

Abstract
Rationale
The large conductance Ca2+-activated K+ (BK) channel, a key determinant of vascular tone, is regulated by angiotensin II (Ang II) type 1 receptor (AT1R) signaling. Upregulation of Ang II functions and downregulation of BK channel activities have been reported in diabetic vessels. However, the molecular mechanisms underlying Ang II-mediated BK channel modulation, especially in diabetes mellitus, have not been thoroughly examined.
Objectives
The aim in this study was to determine whether caveolae-targeting facilitates BK channel dysfunction in diabetic vessels.
Results
We found that BK channels, AT1R, Gαq/11, non-phagocytic NAD(P)H oxidases (NOX-1) and c-Src kinases (c-Src) were co-localized in the caveolae of rat arterial smooth muscle cells (SMC) and the integrity of caveolae in SMC was critical for Ang II-mediated BK channel regulation. Most importantly, membrane microdomain targeting of these proteins was upregulated in the caveolae of streptozotocin (STZ)-induced rat diabetic vessels, leading to enhanced Ang II-induced redox-mediated BK channel modification and causing BK channel and coronary dysfunction. The absence of caveolae abolished the effects of Ang II on vascular BK channel activity and preserved BK channel function in diabetes.
Conclusion
These results identified a molecular scheme of receptor-enzyme-channel-caveolae microdomain complex, which facilitates the development of vascular BK channel dysfunction in diabetes.
Keywords: BK channel, caveolin-1, Angiotensin II, reactive oxygen species, coronary smooth muscle cells
Diabetic vascular complications account for a 2- to 4-fold increase in the risk of heart attack, heart failure and stroke, causing more than 200,000 deaths per year in the United States. Diabetic patients with acute coronary syndrome have a significant increase in mortality due to poor microcirculation and vascular dysfunction 1. The large conductance Ca2+-activated K+ (BK) channel is an important determinant of vascular tone. Activation of vascular BK channel hyperpolarizes the membrane potential of smooth muscle cells (SMC), closes the voltage-gated Ca2+ channels and produces vasorelaxation. However, BK channel function is impaired in diabetes mellitus due to oxidative stress in the vascular wall with enhanced production of reactive oxygen species (ROS), such as superoxide anion (O2•−), hydrogen peroxide (H2O2) and peroxynitrite (OONO−) 2, 3, accompanied by a decrease in the production and bioavailability of vasodilators including nitric oxide and prostaglandin 4, 5. NAD(P)H oxidase activity is thought to be the major source of O2•− generation in vascular SMC 6, 7. Vascular NAD(P)H oxidases are structurally different from those in phagocytic cells, and the non-phagocytic NAD(P)H oxidases (NOXs) include NOX-1, NOX-4, p22phox, NOXO1 (or p47phox), NOXA1 (or p67phox) and Rac-1 subunits 8. In vessels from patients with diabetes, expression and activity of NOXs are significantly increased, whereas those of antioxidant enzymes are reduced 9, 10. Hence, a misbalance between ROS generation and scavenging represents a fundamental mechanism underlying the development of intracellular oxidative stress in diabetes.
Angiotensin II (Ang II) plays a key role in the regulation of cardiovascular homeostasis through binding to the type 1 (AT1R) and type 2 (AT2R) receptors. AT1R is a G-protein-coupled receptor, activating Gαq and Gβγ. The Gαq-mediated phospholipase C/inositol-1,4,5-triphosphate/Ca2+ signaling activates protein kinase C (PKC) and is a primary mechanism through which Ang II exerts its physiological and pathological effects 11, 12. In addition, Gβγ activates c-Src kinase (c-Src), which in turn activates c-Abl tyrosine kinase, causes tyrosine 14 phosphorylation of caveolin-1 (cav-1) and facilities the AT1R translocation into caveolae 13, 14. Since c-Src is also activated by ROS, these steps result in a self-perpetuated activation loop promoting sustained ROS generation in response to Ang II stimulation. Interestingly, NOX-1 is localized in the caveolae of SMC 15 and activation of AT1R by Ang II is accompanied by receptor translocation into the caveolae of vascular SMC 16. The physiological importance of NOX-1 is underscored by studies using NOX-1 knockout mice, which lack Ang II-induced ROS generation and have reduced blood pressure 14, 17.
Caveolae are unique flask-shaped nonclathrin-coated plasma membrane microdomains, 50–100 nm in diameter, and are characterized by their signature structural protein, caveolin (cav) 18,19. Cav-1 is the primary isoform in vascular SMC. The N-terminus of cav-1 (residue 1-101) contains an important functional structure: the caveolin scaffolding domain (residues 82-101), which is essential for membrane binding and for interaction with signaling proteins that contain the caveolin binding motifs (ΦXXXXΦXXΦ and ΦXΦXXXXΦ, where, Φ represents an aromatic amino acid, X is any amino acid), including those of AT1R signaling proteins 13 and BK channels 20, 21. However, the functional role of caveolae targeting for BK channel regulation is unknown, especially in diabetic vessels.
In this study, we hypothesized that caveolae-Ang II signaling complexes may play an important role in the ROS-associated BK channel modulation, including cysteine oxidation, tyrosine nitration, and tyrosine phosphorylation, leading to vascular BK channel dysfunction in diabetes. We found that BK channels, AT1R, Gαq/11, c-Src and NOX-1 were physically associated in caveolae and the integrity of caveolae in SMC was critical for mediating the regulation of BK channel function by Ang II. In addition, cav-1 expression was upregulated in the vasculature of streptozotocin (STZ)-induced diabetic rats, accompanied by increased physical association between BK channels and AT1R signaling complex, resulting in enhanced AT1R-mediated oxidative modification and dysfunction of BK channels. These results highlight the critical role of caveolae in the inhibition of BK channel function by Ang II, which leads to abnormal vascular function in diabetes.
Methods
An expanded Materials and Methods section is available in the online Data Supplement at http://circres.ahajournals.org.
Type I diabetic animal development and vascular SMC isolation
Diabetic rats and mice were produced by STZ injection. Handling and care of animals, as well as animal procedures, were approved by the Institutional Animal Care the Use Committee, Mayo Clinic.
SMC from rat coronary arteries and mouse aortas were enzymatically isolated as previously reported 22.
Electrophysiology
Whole-cell K+ currents were recorded using standard patch clamp techniques 22 and BK currents were defined as the 0.1 μmol/L iberiotoxin (IBTX)-sensitive component.
Measurement of coronary ring tension
Rat coronary left anterior descending artery was used for contraction and relaxation experiments.
