

Abstract
Objective
The objective of this study was to determine the effects and potential mechanisms of C-reactive protein (CRP) on cholesterol efflux from human macrophage foam cells, which may play a critical role in atherogenesis.
Methods and Results
Human THP-1 monocytes and peripheral blood mononuclear cells (PBMC) were pre-incubated with acetylated LDL and [3H]-cholesterol to form foam cells, which were then treated with apolipoprotein A-I (apoA-I) or HDL for cholesterol efflux assay. Clinically relevant concentrations of CRP significantly reduced cholesterol efflux from THP-1 and PBMC to apoA-I or HDL. CRP significantly decreased the expression of ATP-binding membrane cassette transporter A- 1 (ABCA-1) and ABCG1, while it increased superoxide anion production. Futhermore, CRP substantially activated ERK1/2 in THP-1-derived foam-like cells. Reducing superoxide anion by antioxidant seleno-L-methionine or SOD mimetic (MnTBAP) effectively abolished the CRP-induced decrease in cholesterol efflux and the expression of ABCA1 and ABCG1. Inhibiting ERK1/2 activation by its specific inhibitor PD98059 or by a dominant negative mutant of ERK2 could also block CRP’s action on THP-1 cells.
Conclusions
CRP inhibits cholesterol efflux from human foam cells derived from THP-1 and PBMC in vitro though oxidative stress, ERK1/2 activation and down-regulation of intracellular cholesterol transport molecules ABCA-1 and ABCG-1.
Keywords: C-reactive protein, macrophage, cholesterol efflux, ATP-binding membrane cassette transporter, superoxide anion, antioxidant
Introduction
The normal plasma level of CRP in a healthy population without evidence of acute inflammation is 2 μg/mL or less. The levels of CRP in plasma are elevated in numerous disease states. Chronic elevation of CRP (>10μg/mL) is associated with increased risk of atherosclerosis and cardiovascular disease.1,2 More interestingly, CRP may play a direct role in initiation and progression of atherosclerosis.2,3 The proinflammatory and proatherogenic properties of CRP have been found in endothelial cells,4 vascular smooth muscle cells (VSMCs),5 and monocyte-macrophages.6 In monocyte-macrophages, CRP could induce reactive oxygen species (ROS) and proinflammatory cytokine release, increase oxidized low-density lipoprotein uptake,6 and induce foam cell formation.7 CRP levels are also associated with oxidative stress in patients with coronary arterydisease.8 Oxidative stress can affect cholesterol efflux in VSMC-derived foam cells,9 and has also been implicated in vascular injury and activation of NADPH oxidase and mitogen-activated protein kinases (MAPKs).8–11
A major event in the progression of atherosclerosis is the differentiation of monocytes to macrophages that accumulate lipoprotein-derived cholesterol to form foam cells in the vessel wall.12 During this process, cholesterol efflux may play a pivotal role in the removal of excess cholesterol from extra hepatic cells including macrophages and VSMCs.13 Thus, decreased cholesterol efflux from the arterial wall may potentially promote the progression of atherosclerosis.14 Cholesterol efflux can be mediated or regulated by several molecular pathways including ATP-binding membrane cassette transport protein A1 (ABCA1), G1 (ABCG1), scavenger receptor B1 (SR-B1), caveolins, and sterol 27-hydroxylase (CYP27A1).12–14 For example, the experiments using ABCA1 knockout cells or animals showed inhibition of cholesterol efflux.15
The objective of this study was therefore to test our hypothesis that CRP could have a direct effect on cholesterol efflux from human macrophage foam cells with unique molecular mechanisms. The roles of some key molecules mediating cholesterol efflux such as ABCA1, ABCG1, ROS and MAPKs were investigated. This study may provide new insight into CRP-associated cardiovascular disease.
