani.
Table 5.
Mc1r polymorphism for breast color morphs in B. j. calurus and B. j. harlani subspecies.
| Phenotype | Subspecies | Specimen no. | 73 | 292 | 346 |
|---|---|---|---|---|---|
| Consensus | G | C | C | ||
| Rufous | calurus | 10094 | . | . | . |
| Rufous | calurus | 13424 | . | . | . |
| Rufous | calurus | 13642 | . | . | . |
| Rufous | calurus | 13651 | . | . | . |
| Rufous | calurus | 13662 | . | . | . |
| Rufous | calurus | 13737 | . | . | . |
| Rufous | calurus | 13775 | . | . | . |
| Rufous | calurus | 13826 | . | . | . |
| Dark | calurus | 11071 | . | . | . |
| Light | calurus | 10101 | G/A | . | . |
| Light | calurus | 11056 | . | . | . |
| Light | calurus | 13670 | . | . | . |
| Light | calurus | 13671 | . | . | . |
| Light | calurus | 13672 | . | C/T | . |
| Light | calurus | 13673 | . | . | C/T |
| Light | calurus | 13674 | G/A | . | . |
| Partial albino | calurus | 6161 | . | . | . |
| Harlan's | harlani | 10983 | . | . | . |
| Harlan's | harlani | 13654 | . | C/T | . |
| Harlan's | harlani | 14133 | . | . | C/T |
| Harlan's | harlani | 14134 | . | . | . |
| Harlan's | harlani | 14138 | . | . | . |
| Harlan's | harlani | 14139 | . | . | . |
| AA | Thr | Ile | Ile | ||
| AA change | - | - | - | ||
Key: The nucleotide position and base for the consensus sequence is listed across the top. Amino acids from the hawk consensus sequence are listed below. No amino acid replacements were observed.
Discussion
Differentiation and genetic diversity
Our results build upon previous phylogeographic work and show limited genetic differentiation among the B. jamaicensis subspecies examined. The data suggest that all three B. jamaicensis subspecies are quite similar genetically, and that B. j. harlani shows closer evolutionary affinities to B. j. borealis than to B. j. calurus. The close relationship and limited differentiation between B. j. harlani and B. j. borealis is supported by lack of significant θST and significant albeit shallow ΦST estimates (0.004 and 0.059 respectively), high levels of estimated of gene flow based on both marker classes, UPGMA microsatellite groupings, Bayesian support in the microsatellite data for a single B. j. harlani/borealis population cluster, and absence of unique B. j. harlani mitochondrial haplotypes. These results indicate both historical and contemporary gene exchange between B. j. borealis and B. j. harlani. Conversely, θST and ΦST estimates between B. j. calurus and the other two subspecies were significant for both marker types. Significant divergence has been observed among other North American raptor populations and subspecies. Eastern and western subspecies of B. lineatus (red-shouldered hawk) displayed significant divergence in microsatellite and mitochondrial data sets, and eastern and western populations of Accipiter striatus (sharp-shinned hawk) were found to be significantly diverged in the absence of any described subspecies [54,55]. Both studies employed the same set of microsatellite and/or mitochondrial markers as used here.
Estimates of rates of gene flow among subspecies based upon mitochondrial and microsatellite data indicate a high level of gene flow among all three B. jamaicensis subspecies examined. Both mitochondrial and microsatellite data sets indicate that over time a higher level of gene flow has occurred from B. j. borealis and B. j. harlani to B. j. calurus than from B. j. calurus to the other subspecies. Estimates of gene flow between B. j. borealis and B. j. harlani appear symmetrical when considering the microsatellite data; however, the mitochondrial estimates indicate greater gene flow from B. j. borealis to B. j. harlani. This pattern suggests that while rates of male migration between B. j. borealis and B. j. harlani may be equivalent, female B. j. borealis migration rates exceed those of B. j. harlani, a result consistent with a northern Midwest source for Yukon and Alaskan populations of B. jamaicensis.
The three recognized subspecies of B. jamaicensis examined here also displayed similar intra-subspecific levels of genetic diversity. Our sample of B. j. harlani showed equivalent levels of observed heterozygosity, allelic richness, nucleotide diversity and haplotype diversity to B. j. calurus and B. j. borealis. We did not find any unique B. j. harlani mitochondrial haplotypes and only identified five private microsatellite alleles; the absence of unique haplotypes and presence of few unique microsatellite alleles is also suggestive of contemporaneous gene exchange among populations representing the sampled subspecies.