Sucrose gradient density centrifugation
The cellular distribution of cav-1 in rat aortas was determined by sucrose density gradient fractionation as previously described 21. Ten fractions of 1.2-ml each were collected and analyzed by Western blotting.
Cav-1 knockdown by small interfering (si)RNA
Cav-1 in SMC was knocked down using human cav-1 siRNA as previously described 20.
Co-immunoprecipitation and immunoblotting
Immunoprecipitation and western blotting were performed as described previously 2, 23.
Results
Type I diabetic animals
8 weeks after the development of hyperglycemia, blood glucose was significantly increased and body weight was significantly reduced in diabetic animals (see results in online Data Supplement).
Regulation of vascular BK channels by Ang II and caveolae-targeting
We first examined the effects of Ang II on BK channel activities in coronary arterial SMC from control rats. K+ currents were continuously recorded at baseline, after superfusion with Ang II (2 μmol/L) and IBTX (0.1 μmol/L). Ang II produced significant inhibition of K+ currents and this effect was reversible upon washout (Figure 1A). BK currents were obtained by subtracting the IBTX-insensitive K+ components from total K+ currents, and the BK current-voltage (I–V) relationships (holding potential HP=−60 mV, testing potentials TP=−40 mV to +160 mV) before and after exposure to Ang II are shown in Figure 1B. Ang II suppressed BK currents by 50.3% in control rats, from 272.3±26.6 pA/pF at baseline to 135.2±14.2 pA/pF (TP=+150 mV, n=7, p<0.05 vs. baseline).
Figure 1. Caveolae integrity is required for Ang II modulating BK channels of rat coronary arterial SMC.
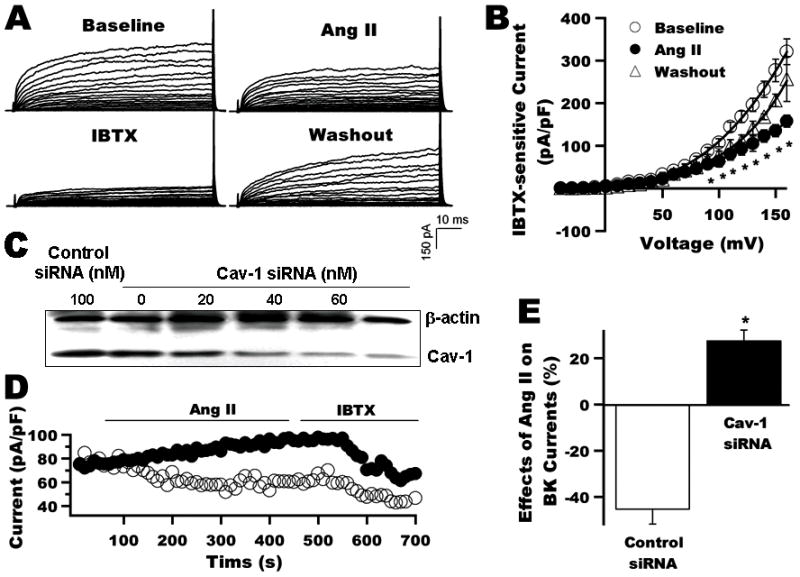
A: Whole-cell K+ currents of rat coronary arterial SMC at baseline, on exposure to 2 μmol/L Ang II and 0.1 μmol/L IBTX and after washout. B: I–V curves of BK currents at baseline, after application of Ang II and washout (n=7). C: Immunoblot shows coronary arterial SMC 48 h after transfection with 0, 20, 40, 60, and 100 nmol/L cav-1 siRNA, and with 100 nmol/L control siRNA. D: Time course of whole-cell K+ currents (HP=-60 mV, TP=+100 mV) before and after the application of 2 μmol/L Ang II and 0.1 μmol/L IBTX. Ang II suppressed the BK currents by about 50% in 100 nmol/L control siRNA treated cells (○), but not in cells treated with 100 nmol/L cav-1 siRNA (●). E: Group results (n=6) were obtained from panel D experiments after the Ang II and IBTX effects have reached steady-state. *: p<0.05 vs. control.
The effect of Ang II on BK channel function was confirmed by outside-out single BK channel recordings (online Figure I in Data Supplement). Extracellular application of 2 μmol/L Ang II, resulted in 49.4 % reduction in channel open probability, from 0.45±0.06 at baseline to 0.23±0.03 with Ang II (p<0.05, n=4).
To determine the role of caveolae in Ang II signal transduction, we examined the effects of cav-1 knockdown in rat coronary arterial SMC using siRNA on BK channel inhibition by Ang II. Figure 1C shows the protein expression of cav-1 in coronary arterial SMC 48 h after transfection with cav-1 siRNA at 0, 20, 40, 60, and 100 nmol/L, and control siRNA at 100 nmol/L. Cav-1 siRNA at 40 nmol/L or higher significantly suppressed cav-1 expression by 80% to 90%, compared with control siRNA. Figure 1D shows the time course of whole-cell K+ currents in rat coronary arterial SMC 48 h after transfection with 100 nmol/L cav-1 siRNA or 100 nmol/L control siRNA, in response to 2 μmol/L Ang II and 0.1 μmol/L IBTX. Ang II suppressed BK currents in cells with control siRNA transfection, but failed to inhibit BK currents in cells with cav-1 siRNA transfection. Instead, a small increase in BK currents was noted. Group data (n=6) are shown in Figure 1E.
Impaired vascular BK channels and coronary vasoreactivity in STZ-induced diabetic rats
To determine the role of AT1R-mediated BK channel regulation in diabetic vessels, we examined the effects of Ang II on BK currents in coronary arterial SMC from STZ-induced diabetic rats. Whole-cell K+ currents were 61.5±10.6 pA/pF at baseline (TP=+150 mV, n=8, p<0.05, vs. control baseline) and 61.2±15.7 pA/pF with 2 μmol/L Ang II (n=8, p=N.S. vs. diabetic baseline), indicating the loss of Ang II effect on vascular BK currents in diabetes. Representative tracings and the BK channel I-V curves before and after exposure to Ang II are shown in Figure 2A. Very little BK currents were present in diabetic coronary arterial SMC and Ang II effects were absent, indicating marked vascular BK channel dysfunction in diabetes.
Figure 2. Impaired BK channel function and vasoreactivity of coronary arteries in STZ-induced diabetic rats.