Materials and Methods
Cholesterol Efflux
Detail information of all chemicals and reagents is provided in the supplementary materials. Human THP-1 monocytes and peripheral blood mononuclear cells (PBMC) were differentiated into macrophages and transformed into foam cells. The cells were treated with CRP (0–20 μg/mL) for 24 hours followed by efflux stage for another 24 hours. The cholesterol efflux and cellular total cholesterol mass were analyzed.16 The time course chosen in the present study was based on the sensitivity of this in vitro model of cholesterol efflux, which was well characterized previously by our group and others.13,16. To be consistent with the time period of cholesterol efflux, we treated cells with CRP for 48 hours for other molecular analysis described below. In some experiments, antioxidants seleno-L-methionine (SeMet) and SOD mimetic (MnTBAP), ERK1/2 inhibitor PD98059, and recombinant adenoviruses encoding dominant negative mutant of ERK2 (ADV-ERK2 ND) were used.16
Real-Time RT-PCR
Total cellular RNA of THP-1-derived foam-like cells was extracted. Primers for genes tested are shown in supplementary Table I. The iQ SYBR green Supermix Kit and iCycler iQ Real-time PCR detection system (Bio-Rad) were used in real-time PCR. A house keeping gene,β-actin, was included for comparison.16
Western Blot Analysis
Cellular proteins were extracted. Equal amount of total proteins (50 μg) were loaded onto 10% SDS-PAGE, fractionated by electrophoresis, and transferred to PVDF membranes. The primary antibodies were used against ABCA1, ABCG1, phosphorylated ERK1/2, total ERK1/2, p22phox, p47phox, and p67phox. Protein bands were visualized with ECL plus chemiluminescent substrate and quantified with density measurements.16
Superoxide Anion Measurement
Macrophage-derived foam cells were incubated with 0.5 mL dihydroethidium (DHE, 10 μM) for 20 minutes. Superoxide anion in the cells was analyzed by FACS Calibure flow cytometry. In addition, macrophage-derived foam cells were directly stained by DHE, covered with DAPI mounting solution, and observed under the florescence microscope.16 To study the sources of superoxide anion, mitochondrial membrane potential was assessed with staining of 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazole-carbocyanide iodine (JC-1). 16 ATP levels were measured with an ATPLite kit.16 Cell lysate proteins were used for MAPK measurement with Luminex multiplex system detecting the amount of phosphorylated and total ERK2, p38, and JNK.16
Statistical Analysis
All data are presented as the mean ± SEM. Inter-group differences were analyzed using one-way ANOVA for the comparison of three or more groups. Student’s t-test was used for the comparison between two groups. A P value < 0.05 was regarded as significant.
Results
CRP Inhibits Cholesterol Efflux in THP-1 Cells and Human PBMC
We determined the effect of CRP on cholesterol efflux from THP-1-derived foam-like cells and human PBMC-derived foam cells by measuring 3H-cholesterol and cholesterol content of THP-1 cells. Foam cells were treated with several concentrations of CRP, and cholesterol efflux was initiated by the addition of apoA-I (50 μg/mL) or HDL (100 μg/mL). In both THP-1 and PBMC, significant decreases of cholesterol efflux to apoA-I were observed in a concentration-dependent manner in response to CRP treatment (Figure 1A and 1B). At 10 μg/mL of CRP, the cholesterol efflux from THP-1 and PBMC to apoA-I was decreased by 27 % and 22%, respectively, compared with controls (apoA-I only) (P<0.05, n=6). A time course study was performed. After the treatment of 10 μg/mL of CRP for 24 hours, cells were then incubated with the addition of 50 μg/mL of apoA-I for 3, 6, 12, 18, 24, and 36 hours, respectively. The differences of cholesterol efflux from THP-1 cells to apoA-I between apoA-I only and CRP treated groups peaked at 18 and 24 hours (Figure 1C). In addition, an enzymatic colorimetric method was used for measuring intracellular cholesterol contents. At 10 μg/mL of CRP, the total cholesterol mass in THP-1 foam-like cells showed increases by 30% compared with the control (ApoA1 only) (P<0.05, n = 6, Figure 1D). Moreover, we performed additional experiments by using human PBMC-derived foam cells for measuring total cholesterol mass. After loaded with acLDL (50 μg/mL), PBMC-derived foam cells were incubated with CRP and total cholesterol mass was measured. Consistent with the data obtained from THP-1 cells, CRP could also contribute to the cholesterol accumulation in PBMC, which may be loaded with relatively high levels of acetylated LDL as previously demonstrated.17 CRP (10 μg/mL) treatment significantly increased total cholesterol mass (91±1.8 μg cholesterol /mg total protein) in PBMC-derived foam cells compared with that in control cells (77± 4.5 μg cholesterol /mg total protein) (P<0.05).
Figure 1.