Our analysis of melanistic individuals sampled in California during autumn migration indicated that they were genetically indistinguishable from B. j. calurus. Additionally, our analysis of sequence data from the melanocortin 1 receptor (Mc1r) showed no differences between melanistic and non-melanistic individuals, or between B. j. harlani and any B. j. borealis or calurus. This suggests that the Mc1r gene does not play the same role in B. jamaicensis melanism as in some other avian species [51,56].
Evolutionary interpretation of genetic data
The genetic data described here do not support the historical designation of B. j. harlani as a distinct species [18,57]. The lack of significant differentiation between B. j. harlani and B. j. borealis in the microsatellite data suggests that gene exchange between the two subspecies occurs at a relatively high level, and the lack of reciprocal monophyly among haplotypes is inconsistent with differences expected at the species level. Furthermore, the absence of field evidence demonstrating assortative mating among B. j. harlani individuals relative to B. j. calurus or B. j. borealis and the apparent interbreeding with neighboring subspecies [19,58,59] also supports recognition of B. j. harlani as a member of B. jamaicensis, and not a distinct species. In fact, under some criteria, the pattern observed between B. j. borealis and B. j. harlani is inconsistent with differentiation expected among subspecies or "evolutionary significant units" [24-26]. Given the observed levels of differentiation based on traditional and Bayesian analyses of population structure and results of the MIGRATE analyses, it is possible that B. j. harlani represents a post-Pleistocene northern extension of B. j. borealis populations; however additional study is required to test this hypothesis.
The failure to detect microsatellite differentiation between B. j. harlani and B. j. borealis is not associated with lack of sensitivity in the genetic tools employed. In other studies, Bayesian analyses were able to identify fine-scale population structure between western North American populations of both B. j. calurus and B. swainsoni (Swainson's hawk) employing the same set of microsatellite markers used here [22,43]. The population divergences recovered in these examples corresponded to θST values of ~0.01 in both cases. In the case of B. j. calurus, within-subspecies genetic structure in western populations included fine-scale substructure between central California and the Intermountain West populations and was associated with significant morphological (overall size) differentiation. The morphological differentiation observed between these populations of a well described subspecies (B. j. calurus) indicates that variation in morphology alone may not accurately reflect speciation events. Given these previous findings using the same marker set, the data set used here is sensitive enough to detect species-level differentiation between B. j. harlani and other B. jamaicensis subspecies if it were present.
Relevance of phenotype
In addition to a high incidence of melanism, B. j. harlani are highly variable in plumage particularly in tail pattern, extent of white below chin, spotting on back, and extent of melanism throughout body feathers [15,16,60]. The extent of variation makes description of definitive B. j. harlani characters difficult and no diagnostic set of B. j. harlani plumage characters has yet been accepted [60]. Whether the variable plumage characteristics within B. j. harlani also occur within other subspecies is also debated. A breeding individual observed in central California displayed many of the typical B. j. harlani plumage characters and many juvenile hawks observed during autumn migration in California have plumage characteristics that have been suggested as belonging to B. j. harlani [AC Hull, pers. comm.; WC Clark pers. comm.], yet these migrant juveniles were found to be genetically similar to the breeding populations of B. j. calurus from central California and the Great Basin [22], not B. j. borealis or harlani. To provide a more thorough understanding of how plumage variation in B. jamaicensis is correlated with nominate subspecies, additional field research is necessary to more completely categorize the range of plumage variation throughout B. jamaicensis and describe the geographic bounds of observed variation. Additional surveys and sampling of individuals from the Yukon, British Columbia, and prairie region would be particularly helpful in resolving the relationship between phenotype, environment, and population structure.
The mechanisms influencing plumage patterns in B. j. harlani require further investigation. Potential mechanisms include: 1) a B. j. harlani-specific gene or gene-complex, or 2) particular environmental, temporal, or physiological conditions within the B. j. harlani range interacting with the standard B. jamaicensis genotype without involvement of a unique B. j. harlani genotype. In light of recent research that has begun to reveal the role of environmental conditions, including temperature and oxidative stress, on melanin-based plumage [11-13,61], a controlled study of B. j. harlani plumage characters would be a fruitful avenue of future research.
More broadly, the underlying factors responsible for the base melanistic plumage in B. j. harlani, and more generally, melanism in B. jamaicensis require additional study. Our data suggest that the Mc1r locus does not play a similar role as in other avian species previously examined [51,56,62]. The gene or genes responsible for melanism in B. jamaicensis remain to be identified.