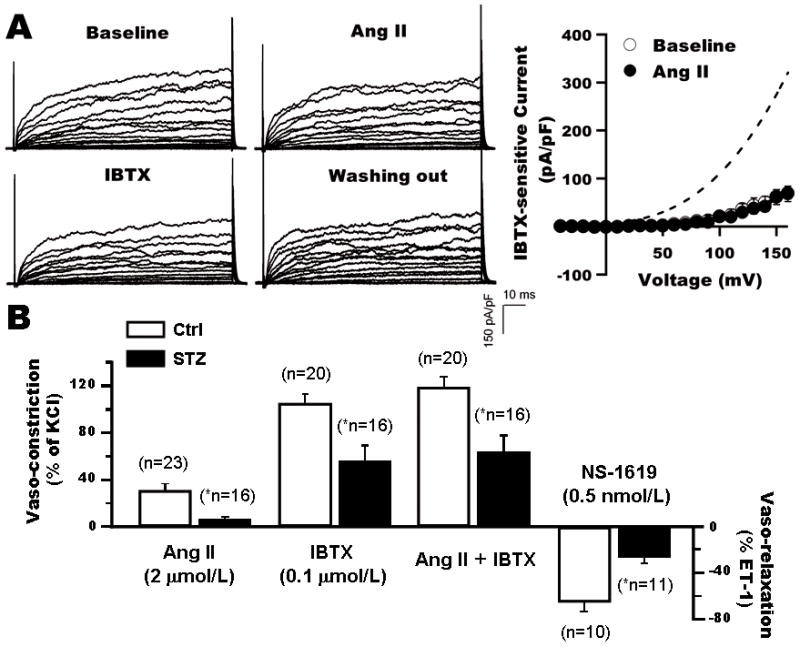
A: Whole-cell K+ currents and BK channel I–V curves from diabetic rats at baseline, after the application of Ang II, IBTX, and washout of chemicals. BK currents in diabetic SMC was significantly reduced with loss of the Ang II effects. Dashed line indicates the I–V curve at baseline of control rats. B: Impaired reactivity of coronary arteries from STZ-induced diabetic rat in response to Ang II, IBTX and NS-1619. *: p<0.05 vs. control.
To determine the physiological relevance of these findings, the effects of Ang II and NS-1619 (BK channel specific activator) on the contraction/relaxation of coronary rings from control and STZ-induced diabetic rats were measured. We found that in diabetic rats, coronary constriction by Ang II (2 μmol/L) and by IBTX (0.1 μmol/L) was reduced by 76.6% and 46.2% respectively; whereas NS-1619-mediated coronary relaxation was reduced by 59.0% (Figure 2B). These results suggest that BK channel-mediated coronary vasoreactivity was abnormal in diabetes.
Co-localization of BK channels, AT1R, Gαq/11, NOX-1 and c-Src in the caveolae of vascular SMC
To better understand the molecular mechanism whereby caveolae-targeting modulates BK channel activity, we determined the cellular distribution of BK channels, AT1R, Gαq/11, NOX-1 and c-Src in vessels from control and STZ-induced diabetic rats by sucrose density gradient fractionation. Since the BK channels in aortic SMC and coronary arterial SMC showed similar abnormalities in STZ-induced diabetic rats (online Figure II in Data Supplement), we used the aorta for further biochemical characterization. Figures 3A and 3B show immunoblots of the cell lysates and the fractions (1 to 10, 1 being the lightest and 10 the heaviest) of control and diabetic rat aortas respectively, blotted against anti-cav-1, anti-BK channel, anti-c-Src, anti-NOX-1, anti-Gαq/11 and anti-AT1R antibodies. BK channels, NOX-1, Gαq/11 and c-Src were detected in the low buoyant density, caveolae-rich fractions of control and diabetic rat aortas. In contrast, very little AT1R was detected in the caveolae-rich fractions of both control and diabetic rats under baseline condition. However, aortas obtained from animals after treatment with Ang II showed that AT1R was readily detected in the low buoyant density fractions, suggesting receptor translocation into caveolae upon agonist activation, similar to previous reports 16. Analysis of the distribution of AT1R in the membrane fractions showed that it was very different between control and STZ-induced diabetic rats after Ang II treatment. In control rats only 28.5% of the total AT1R was in the low buoyant density fractions (fractions 1 to 6), whereas 83.4% of the total AT1R was found in the low buoyant density fractions of STZ-induced diabetic rats (Figure 3C). These results suggest that caveolae targeting of AT1R is enhanced in diabetic vessels.
Figure 3. Co-localization of BK channels with AT1R and its signaling proteins in rat aortic SMC caveolae.
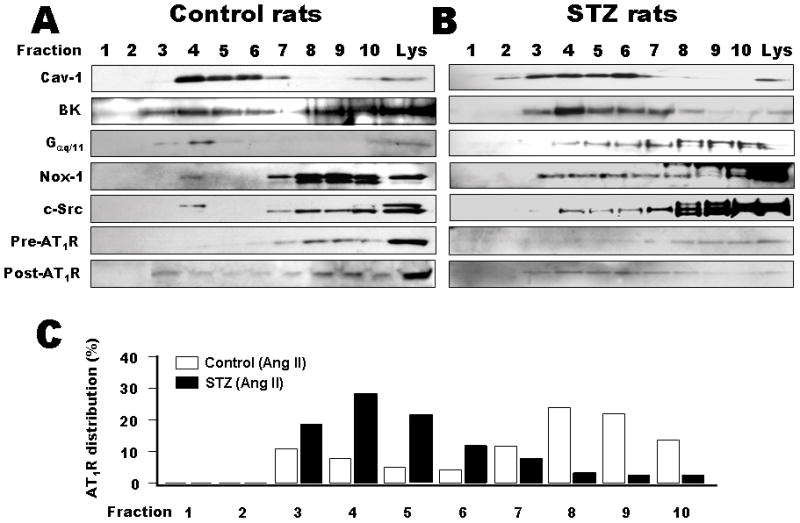
Distribution of cav-1, AT1R, BK channels, c-Src, NOX-1 and Gαq/11 in control (A) and diabetic rat aortas (B) by density gradient fractionation analysis with 1 being the lightest and 10 the heaviest fraction. Immunoblotting of the fractions against anti-BK channel, anti-AT1R, anti-c-Src, -NOX-1 and -Gαq/11 antibodies showed their presence in the low buoyant density, caveolae-rich fractions. There was very little AT1R in the caveolae-rich fractions in control and diabetic rats at baseline (pre-Ang II), but their presence was readily detected after treatment with Ang II (post-Ang II). The patterns of AT1R distribution in the density gradient fractions are compared between control and diabetic rat aortas (C). In control rats, only 28.5 % of the total AT1R translocated to the low buoyant density fractions (No. 1 to 6), upon agonist stimulation, whereas 83.4% of the AT1R did so in diabetic rats.