Effects of CRP on cholesterol efflux and cellular cholesterol contents in THP-1 and human PBMC foam cells. (A) and (B). Cholesterol efflux to apoA-I from THP-1 and human PBMC foam cells, respectively, in response to different concentrations of CRP (5, 10 and 20 μg/mL) treatment for 24 hours. (C). Cholesterol efflux to apoA-I in response to 10 μg/mL CRP treatment and different exposure time (3, 6, 12, 18, 24, and 32 hours). (D). Total cholesterol mass in THP-1 foam cells in response to CRP and apoA-I treatment for 24 hours. (E). Heat-inactivated CRP had no effect on cholesterol efflux, while anti-CRP antibody effectively blocked CRP-induced inhibition of cholesterol efflux (24 hours). (F). Another recombinant CRP without sodium azide (R & D Systems Inc., Minneapolis, MN) was used cholesterol efflux to apoA-I from THP-1 foam cells (24 hours). (G). Cholesterol efflux to HDL from THP-1 foam-like cells in response to different concentrations of CRP for 24 hours. (H). Totol cholesterol mass in foam cells in response to CRP and HDL treatment for 24 hours. Data represent mean ± SEM. *P<0.05, **P<0.001 versus controls, n=6.
In order to determine the specificity of the CRP action, a neutralizing antibody against CRP was used along with CRP and completely abolished CRP-induced inhibition of cholesterol efflux compared with CRP treatment (P =0.03, Figure 1E). Heat-inactivated CRP was used as controls and showed no effects on cholesterol efflux from THP-1 foam cells to ApoA-I (Figure 1E). Furthermore, another recombinant CRP preparation without sodium azide (R & D Systems Inc., Minneapolis, MN) was used instead of CRP from Calbiochem and also induced a significant decrease of cholesterol efflux to apoA-I in a concentration-dependent manner (Figure 1F).
Another cholesterol acceptor, HDL, was used to evaluate the effect of CRP on the cholesterol efflux in THP-1 as well. Consistently with apoA-I, CRP treatment also induced a significant decrease of cholesterol efflux from foam cells to HDL in a concentration-dependent manner (Figure 1G). At 10 μg/mL of CRP, the cholesterol efflux from THP-1 showed a decrease by 23% compared with controls (HDL only) (P=0.01, n = 6). For directly measuring the intracellular cholesterol content, CRP-treated THP-1 foam-like cells also showed an increase by 34% compared with the control (HDL only) (P<0.05, n = 6, Figure 1H).
CRP Decreases the Expression of ABCA1 and ABCG1 in THP-1 Cells
To study possible mechanisms that are responsible for CRP action, we analyzed the expression of several key molecules which are involved in cholesterol efflux including ABCA1, ABCG1, caveolin-1, caveolin-2 and CYP27A1 in foam cells. After 48 hours incubation with CRP, THP-1-derived foam-like cells showed significant decreases of ABCA1 and ABCG1 at both mRNA and protein levels in a concentration-dependent manner. At 10 and 20 μg/mL of CRP, ABCA1 mRNA had a decrease of 34% and 60%, respectively, compared with controls (P<0.05, n=4, Figure 2A). Similarly at the same concentrations of CRP, ABCG1 mRNA was decreased by 36% and 56%, respectively, compared with controls (P<0.05, n=4, Figure 2B). Protein levels of ABCA1 and ABCG1 had a trend of decrease similar to mRNA levels when treated by CRP (Figure 2C and 2D). Furthermore, treatment of CRP for 12 hours decreased the mRNA levels of ABCA1 and ABCG1 in a concentration-dependent manner (Figure 2E and 2F). However, the expression of caveolin-1, caveolin-2 and CYP27A1 did not show any obvious changes between the CRP-treated and control groups (data not shown).
Figure 2.

Effects of CRP on the expression of ABCA1 and ABCG1 in THP-1 foam-like cells. Cells were treated with CRP (5, 10 and 20 μg/mL) for 48 hours. The expression of ABCA1 and ABCG1 were measured in mRNA levels by real-time PCR and in the protein levels by Western blot. CRP significantly decreased both the mRNA levels and the protein levels of ABCA1 (A, C) and ABCG1 (B, D). THP-1 foam-like cells were treated with CRP (5, 10 and 20 μg/ml) for 12 hours. CRP decreased the mRNA levels of ABCA1 (E) and ABCG1 (F). Data represent mean ± SEM. *P<0.05, **P<0.01 versus controls (without CRP). n=4 for real time PCR. n=3 for western blot.
CRP Increases Superoxide Anion Production in THP-1 Cells
To investigate whether ROS could be involved in the CRP-induced decrease of cholesterol efflux, superoxide anion production from THP-1-derived foam-like cells was analyzed with DHE staining. Cells treated with 10 μg/mL CRP revealed a significant increase of superoxide anion production by 77% compared with controls (P<0.05, Figure 3A and 3B). Meanwhile, DHE direct staining under florescent microscope also revealed an increase of superoxide anion production in CRP treated cells compared with untreated cells. Furthermore, CPR-induced increase of superoxide anion production in THP-1-derived foam-like cells was effectively blocked by co-incubation with an antioxidant SeMet (Figure 3C).