Conclusions
These genetic data indicate low levels of differentiation among three recognized subspecies of B. jamaicensis examined, particularly between B. j. borealis and B. j. harlani. In light of previous population genetic structure investigations of species within the genus Buteo, our findings are inconsistent with the historical designation of B. j. harlani as a separate species. Rather, the mtDNA data are consistent with contemporary subspecies nomenclature and data from both marker classes suggest that gene flow is occurring at a relatively high level among these subspecies. Additional research is necessary to fully describe the geographical variation in plumage observed in B. jamaicensis and to determine the underlying mechanisms responsible for the variation seen in B. j. harlani.
Authors' contributions
JH, ST, DM, HH, and HE designed the study. JH and ST obtained the genetic samples. JH, ST, and EK did the molecular work and obtained the sequence data. JH and ST did the phylogenetic analyses. JH, ST, and EK wrote a first draft. The final version was completed by JH, DM, ST, EK, HH, and HE. All authors read and approved the final manuscript.
Contributor Information
Joshua M Hull, Email: jmhull@ucdavis.edu.
David P Mindell, Email: dmindell@calacademy.org.
Sandra L Talbot, Email: sandy_talbot@usgs.gov.
Emily H Kay, Email: ekay@oeb.harvard.edu.
Hopi E Hoekstra, Email: hoekstra@oeb.harvard.edu.
Holly B Ernest, Email: hbernest@ucdavis.edu.
Acknowledgements
We thank William Clark, Angus Hull, Ted Swem, Clayton White, and the Golden Gate Raptor Observatory for assisting in sample collection. Support was provided by the UC Davis Veterinary Genetics Laboratory, USGS Alaska Science Center, and a National Science Foundation Graduate Research Fellowship to Emily Kay. Joshua Israel, Jessica Petersen, Molly Stephens, John Martin and Travis Booms provided valuable comments on earlier versions of this manuscript. Two anonymous reviewers contributed important suggestions and guidance. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
References
- Sites JW, Marshall JC. Operational criteria for delimiting species. Annu Rev Ecol Evol Syst. 2004;35:199–227. doi: 10.1146/annurev.ecolsys.35.112202.130128. [DOI] [Google Scholar]
- de Queiroz K. Different species problems and their resolution. BioEssays. 2005;27:1263–1269. doi: 10.1002/bies.20325. [DOI] [PubMed] [Google Scholar]
- Hey J. On the failure of modern species concepts. Trends Ecol Evol. 2006;21:447–450. doi: 10.1016/j.tree.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Omland KE, Tarr CL, Boarman WI, Marzluff JM, Fleischer RC. Cryptic genetic variation and paraphyly in ravens. Proc R Soc Lond B Biol Sci. 2000;267:2475–2482. doi: 10.1098/rspb.2000.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull JM, Savage WK, Bollmer JL, Kimball RT, Parker PG, Whiteman NK, Ernest HB. On the origin of the Galapagos hawk: an examination of phenotypic differentiation and mitochondrial paraphyly. Biol J Linn Soc. 2008;95:779–789. doi: 10.1111/j.1095-8312.2008.01082.x. [DOI] [Google Scholar]
- Zink RM. The role of subspecies in obscuring avian biological diversity and misleading conservation policy. Proc R Soc Lond B Biol Sci. 2004;271:561–564. doi: 10.1098/rspb.2003.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Ornithologists' Union. Forty-second supplement to the American Ornithologists' Union Check-list of North American Birds. Auk. 2000;117:847–858. doi: 10.1642/0004-8038(2000)117[0847:FSSTTA]2.0.CO;2. [DOI] [Google Scholar]
- Zink RM, Barrowclough GF, Atwood JL, Blackwell-Rago RC. Genetics, taxonomy, and conservation of the threatened California Gnatcatcher. Conserv Biol. 2000;5:1394–1405. doi: 10.1046/j.1523-1739.2000.99082.x. [DOI] [Google Scholar]
- Johnson JA, Burnham KK, Burnham WA, Mindell DP. Genetic structure identified among continental and island populations of gyrfalcons. Mol Ecol. 2007;16:3145–3160. doi: 10.1111/j.1365-294X.2007.03373.x. [DOI] [PubMed] [Google Scholar]
- Phillimore AB, Owens IPF. Are subspecies useful in evolutionary and conservation biology? Proc R Soc Lond B Biol Sci. 2006;1590:1049–1053. doi: 10.1098/rspb.2005.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith SC, Parker TH, Olson VA. Melanin versus carotenoid-based sexual signals: is the difference really so black and red? Anim Behav. 2006;71:749–763. doi: 10.1016/j.anbehav.2005.07.016. [DOI] [Google Scholar]