Inhibition of BK channel activity by Ang II-induced post-translational modulation
We proceeded to determine the roles of PKC, NOX-1 and c-Src on the Ang II-mediated development of BK channel dysfunction. We found that after a 1-h incubation with the membrane permeable PKC peptide inhibitor (50 μmol/L), BK currents in coronary arterial SMC were no longer suppressed by Ang II. BK current densities were 229.9±67.5 pA/pF at baseline and 274.8±64.5 pA/pF (TP=+150 mV, n=10, p=N.S. vs. baseline) after exposure to 2 μmol/L Ang II (Figure 4A). Similarly, a 1-h pretreatment with the c-Src inhibitor Lavendustin A (LavA, 10μmol/L) or with the NOX-1 inhibitor diphenylene iodonium (DPI, 20 μmol/L) abolished the Ang II effects on BK channel activity. After pretreatment with LavA, BK current densities were 210.1±58.8 pA/pF at baseline vs. 249.0±67.2 pA/pF (n=11, p=N.S. vs. baseline) with Ang II (Figure 4B). After pretreatment with DPI, BK currents were 180.7±31.3 pA/pF at baseline vs. 182.0±42.3 pA/pF (TP=+150 mV, n=8, p=N.S.) with Ang II (Figure 4C). In contrast, pretreatment with 10 μmol/L Lavendustin B (LavB, the negative control of LavA) did not inhibit the Ang II effects (data not shown). Hence, these results suggest that Ang II inhibits BK channels through PKC-, c-Src- and NOX-mediated mechanisms.
Figure 4. Effects of inhibitors of PKC, c-Src, and NOX-1 on the inhibition of BK channels by Ang II.
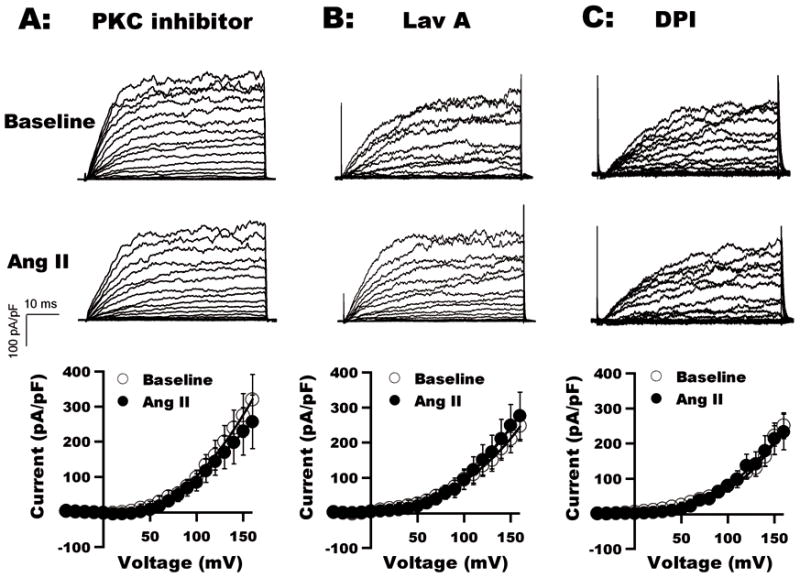
BK currents in coronary arterial SMC after a 1-h incubation with membrane permeable PKC peptide inhibitor (A), LavA (B) and DPI (C) at baseline and on exposure to 2 μmol/L Ang II. The Ang II effects were abrogated by these inhibitors. I–V curves from group results (n=8 to 10, p=N.S. vs. baseline) are shown.
Comparison of BK channel AT1R and cav-1 expression in the aortas between normal and STZ-induced diabetic rats
We determined the expression of BK channels, AT1R and cav-1 in diabetic vessels. Figure 5A shows the immunoblot of aortic homogenates from control and diabetic rats against anti-BK channel, anti-AT1R and anti-cav-1 antibodies, as well as anti-β-actin antibodies as loading control. There was no significant difference in BK channel and AT1R expression between control and diabetic rats, but cav-1 expression was increased by 3.1±0.2 fold (n=3, p<0.05 vs. control) in diabetic rats. These results suggest that reduction of BK channel activity in diabetic vessels was not due to down-regulation of channel expression, but was associated with altered channel function in the presence of increased caveolae abundance.
Figure 5. Upregulation of cav-1 expression and caveolae-targeting in the aortas of STZ-induced diabetic rats.
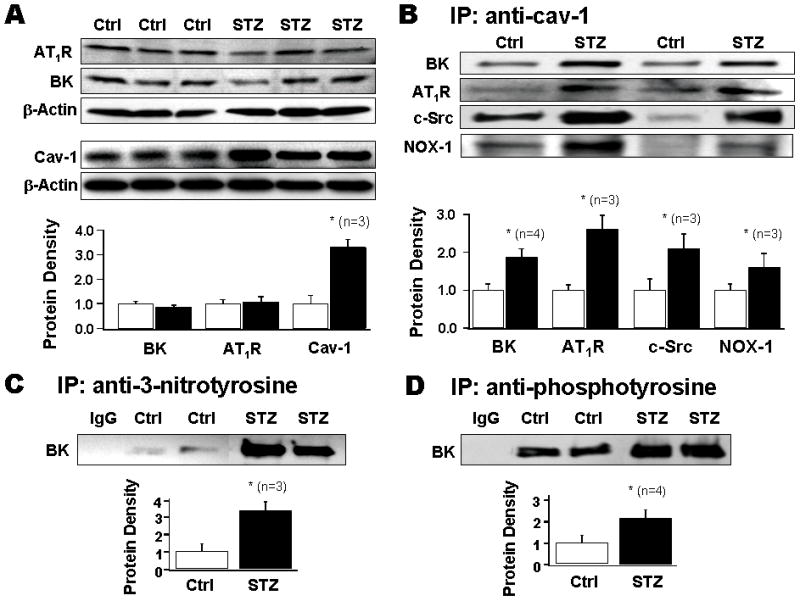
A: Immunoblotting against BK channels, AT1R and cav-1 from three pairs of aortas from control and diabetic rats. Cav-1 expression was significantly increased but not BK channel or AT1R. B: Immunoprecipitates (IPs) of anti-cav-1 antibody on aorta homogenate from control and diabetic rats were analyzed using anti-AT1R, anti-NOX-1, anti-c-Src antibodies. C: IPs of anti-3-nitrotyrosine antibody were blotted against anti-BK channel antibody. D: IPs of anti-phosphotyrosine antibody were blotted against anti-BK channel antibody.