Figure 3.

Effect of CRP on superoxide anion production in THP-1 foam-like cells. (A). Superoxide anion in the foam cells treated with CRP was analyzed with DHE staining and flow cytometric analysis. (a). Foam cells without DHE staining served as a negative control. (b). DHE-stained cells without CRP served as a staining control. (c). Cells were treated with CRP. (B). Results from 3 separate experiments were averaged. Data represent mean ± SEM. *P<0.05, versus controls (staining). (C) Representative fluorescence macroscope images showed an increased superoxide anion production (red) in foam cells when treated with 10 μg/mL CRP compared with the control. SeMet abolished the CRP-induced increase of superoxide anion production. DAPI was counterstained for nuclei (blue). Magnification: ×400.
CRP Causes Mitochondrial Dysfunction in THP-1 Cells
During the transfer of electrons to molecular oxygen, an estimated 1 to 5% of electrons in the respiratory chain “leak” to form superoxide radicals. Mitochondrial membrane potential can serve as an indicator for the function of mitochondrial respiration chain. To determine whether mitochondria could be involved in CRP-induced increase of superoxide anion, THP-1-derived foam-like cells were treated with 10 μg/mL CRP and showed a substantial reduction of mitochondrial membrane potential by 57% compared with untreated cells (P<0.005, n=3, Figure 4A and 4B). Decreased mitochondrial membrane potential could result in a decrease in ATP production. Indeed, treatment of CRP significantly reduced ATP levels by 11% compared with controls in THP-1 foam-like cells (P<0.05, n=6, Figure 4C).
Figure 4.
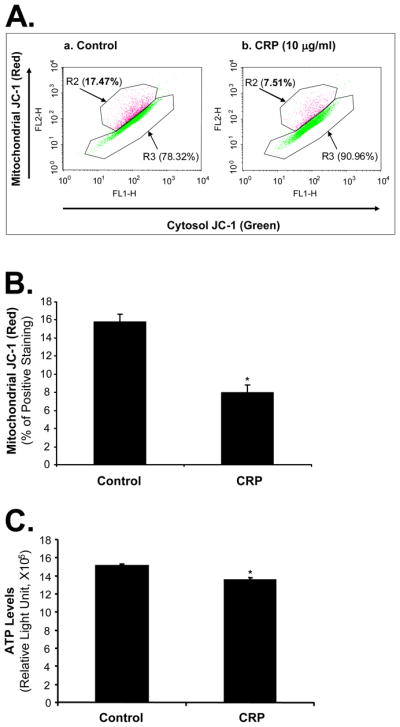
Effects of CRP on mitochondrial dysfunction in THP-1 foam-like cells. Cells were treated with 10 μg/mL CRP for 48 hours. (A) and (B). Mitochondrial membrane potential was assessed with JC-1 staining and flow cytometry analysis. CRP significantly decreased the normal potential (Red) in THP-1 cells compared with controls. n= 3. (C). Cellular ATP levels were measured with an ATPLite kit. Data represent mean ± SEM. n=6. *P<0.05.
CRP Increases the Expression of NADPH Oxidase Subunits in THP-1 Cells
Oxidative stress could result from up-regulation of ROS generating enzymes and/or down-regulation of internal antioxidant enzymes. To test whether these molecules could play a role in CRP-induced oxidative stress, we determined expressions of NADPH oxidase subunits, major ROS generating enzymes, and several major internal antioxidant enzymes such as SOD, catalase, and glutathione peroxidase (GPX) in THP-1 foam-like cells. Treatment with CRP increased the expression of NADPH oxidase subuntis p22phox, p47phox and p67phox in foam cells in a concentration-dependent manner (Figure 5). At 10 μg/mL of CRP, mRNA levels of p22phox, p47phox and p67phox were significantly increased by 3.8, 5.6, and 6.4-fold, respectively, compared with untreated cells (P<0.01, n=4, Figure 5A). Consistent with mRNA data, CRP-treated THP-1 cells increased protein levels of p22phox, p47phox and p67phox in a concentration-dependent manner (Figure 5B and 5C). However, the expression of catalase, cyclooxygenase 1, GPX1, NADPH oxidase 2 and SOD1 in mRNA levels did not show any obvious changes between the CRP-treated and untreated cells (data not shown).
Figure 5.