- McGraw KJ. An update on the honesty of melanin-based color signals in birds. Pigment Cell and Melanoma Res. 2008;21:133–138. doi: 10.1111/j.1755-148X.2008.00454.x. [DOI] [PubMed] [Google Scholar]
- Galvan I, Alonso-Alvarez C. The expression of melanin-based plumage is separately modulated by exogenous oxidative stress and a melanocortin. Proc R Soc Lond B Biol Sci. 2009;1670:3089–3097. doi: 10.1098/rspb.2009.0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara P, Fargallo JA, Martinez-Padilla J, Lemus JA. Inter-annual variation and information content of melanin-based coloration in female Eurasian kestrels. Biol J Linn Soc. 2009;97:781–790. doi: 10.1111/j.1095-8312.2009.01263.x. [DOI] [Google Scholar]
- Preston CR, Beane RD. In: The Birds of North America. Poole A, Gill F, editor. Philadelphia: The Academy of Natural Sciences; Washington, DC: The American Ornithologists' Union; 1993. Red-tailed Hawk (Buteo jamaicensis) p. 52. [Google Scholar]
- Clark WS, Wheeler BK. A Field Guide to Hawks of North America, Revised. Boston: Houghton Mifflin; 2001. [Google Scholar]
- Clark WS. Extreme variation in the tails of adult Harlan's Hawks . Birding; in press . [Google Scholar]
- Peters JL. Check-list of birds of the world. Vol. 1. Cambridge: Harvard University Press; 1931. [Google Scholar]
- Ridgway R. Harlan's Hawk a race of the Red-tail, and not a distinct species. Auk. 1890;7:205. [Google Scholar]
- Mindell DP. Harlan's Hawk (Buteo jamaicensis harlani): a valid subspecies. Auk. 1983;100:161–169. [Google Scholar]
- Mindell DP. Plumage variation and winter range of Harlan's Hawk (Buteo jamaicensis harlani) Amer Birds. 1985;39:127–133. [Google Scholar]
- Hull JM, Hull AC, Sacks BN, Smith JP, Ernest HB. Landscape characteristics influence morphological and genetic differentiation in a widespread raptor. Mol Ecol. 2008;17:810–824. doi: 10.1111/j.1365-294X.2007.03632.x. [DOI] [PubMed] [Google Scholar]
- Pearlstine EV. Variation in mitochondrial DNA of four species of migratory raptors. J Raptor Res. 2004;38:250–255. [Google Scholar]
- Ball RM, Avise JC. Mitochondrial-DNA phylogeographic differentiation among avian populations and the evolutionary significance of subspecies. Auk. 1992;109:626–636. [Google Scholar]
- Mortiz C. Defining evolutionarily significant units for conservation. Trends Ecol Evol. 1994;9:373–375. doi: 10.1016/0169-5347(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Avise JC, Walker D. Pleistocene phylogeographic effects on avian populations and the speciation process. Proc R Soc Lond B Biol Sci. 1998;265:457–463. doi: 10.1098/rspb.1998.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull JM, Tufts D, Topinka JR, May B, Ernest HB. Development of 19 microsatellite loci for Swainson's hawks (Buteo swainsoni) and other buteos. Mol Ecol Notes. 2007;7:346–349. doi: 10.1111/j.1471-8286.2006.01604.x. [DOI] [Google Scholar]
- Toonen RJ, Hughes S. Increased Throughput for Fragment Analysis on an ABI Prism® 377 Automated Sequencer Using a Membrane Comb and STRand Software. Biotechniques. 2001;31:1320–1324. [PubMed] [Google Scholar]
- Raymond M, Rousset F. GENEPOP Version 1.2: population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–249. [Google Scholar]
- Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.2307/2409177. [DOI] [PubMed] [Google Scholar]
- van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004;4:535–538. doi: 10.1111/j.1471-8286.2004.00684.x. [DOI] [Google Scholar]
- Glaubitz JC. CONVERT: a user friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol Ecol Notes. 2004;4:309–310. doi: 10.1111/j.1471-8286.2004.00597.x. [DOI] [Google Scholar]
- Park SDE. PhD Thesis. University of Dublin; 2001. Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. [Google Scholar]
- Goudet J. FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered. 1995;86:485–486. [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Felsenstein J. PHYLIP - Phylogenetic Inference Package, Version 3.2. University of Washington; 1989. [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Wen W. Documentation for Structure Software Version 2. http://pritch.bsd.uchicago.edu.