Enhanced caveolae-targeting of BK channels, AT1R, NOX-1 and c-Src and oxidative modification of BK channels in diabetic vessels
Figure 5B shows results from two representative pairs of control and STZ-induced diabetic rat aorta homogenates, in which immunoprecipitates with anti-cav-1 antibody were analyzed for the presence of BK channels, c-Src, and NOX-1. Cav-1-associated BK channels, AT1R, c-Src and NOX-1 were significantly increased in diabetic rats, by 1.9±0.2 (n=4), 2.6±0.4 (n=3), 2.1±0.4 (n=3) and 1.6±0.1 fold (n=3) respectively, compared to control rats.
Since ROS are prominent products of AT1R signaling and the machinery for producing oxidative modulation are in the vicinity of BK channels in caveolae of SMC, we examined the potential consequence of the augmented caveolae-mediated association between BK channels and the AT1R signaling cascade in diabetes by determining BK channel oxidative modification. We found that in diabetic vessels, there was a 3.3±0.4 fold (n=3, p<0.05 vs. control) increase in the BK channel tyrosine nitration (Figure 5C), and a 2.1±0.3 fold (n=4, p<0.05 vs. control) increase in BK channel tyrosine phosphorylation (Figure 5D). These results suggest that the enhanced caveolae-targeting of BK channels and AT1R signaling molecules in diabetic vessels may underlie the enhanced BK channel protein post-translational oxidative modification, accounting for the molecular mechanisms of BK channelopathy in diabetes.
Preserved BK channel activity in cav-1 knockout (KO) diabetic mice
To determine the role of caveolae on BK channel function in control and diabetes, we used cav-1 KO mice for further studies. Figures 6A and 6B show the time course and representative tracings of K+ currents in aortic SMC from non-diabetic WT and KO mice at baseline, after exposure to Ang II and to IBTX, and upon washout of chemicals. The I–V curves of BK currents before and after the application of 2 μmol/L Ang II are illustrated in Figure 6C. Ang II produced 50% inhibition of BK currents in WT mice; whereas there was no Ang II effect in cav-1 KO mice. The results are similar to those from rat coronary arterial SMC after cav-1 siRNA treatment. In diabetic WT mice, vascular BK channels showed very little response to Ang II (Figure 7). The current density was 27.2±6.9 pA/pF at baseline and was 26.8±12.8 pA/pF after exposure to 2 μmol/L Ang II (TP=+150 mV, n=9, p=N.S vs. baseline). In diabetic cav-1 KO mice, BK channels were also insensitive to Ang II; BK current density was 155.5±28.3 pA/pF at baseline (TP=+150 mV, n=9, p<0.05 vs. WT) and 133.1± 30.7 pA/pF after Ang II treatment (TP=+150 mV, n=9, p=N.S vs. baseline). These results indicate that in diabetes there was profound loss of vascular BK channel activity with no further suppression by Ang II. However, in KO mice, vascular BK channel function is preserved in diabetes because the absence of caveolae spared the channels from Ang II-mediated inactivation.
Figure 6. Effects of Ang II on BK currents in aortic SMC from WT and cav-1 KO mice.
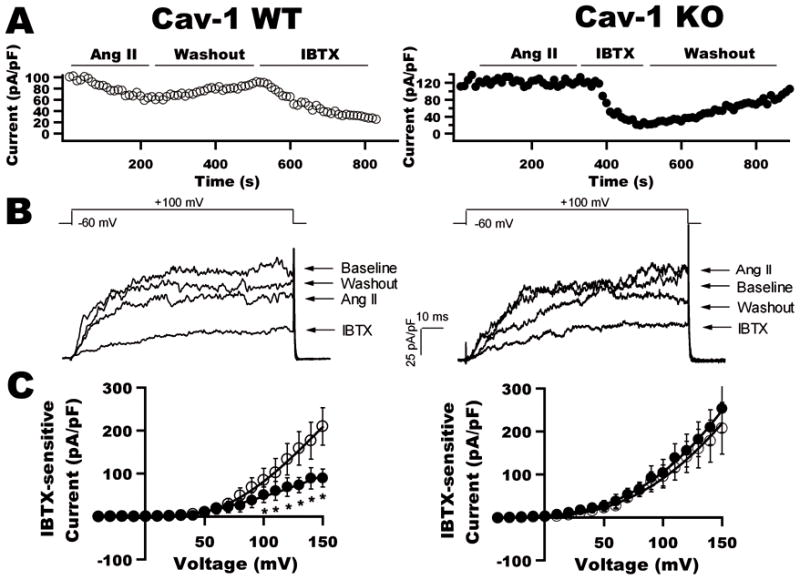
A: Time course of BK currents in aortic SMC from WT and cav-1 KO mice at baseline and after exposure to 2 μmol/L Ang II, and to 0.1 μmol/L IBTX. B: K+ currents in aortic SMC from WT and KO mice at each phase of the experiment in A. Ang II had no effect on BK currents in KO mice. C: I–V curves of BK currents in WT and KO mice before (○) and after (●) the application of Ang II (n=10 for each group).
Figure 7. Effects of Ang II on BK channels of aortic SMC from STZ-induced diabetic WT and cav-1 KO mice.
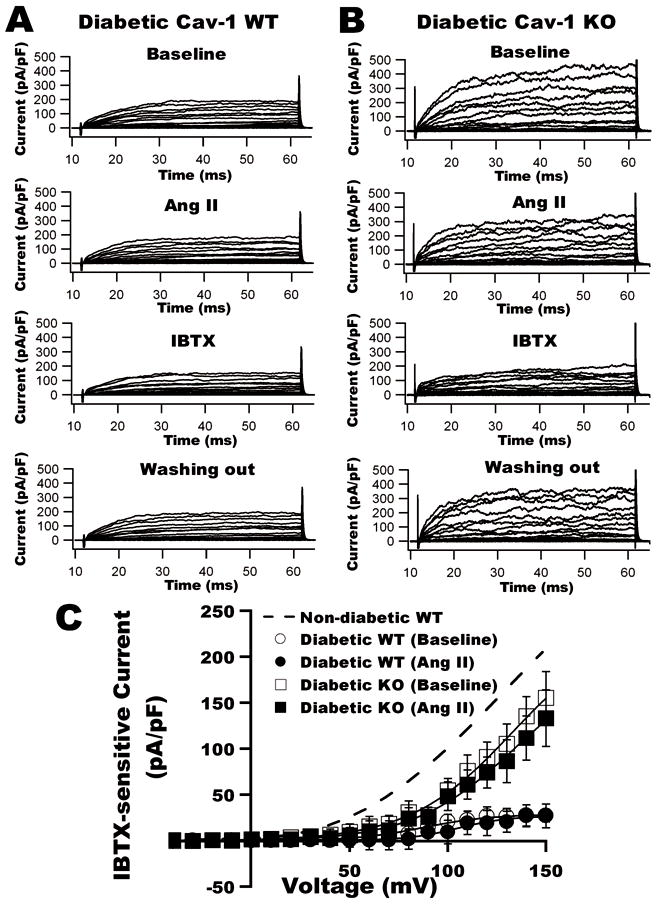
A: Whole-cell K+ currents in aortic SMC from diabetic WT (left) and KO mice (right) at baseline, after the application of Ang II, IBTX and washout of chemicals. B: I–V curves of BK currents in diabetic WT and KO mice before and after exposure to Ang II. Dashed line represents the BK current density in WT control.