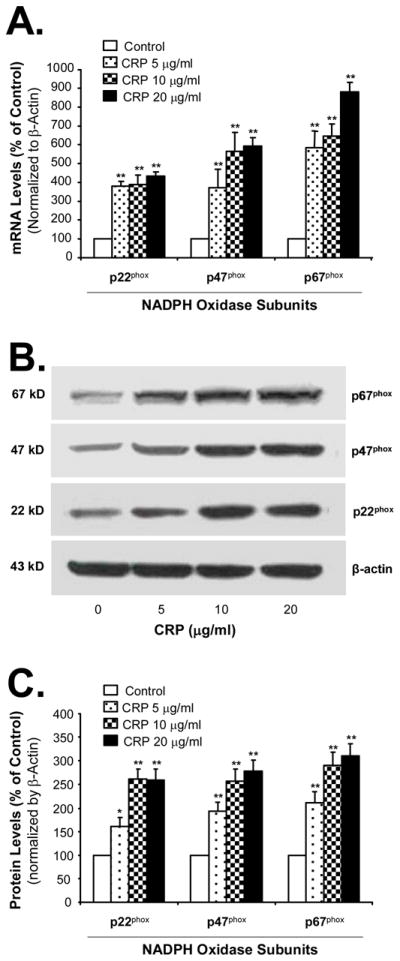
Effects of CRP on the expression of NADPH oxidase subunits in THP-1 foam-like cells. Cells were treated with different concentrations of CRP for 48 hours. The NADPH oxidase subunits were measured in mRNA levels by real-time PCR (A) and in protein levels by western blot (B, C). Data represent mean ± SEM. **P<0.01 versus controls (without CRP). n=4 for real time PCR. n=3 for western blot.
CRP Activates ERK1/2 in THP-1 Cells
Since MAPKs are redox sensitive, we hypothesized that MAPKs could be involved in the CRP-induced inhibition of cholesterol efflux from THP-1-derived foam-like cells. The activation status of three major MAPKs (ERK2, JNK, and p38) was analyzed by Bio-Plex immunoassay. CRP treatment (10 μg/mL) for 90 minutes substantially increased the phosphorylation of ERK2 in macrophage-derived foam cells by 2.5-fold compared with untreated cells (at 0 minute) (Figure 6A). Meanwhile, western blot also revealed a significant increase of phosphorylation of ERK1/2 in macrophages at 90 minutes of CRP treatment compared with untreated cells. Moreover, such increase of phosphorylation of ERK1/2 was effectively blocked by co-incubation with the specific ERK1/2 inhibitor PD98059 (P<0.05, n=3, Figure 6B). However, there were no changes of phosphorylation in JNK and p38 in response to CRP treatment (data not shown).
Figure 6.

Roles of ERK1/2 and antioxidants in CRP activities in THP-1 foam-like cells. (A) and (B). ERK1/2 activation. THP-1 foam cells were treated with 10 μg/mL CRP for different durations. The phosphorylated and total ERK1/2 proteins were detected by the Bio-Plex immunoassay kit (A) and western blot (B). ERK inhibitor PD98059 (20 μM) were used. Data represent mean ± SEM. *P<0.05. n=3. (C) and (D). Cholesterol efflux to apoA-I. Foam cells were treated with 10 μg/mL CRP in the presence or absence of antioxidant SeMet (40 μM), ERK1/2 inhibitor PD98059 (20 μM) or SOD mimetic MnTBAP for 24 hours, followed by an addition of apoA-I (50 μg/mL) for 24 hours. Cholesterol efflux was measured. (E). Effects of MnTBAP on ABCA1 and G1 protein levels measured by western blot analysis. (F). Before treated by 10 μg/mL CRP, THP-1 cells were infected with ADV ERK2 DN or ADV-GFP. Cholesterol efflux was measured. Data represent mean±SEM. *P<0.05 versus CRP treated group, **P<0.001 versus controls or ADV-GFP control. n=6.