- Waples RS, Gaggiotti O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol. 2006;15:1419–1439. doi: 10.1111/j.1365-294X.2006.02890.x. [DOI] [PubMed] [Google Scholar]
- Kimball RT, Braun EL, Zwartjes PW, Crowe TM, Ligon JD. A molecular phylogeny of the pheasants and partridges suggests that these lineages are not monophyletic. Mol Phylogenet Evol. 1999;11:38–54. doi: 10.1006/mpev.1998.0562. [DOI] [PubMed] [Google Scholar]
- Bollmer JL, Kimball RT, Whiteman NK, Sarasola JH, Parker PG. Phylogeography of the Galapagos hawk (Buteo galapagoensis): A recent arrival to the Galapagos Islands. Mol Phylogenet Evol. 2006;39:237–247. doi: 10.1016/j.ympev.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Hull JM, Anderson R, Bradbury M, Estep J, Ernest HB. Population structure and genetic diversity of Swainson's Hawks (Buteo swainsoni): implications for conservation. Conservat Genet. 2008;9:305–316. doi: 10.1007/s10592-007-9342-y. [DOI] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland: Sinauer; 2000. [Google Scholar]
- Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Akaike H. A new look at the statistical model identification. IEEE Transactions on Automatic Control. 1974;19:716–723. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
- Rozas J, Sánchez-De JC, Barrio I, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Beerli P, Felsenstein J. Maximum likelihood estimation of migration rates and population numbers of two populations using a coalescent approach. Genetics. 1999;152:763–773. doi: 10.1093/genetics/152.2.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli P. MIGRATE: documentation and program, part of LAMARC. Version 2.0. Revised 23 December 2004. http://popgen.sc.fsu.edu/Migrate/Migrate-n.html
- Beerli P, Felsenstein J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA. 2001;98:4563–4568. doi: 10.1073/pnas.081068098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy NI, Badcock NS, Hart T, Scribner K, Jansen K, Nadeau NJ. Conserved genetic basis of a quantitative plumage trait involved in mate choice. Science. 2004;303:1870–1873. doi: 10.1126/science.1093834. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino KT. Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Kimura M. Estimation of evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1981;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Hull JM, Strobel BN, Boal CW, Hull AC, Dykstra CR, Irish AM, Fish AM, Ernest HB. Comparative phylogeography and population genetics within Buteo lineatus reveals evidence of distinct evolutionary lineages. Mol Phylogenet Evol. 2008;49:988–996. doi: 10.1016/j.ympev.2008.09.010. [DOI] [PubMed] [Google Scholar]
- Hull JM, Girman DJ. Effects of Holocene climate change on the historical demography of migrating sharp-shinned hawks (Accipiter striatus velox) in North America. Mol Ecol. 2005;14:159–170. doi: 10.1111/j.1365-294X.2004.02366.x. [DOI] [PubMed] [Google Scholar]
- Theron E, Hawkins K, Bermingham E, Ricklefs RE, Mundy NI. The molecular basis of an avian plumage polymorphism in the wild: a melanocortin-1-receptor point mutation is perfectly associated with the melanic plumage morph of the bananaquit, Coereba flaveola. Curr Biol. 2001;11:550–557. doi: 10.1016/S0960-9822(01)00158-0. [DOI] [PubMed] [Google Scholar]
- Audubon JJ. Ornithological Biography. Edinburgh: Adam and Charles Black; 1831. [Google Scholar]
- Taverner PA. A study of Buteo borealis, the Red-tailed Hawk, and its varieties in Canada. Victoria Memorial Museum Bulletin. 1927;48 [Google Scholar]
- Lowe CM. MS thesis. University of Alaska, Fairbanks; 1978. Certain life history aspects of the Red-tailed Hawk in central Oklahoma and interior Alaska. [Google Scholar]
- Liguori J, Sullivan BL. Comparison of Harlan's with western and eastern red-tailed hawks. Birding. 2010;42:31–37. [Google Scholar]
- Galeotti P, Rubolini D, Sacchi R, Fasola M. Global changes and animal phenotypic responses: melanin-based plumage redness of scops owls increased with temperature and rainfall during the last century. Biol Letters. 2009;5:532–534. doi: 10.1098/rsbl.2009.0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheviron ZA, Hackett SJ, Brunfield RT. Sequence variation in the coding region of the melanocortin-1-receptor (MC1R) is not associated with plumage variation in the blue-crowned manakin (Lepidothrix coronata) Proc R Soc Lond B Biol Sci. 2006;273:1613–1318. doi: 10.1098/rspb.2006.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]