Discussion
In this study, we have provided compelling evidence on the critical role of caveolae in mediating the inhibition of vascular BK channels by Ang II. First, we have shown that BK channels and the AT1R signaling cascade, including Gαq/11, c-Src and NOX-1, are co-localized in the caveolae of vascular SMC. Ang II inhibits BK channel function through activation of its downstream pathways. Second, Ang II loses its effects on BK channel function with lack of caveolae by cav-1 knockdown using siRNA in vascular SMC and by cav-1 gene ablation in cav-1 KO mice. Third, coronary arterial BK channel activity and coronary vasoreactivity are impaired in STZ-induced diabetic rats. With the development of diabetes, cav-1 expression is up-regulated and this is accompanied by increased oxidative modification of BK channels by tyrosine phosphorylation and tyrosine nitration. Fourth, the absence of caveolae preserves BK channel function in diabetes. These results indicate that vascular BK channel functions are importantly modulated by Ang II-AT1R signaling and caveolae targeting is critical in facilitating the Ang II-AT1R-mediated effects. BK channel function is significantly compromised by the heightened Ang II-AT1R-caveolae oxidative stress signaling in diabetes.
Caveolae have emerged as important membrane microdomains where signal transduction mechanisms are facilitated because a wide variety of signaling molecules are found to reside in caveolae of vascular SMC, including BK channels and AT1R signaling proteins. However, the functional consequence of caveolae targeting and the molecular mechanisms of caveolae-mediated modulation of vascular BK channel function are unclear. In cultured bovine aortic endothelial cells, BK channels are quiescent but can be activated upon isoproterenol stimulation or by dissolution of caveolae using β-cyclodextrin 20. Based on BK current measurements in inside-out macropatches with similar pipette resistance (assuming that each one has a similar surface area of pipette tip), Alioua et al. 21 reported that cav-1 affects Slo surface expression. Specifically, co-expression with cav-1 in HEK293 cells reduced Slo currents (in nA*MΩ) by 70% while the channel sensitivity to voltage- and Ca2+ was unaltered. Further deletion of the YNMLCFGIY caveolin binding motif in Slo abolished channel surface expression. Using whole-cell recording technique, however, we did not find any difference in IBTX-sensitive K+ currents (in pA/pF) in SMC between WT and cav-1 KO mice. These results suggest that the mechanism of BK channel modulation by cav-1 in native vascular SMC may be different from those in heterologous expression systems.
A key finding in this study is that caveolae integrity is crucial for AT1R-mediated BK channel regulation. Inhibition of BK channels by Ang II was abolished in rat coronary SMC with cav-1 knock-down and in aortic SMC of cav-1 KO mice. Our finding that targeting of AT1R to vascular caveolae requires agonist stimulation suggests the presence of an elegant balance and control mechanism that precludes nonspecific incidental activation of AT1R signaling cascade, consistent with previous observations 16. Furthermore, our finding that BK channels and AT1R, signaling proteins are co-localized in the caveolae of SMC, demonstrated by sucrose density gradient fractionation, confocal imaging analysis (online Figure III in Data Supplement), and by co-immunoprecipitation with cav-1, carries physiological significance, as such an organization brings BK channels to the vicinity and in close proximity with c-Src, PKC and NOX-1 which are downstream effectors of AT1R signaling. Stimulation of AT1R by Ang II activates two major downstream pathways with activation of c-Src and PKC, which would lead to activation of NOXs. NOX-1 and NOX-4 are the major NOX isoforms in human coronary arterial SMC and they have distinct subcellular distributions with NOX-1 localized in caveolae and NOX-4 in the cytoplasm 15. Most importantly, we found a 3.1-fold increase in cav-1 expression in diabetic rat aortas, similar to previous report 24. Furthermore, upon Ang II stimulation, 83.4% of the total AT1R moved to the low buoyant density fractions in diabetic rats, compared to 28.5% in control rats. Such cellular remodeling has contributed a 1.6- to 2.6-fold increase in the abundance of BK channels, AT1R, NOX-1 and c-Src in caveolae.
Ang II-mediated effects are dependent on ROS generation and the renin-angiotensin system is activated in diabetes 25. Yoshimoto et al. 26 reported that a 2-h incubation with 10 μmol/L Ang II produced a significant increase in ROS generation and NOX-1 expression in SMC of rat aortas. We have confirmed that production of intracellular ROS in cultured human coronary SMC is significantly reduced after treatment with a NOX inhibitor, but not with a mitochondrial electron transport complexes II inhibitor (online Figure IV in Data Supplement). Moreover, incubation with 2 μmol/L Ang II significantly increases ROS generation in freshly isolated aortic SMC of WT mice, but not cav-1 KO mice (online Figure V in Data Supplement). These results suggest that in arterial SMC, caveolae-associated NOX-1 constitutes the major source of intracellular O2•− generation in response to AT1R stimulation. However, the elevated activities of the AT1R signaling cascade in diabetes and the proximity to ROS generating enzymes has rendered BK channels particularly vulnerable to redox modulation. We have previously reported hSlo expressed in HEK293 cells in the presence of high glucose is susceptible to the inhibitory modulation by H2O2 and OONO− 2. In addition, O2•− could further enhance c-Src activity 27, leading to channel tyrosine phosphorylation, and O2•− could also react with NO to generate ONOO−, resulting in channel tyrosine nitration 28. Tyrosine phosphorylation and nitration are two important mechanisms of protein post-translational modification. Indeed, there is 2.1- and 3.3-fold increase in BK channel tyrosine phosphorylation and tyrosine nitration respectively in STZ-induced diabetic rat arteries. ONOO− is known to directly inhibit BK channel function 2, 29, and we found that BK channel activity in coronary arterial SMC was lost after incubation with 0.1μmol/L ONOO− for 2 h (data not shown), a time when protein nitration has reached a maximal effect 28. Incubation with LavA abolished the Ang II effects on BK currents, which is in agreement with Alioua’s report 30. With the upregulation of cav-1 expression and increased localization of BK channels and AT1R signaling proteins in the caveolae of diabetic vessels, we believe that Ang II-mediated ROS generation via caveolae-associated NOX-1 activation plays a central role in BK channel regulation. In contrast, the absence of caveolae prevents the deleterious effects of Ang II on BK channel activities in diabetes.