Effects of antioxidants and ERK inhibitor on CRP-induced Inhibition of Cholesterol Efflux in THP-1 Cells
To confirm the functional significance of superoxide anion and ERK1/2 in the CRP action, we evaluated the blocking effects of two antioxidants, SeMet and SOD mimetic (MnTBAP), and a specific ERK1/2 inhibitor, PD98059, on the CRP-induced inhibition of cholesterol efflux in THP-1 cells. SeMet (40 μM) significantly increased cholesterol efflux from THP-1 cells to apoA-I by 29% compared with the treatment of 10 μg/mL CRP alone group (P<0.05, n=6, Figure 6C). In a separate experiment, the effect of antioxidant SeMet on the cholesterol efflux from THP-1-derived foam-like cells to HDL was determined. CRP-induced inhibition of cholesterol efflux to HDL was also effectively blocked by antioxidant SeMet (40 μM) (24±1.6% for CRP treatment, and 29±2.8% for co-culture with SeMet) (n=6). Specific ERK1/2 inhibitor PD98059 (20 μM) effectively abolished CRP-induced inhibition of cholesterol efflux from THP-1 cells by 49% compared with CRP alone group (P<0.05, n=6, Figure 6C). CRP-induced inhibition of cholesterol efflux was also significantly abolished by co-incubation with 3 μM MnTBAP (P=0.03, n=6, Figure 6D). Moreover, the effects of antioxidant MnTBAP on the expression of ABCA1 and ABCG1 from THP-1 cells were determined by western blot analysis. Antioxidant MnTBAP effectively blocked CRP-induced downregulation of ABCA1 and ABCG1 in THP-1 foam-like cells (Figure 6E). The involvement of the ERK1/2 pathway in cholesterol efflux in CRP-treated THP-1 cells was also tested with infection of ADV-EKR2 DN. ADV-GFP was used as a control. ADV-ERK2 DN effectively blocked CRP-induced inhibition of cholesterol efflux in THP-1-derived foam-like cells compared with ADV-GFP treated cells (P<0.05, n=6, Figure 6F). These data indicate that oxidative stress and phosphorylation of ERK1/2 play a critical role in CRP-induced cholesterol efflux in macrophages.
Discussion
Both clinical data and basic science studies suggest that CRP may contribute to the progression of atherosclerosis, and the current study provides further mechanisms for the effect of CRP on the vascular system. To our best knowledge, the current study reports three novel findings: 1). CRP inhibits cholesterol efflux from human macrophage-derived foam cells; 2). CRP decreases expression levels of ABCA1 and ABCG1, which are key molecules in mediating cholesterol efflux; and 3). CRP increases superoxide anion production through mitochondrial dysfunction and upregulation of NADPH oxidase subunits. In turn, increased superoxide anion may induce ERK1/2 activation, which may function as a signal transduction pathway for CRP’s action in macrophage-derived foam cells. The inhibition of cholesterol efflux from human macrophage-derived foam cells by CRP could contribute to the accumulation of cholesterol in the foam cells, promoting progression of atherosclerosis on the arterial wall.
Since peripheral cells are unable to catabolize cholesterol, excess cholesterol in these cells is effluxed to extracellular acceptors such as HDL and apoA-I, and is transported to the liver for degradation and excretion, which is referred as reverse cholesterol transport (RCT).12 The first and most likely rate-limiting step of RCT is cholesterol efflux, which removes excess cholesterol from cells and tissues including the arterial wall, thus preventing the development of atherosclerosis.12 The formation of foam cells within the arterial wall is thought to play a central role in the development of atherosclerotic lesions.11 The human monocytic cell line THP-1 have been extensively used in cholesterol efflux to apoA-I or HDL in many studies.13 In the current study, both THP-1 and human PBMC were used to evaluate the effect of CRP on cholesterol efflux to apoA-I or HDL.
Our data show that CRP at the clinical relevant concentrations (10 and 20μg/mL) induced a significant inhibition of cholesterol efflux in THP-1- and human PBMC-derived foam cells compared with controls. There is a concern about the potential contamination of commercial CRP preparations with sodium azide and endotoxin lipopolysaccharide (LPS).18 In the current study, we used the recombinant CRP from CALBIOCHEM containing a low level of sodium azide (0.0005%). In order to rule out the possible effect of sodium azide on THP-1 cells, we have performed a separate experiment by using an azide-free CRP preparation from R & D Systems Inc. Azide-free CRP significantly reduced cholesterol efflux in THP-1-derived foam-like cells, and this effect is similar to the that of CPR preparation from Calbiochem. With regard to LPS, we determined the LPS levels in two commercial sources of CRP preparations (Calbiochem and R & D Systems, Inc.). For CRP (10 μg) from the Calbiochem and the R & D Systems Inc., the LPS level was 0.05 EU and 0.2 EU, respectively.19 The LPS level in both CRP preparations is below 0.5 EU, which is the acceptable LPS content for the animal study.19 We have previously shown that such low endotoxin levels in CRP preparations had no impact on human monocyte derived dendritic cells.19 To support this idea, we have performed two critical experiments. Firstly, anti-CRP antibody effectively blocked CRP-induced decrease of cholesterol efflux. Secondly, heat-inactivated CRP failed to induce the decrease of cholesterol efflux in THP-1 cells. CRP is heat sensitive while LPS is heat resistant.20 Therefore, these data strongly suggest that the inhibition of cholesterol efflux in response to recombinant CRP preparations was specific to CRP action, but not LPS or sodium azide contamination.