Our study has potential limitations. First, density gradient fractionation and immunoprecipitation experiments require a lot of proteins and these experiments were done using aortas instead of coronary arteries. Second, BK channel activity in WT and cav-1 KO mice was characterized in aortic SMC. However, we have shown that Ang II-BK channel regulation is the same in SMC from rat coronaries, rat aortas, and mouse aortas. Hence, we believe the conclusions derived from these experiments are valid as they all bear the same results.
In summary, we have shown that BK channels and AT1R signaling proteins are co-localized in vascular caveolae microdomains and caveolae-targeting is critical for mediating the regulation of BK channel function by Ang II. In Figure 8, we present a working model to illustrate that caveolae of vascular SMC facilitate the assembly of BK channels and AT1R signaling proteins into a molecular complex, leading to increase of BK channel post-translational modulation Hence, our results delineated a molecular mechanism through which caveolae microdomain organization facilitates the inhibition of BK channel function in diabetic vessels, which in turn regulates coronary blood flow and may affect the clinical outcome of diabetic patients with acute coronary syndrome.
Figure 8. Model of caveolae facilitating Ang II-induced vascular BK channel dysfunction in diabetes.
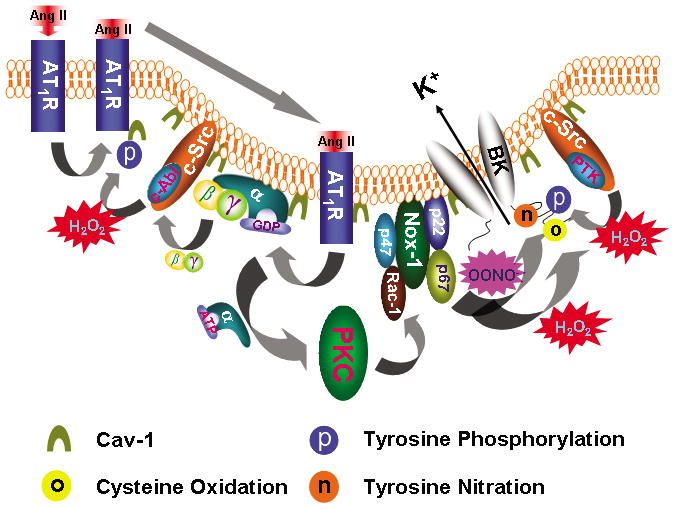
Upon Ang II stimulation, AT1R translocates into caveolae, which requires c-Abl-mediated cav-1 tyrosine 14 phosphorylation. In caveolae, AT1R interacts with Gαq that activates the PKC and enhances NOX-1 complex activity through p47phox phosphorylation and Rac-1 phosphorylation. Binding of Ang II to AT1R releases Gβγ which activates c-Src/c-Abl, resulting in increase of cav-1 tyrosine 14 phosphorylation and AT1R translocation. ROS enhances c-Src/protein tyrosine kinase (PTK) activity and inhibits protein tyrosine phosphatase (PTP) activity, which leads to increase in protein tyrosine phosphorylation. Both H2O2 and OONO− directly cause BK channel cysteine oxidation and OONO− produces BK channel tyrosine nitration. These mechanisms of Ang II-induced oxidative post-translational modification impair vascular BK channel function.
Novelty and Significance.
What is known?
Caveolae provide a central platform for signaling transduction, including that of AT1R signaling.
The effects of Ang II are mediated through reactive oxygen species generation.
Vascular large conductance Ca2+-activated K+ (BK) channel activity is impaired in diabetes, contributing to diabetic vascular dysfunction.
What new information does this article contribute?
BK channels are colocalized with AT1R and its signaling proteins such as PKC, NOX-1 and c-Src in the caveolae of vascular smooth muscle cells, forming a channel-receptor-enzyme-caveolae microdomain complex.
Cav-1 expression is upregulated in diabetic vessels with enhanced BK channel-AT1R signaling caveolae targeting, resulting in increased redox-mediated BK channel modification.
Cav-1 gene ablation protects vascular BK channel function in diabetes.
This is the first report that caveolae-Ang II signaling participates in vascular BK channel regulation and facilitates BK channel and coronary dysfunction in diabetes. Our results delineate a fundamental mechanism underlying diabetic vascular dysfunction and may help to establish the BK channels as a therapeutic target in the treatment of diabetic vascular complications.
Supplementary Material
Acknowledgments
Sources of Funding
This work is supported by grants from American Diabetes Association (ADA-JFA-07-39) and the National Institutes of Health (HL74180 and HL080118).
Non-standard Abbreviations and Acronyms
- Ang II
angiotensin II
- AT1R
angiotensin II type 1 receptor
- BK channel
the large conductance Ca2+-activated K+ channel
- Cav-1
caveolin-1
- c-Src
c-Src kinase
- Gαq/11
G protein q/11 alpha subunit
- HP
holding potential
- IP
immunoprecipitate
- IBTX
iberiotoxin
- NOX-1
non-phagocytic NAD(P)H oxidase-1
- PKC
protein kinase C
- ROS
reactive oxygen species
- Peroxynitrite
OONO−, STZ, streptozotocin
- SMC
smooth muscle cells
- TP
testing potential
Footnotes
Disclosures
None.