ApoA-I and HDL can act as cholesterol acceptors, approach macrophages in subintimal space, and carry cholesterol for excretion. In this pathway, several key molecules including ABCA1, ABCG1, SR-B1, caveolins, and CYP27A1 have been known to mediate cholesterol efflux.8–12 The current study shows that treatment with CRP decreases expression levels of ABCA1 and ABCG1, but not caveolin-1, caveolin-2, and CYO27A1, in THP-1-dervied foam-like cells. These data indicate that CRP may inhibit cholesterol efflux through the down-regulation of ABCA1 and ABCG1 in macrophages.
Increasing evidence suggests that ROS could have direct detrimental effects on atherogenesis and plaque instability, and they may also act as novel signal mediators that regulate signal transduction events including MAPKs in many types of cells such as macrophages.21 The current study demonstrates that CRP is able to increase superoxide anion production in human THP-1-derived foam-like cells. Over-production of superoxide anion may directly contribute to the inhibition of cholesterol efflux because SOD mimetic MnTBAP or antioxidant SeMet can effectively block the effects of CRP on both superoxide production and cholesterol efflux.
ROS are generated mainly from normal metabolism such as mitochondrial respiratory chain wherein excess electrons are donated to molecular oxygen to generate superoxide anion. Other major sources for ROS are enzymes including NADPH oxidase. In the current study, we detected a significant decrease in both the mitochondrial membrane potential and ATP production in CRP-treated THP-1 cells. In addition, treatment with CRP increased expression levels of p22phox, p47phox and p67phox in foam cells. These data indicate that increased superoxide anion production in CRP-treated foam cells may result from dysfunction of mitochondria and upregulation of NADPH oxidase. A transient elevation of ROS could result in the activation of MAPK.22 Indeed, our data show that CRP activates ERK1/2 of THP-1-derived foam-like cells, and ERK1/2 specific inhibitor PD98059 and ADV-ERK2 DN effectively block CRP-induced inhibition of cholesterol efflux in THP-1 cells. Thus, ERK1/2 activation is involved in the action of CRP.
Numerous studies have now emerged in support of the role of CRP in atherogenesis. Current in vitro data support the general hypothesis that CRP contributes to atherogenesis. However, in vivo studies are warranted to further support this important hypothesis. Further studies with appropriate animal models such as CRP knockout or CRP transgenetic mice may elucidate the effects of CRP on reverse cholesterol transport and vascular lesion formation. Indeed, a recent study in ApoE knockout mice showed that CRP accelerated atherosclerosis and endothelial dysfunction.23
In conclusion, accumulation of lipid in macrophages resulting from inhibition of cholesterol efflux could be one of the most important factors contributing to the progression of atherosclerosis. We describe here that the interaction of CRP with cholesterol-loaded macrophages decreases the cholesterol efflux and that downregulation of ABCA1 and ABCG1 in these cells may be responsible for this effect. Furthermore, this study demonstrates a clear link between CRP and in vitro cholesterol efflux, for which the increase of oxidative stress and ERK1/2 activation may be the molecular mechanism. Consequently, antioxidants or ERK1/2 inhibitors may have the potential to prevent CRP-associated cardiovascular disease.
Supplementary Material
Acknowledgments
This work is partially supported by research grants from the National Institutes of Health (Yao: DE15543 and AT003094; and Chen: HL65916, HL72716, EB-002436 and HL083471) and by the Michael E. DeBakey Department of Surgery, Baylor College of Medicine.
References
- 1.Torres JL, Ridker PM. Clinical use of high sensitivity C-reactive protein for the prediction of adverse cardiovascular events. Curr Opin Cardiol. 2003;18:471–478. doi: 10.1097/00001573-200311000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Jialal I, Devaraj S, Venugopal SK. C-reactive protein: risk marker or mediator in atherothrombosis? Hypertension. 2004;44:6–11. doi: 10.1161/01.HYP.0000130484.20501.df. [DOI] [PubMed] [Google Scholar]
- 3.Schwedler SB, Amann K, Wernicke K, Krebs A, Nauck M, Wanner C, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2005;112:1016–1023. doi: 10.1161/CIRCULATIONAHA.105.556530. [DOI] [PubMed] [Google Scholar]
- 4.Calabro P, Willerson JT, Yeh ET. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation. 2003;108:1930–1932. doi: 10.1161/01.CIR.0000096055.62724.C5. [DOI] [PubMed] [Google Scholar]
- 5.Devaraj S, Xu DY, Jialal I. C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: implications for the metabolic syndrome and atherothrombosis. Circulation. 2003;107:398–404. doi: 10.1161/01.cir.0000052617.91920.fd. [DOI] [PubMed] [Google Scholar]
- 6.Han KH, Hong KH, Park JH, Ko J, Kang DH, Choi KJ, et al. C-reactive protein promotes monocyte chemoattractant protein-1--mediated chemotaxis through upregulating CC chemokine receptor 2 expression in human monocytes. Circulation. 2004;109:2566–2571. doi: 10.1161/01.CIR.0000131160.94926.6E. [DOI] [PubMed] [Google Scholar]
- 7.Chang MK, Binder CJ, Torzewski M, Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci U S A. 2002;99:13043–13048. doi: 10.1073/pnas.192399699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fichtlscherer S, Breuer S, Schachinger V, Dimmeler S, Zeiher AM. C-reactive protein levels determine systemic nitric oxide bioavailability in patients with coronary artery disease. Eur Heart J. 2004;25:1412–1418. doi: 10.1016/j.ehj.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 9.Gesquiere L, Loreau N, Minnich A, Davignon J, Blache D. Oxidative stress leads to cholesterol accumulation in vascular smooth muscle cells. Free Radic Biol Med. 1999;27:134–145. doi: 10.1016/s0891-5849(99)00055-6. [DOI] [PubMed] [Google Scholar]
- 10.Qamirani E, Ren Y, Kuo L, Hein TW. C-reactive protein inhibits endothelium-dependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2005;25:995–1001. doi: 10.1161/01.ATV.0000159890.10526.1e. [DOI] [PubMed] [Google Scholar]
- 11.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 12.Ohashi R, Mu H, Wang X, Yao Q, Chen C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM. 2005;98:845–856. doi: 10.1093/qjmed/hci136. [DOI] [PubMed] [Google Scholar]
- 13.Palmer AM, Murphy N, Graham A. Triglyceride-rich lipoproteins inhibit cholesterol efflux to apolipoprotein (apo) A1 from human macrophage foam cells. Atherosclerosis. 2004;173:27–38. doi: 10.1016/j.atherosclerosis.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Klucken J, Buchler C, Orso E, Kaminski WE, Porsch-Ozcurumez M, Liebisch G, et al. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci U S A. 2000;97:817–822. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Mu H, Chai H, Liao D, Yao Q, Chen C. Human immunodeficiency virus protease inhibitor ritonavir inhibits cholesterol efflux from human macrophage-derived foam cells. Am J Pathol. 2007;171:304–314. doi: 10.2353/ajpath.2007.060965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dressman J, Kincer J, Matveev SV, Guo L, Greenberg RN, Guerin T, et al. HIV protease inhibitors promote atherosclerotic lesion formation independent of dyslipidemia by increasing CD36-dependent cholesteryl ester accumulation in macrophages. J Clin Invest. 2003;111:389–397. doi: 10.1172/JCI16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pepys MB, Hawkins PN, Kahan MC, Tennent GA, Gallimore JR, Graham D, et al. Proinflammatory effects of bacterial recombinant human C-reactive protein are caused by contamination with bacterial products, not by C-reactive protein itself. Circ Res. 2005;25:e97–e103. doi: 10.1161/01.RES.0000193595.03608.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang R, Becnel L, Li M, Chen C, Yao Q. C-reactive protein impairs human CD14+ monocyte-derived dendritic cell differentiation, maturation and function. Eur J Immunol. 2006;36:2993–3006. doi: 10.1002/eji.200635207. [DOI] [PubMed] [Google Scholar]
- 20.Blutt SE, Crawford SE, Warfield KL, Lewis DE, Estes MK, Conner ME. The VP7 outer capsid protein of rotavirus induces polyclonal B-cell activation. J Virol. 2004;78:6974–6981. doi: 10.1128/JVI.78.13.6974-6981.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC. Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med. 1993;328:1450–1456. doi: 10.1056/NEJM199305203282004. [DOI] [PubMed] [Google Scholar]
- 22.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 23.Schwedler SB, Kuhlencordt PJ, Ponnuswamy PP, Hatiboglu G, Quaschning T, Widder J, et al. Native C-reactive protein induces endothelial dysfunction in ApoE(−/−) mice: Implications for iNOS and reactive oxygen species. Atherosclerosis. 2007 Jul 30; doi: 10.1016/j.atherosclerosis.2007.06.013. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.