References
- 1.Kurisu S, Inoue I, Kawagoe T, Ishihara M, Shimatani Y, Nishioka K, Umemura T, Nakamura S, Yoshida M. Diabetes mellitus is associated with insufficient microvascular reperfusion following revascularization for anterior acute myocardial infarction. Intern Med. 2003;42:554–559. doi: 10.2169/internalmedicine.42.554. [DOI] [PubMed] [Google Scholar]
- 2.Lu T, He T, Katusic ZS, Lee HC. Molecular mechanisms mediating inhibition of human large conductance Ca2+-activated K+ channels by high glucose. Circ Res. 2006;99:607–616. doi: 10.1161/01.RES.0000243147.41792.93. [DOI] [PubMed] [Google Scholar]
- 3.Brzezinska AK, Gebremedhin D, Chilian WM, Kalyanaraman B, Elliott SJ. Peroxynitrite reversibly inhibits Ca2+-activated K+ channels in rat cerebral artery smooth muscle cells. Am J Physiol. 2000;278:H1883–1890. doi: 10.1152/ajpheart.2000.278.6.H1883. [DOI] [PubMed] [Google Scholar]
- 4.Pieper GM. Superoxide dismutase plus catalase improves post-ischaemic recovery in the diabetic heart. Cardiovasc Res. 1988;22:916–926. doi: 10.1093/cvr/22.12.916. [DOI] [PubMed] [Google Scholar]
- 5.Lu T, Wang XL, He T, Zhou W, Kaduce TL, Katusic ZS, Spector AA, Lee HC. Impaired arachidonic acid-mediated activation of large-conductance Ca2+-activated K+ channels in coronary arterial smooth muscle cells in Zucker Diabetic Fatty rats. Diabetes. 2005;54:2155–2163. doi: 10.2337/diabetes.54.7.2155. [DOI] [PubMed] [Google Scholar]
- 6.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 7.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 8.Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun. 2005;338:677–686. doi: 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- 9.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 10.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 11.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- 12.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 13.Ushio-Fukai M, Alexander RW. Caveolin-dependent angiotensin II type 1 receptor signaling in vascular smooth muscle. Hypertension. 2006;48:797–803. doi: 10.1161/01.HYP.0000242907.70697.5d. [DOI] [PubMed] [Google Scholar]
- 14.Basset O, Deffert C, Foti M, Bedard K, Jaquet V, Ogier-Denis E, Krause KH. NAD(P)H oxidase 1 deficiency alters caveolin phosphorylation and angiotensin II receptor localization in vascular smooth muscle. Antioxid Redox Signal. 2009;11:2371–2384. doi: 10.1089/ars.2009.2584. [DOI] [PubMed] [Google Scholar]
- 15.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscle Thromb Vacs Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 16.Zuo L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW. Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after angiotensin II type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arterioscler Thromb Vasc Biol. 2005;25:1824–1830. doi: 10.1161/01.ATV.0000175295.09607.18. [DOI] [PubMed] [Google Scholar]
- 17.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS letters. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 18.Krajewska WM, Maslowska I. Caveolins: structure and function in signal transduction. Cell Mol Biol Lett. 2004;9:195–220. [PubMed] [Google Scholar]
- 19.Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes. 2004;53:1261–1270. doi: 10.2337/diabetes.53.5.1261. [DOI] [PubMed] [Google Scholar]
- 20.Wang XL, Ye D, Peterson TE, Cao S, Shah VH, Katusic ZS, Sieck GC, Lee HC. Caveolae targeting and regulation of large conductance Ca2+-activated K+ channels in vascular endothelial cells. J Biol Chem. 2005;280:11656–11664. doi: 10.1074/jbc.M410987200. [DOI] [PubMed] [Google Scholar]
- 21.Alioua A, Lu R, Kumar Y, Eghbali M, Kundu P, Toro L, Stefani E. Slo1 caveolin-binding motif, a mechanism of caveolin-1-Slo1 interaction regulating Slo1 surface expression. J Biol Chem. 2008;283:4808–4817. doi: 10.1074/jbc.M709802200. [DOI] [PubMed] [Google Scholar]
- 22.Lu T, Ye D, Wang X, Seubert JM, Graves JP, Bradbury JA, Zeldin DC, Lee HC. Cardiac and vascular KATP channels in rats are activated by endogenous epoxyeicosatrienoic acids through different mechanisms. J Physiol. 2006;575:627–644. doi: 10.1113/jphysiol.2006.113985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liou JY, Deng WG, Gilroy DW, Shyue SK, Wu KK. Colocalization and interaction of cyclooxygenase-2 with caveolin-1 in human fibroblasts. J Biol Chem. 2001;276:34975–34982. doi: 10.1074/jbc.M105946200. [DOI] [PubMed] [Google Scholar]
- 24.Lam TY, Seto SW, Lau YM, Au LS, Kwan YW, Ngai SM, Tsui KW. Impairment of the vascular relaxation and differential expression of caveolin-1 of the aorta of diabetic +db/+db mice. Eur J Pharmacol. 2006;546:134–141. doi: 10.1016/j.ejphar.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Connelly KA, Boyle AJ, Kelly DJ. Angiotensin II and the cardiac complications of diabetes mellitus. Curr Pharm Des. 2007;13:2721–2729. doi: 10.2174/138161207781662984. [DOI] [PubMed] [Google Scholar]
- 26.Yoshimoto T, Fukai N, Sato R, Sugiyama T, Ozawa N, Shichiri M, Hirata Y. Antioxidant effect of adrenomedullin on angiotensin II-induced reactive oxygen species generation in vascular smooth muscle cells. Endocrinology. 2004;145:3331–3337. doi: 10.1210/en.2003-1583. [DOI] [PubMed] [Google Scholar]
- 27.Aslan M, Ozben T. Oxidants in receptor tyrosine kinase signal transduction pathways. Antioxid Redox Signal. 2003;5:781–788. doi: 10.1089/152308603770380089. [DOI] [PubMed] [Google Scholar]
- 28.Herrero MB, de Lamirande E, Gagnon C. Tyrosine nitration in human spermatozoa: a physiological function of peroxynitrite, the reaction product of nitric oxide and superoxide. Mol Hum Reprod. 2001;7:913–921. doi: 10.1093/molehr/7.10.913. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Terata K, Chai Q, Li H, Kleinman LH, Gutterman DD. Peroxynitrite inhibits Ca2+-activated K+ channel activity in smooth muscle of human coronary arterioles. Circ Res. 2002;91:1070–1076. doi: 10.1161/01.res.0000046003.14031.98. [DOI] [PubMed] [Google Scholar]
- 30.Alioua A, Mahajan A, Nishimaru K, Zarei MM, Stefani E, Toro L. Coupling of c-Src to large conductance voltage- and Ca2+-activated K+ channels as a new mechanism of agonist-induced vasoconstriction. Proc Natl Acad Sci U S A. 2002;99:14560–14565. doi: 10.1073/pnas.222348099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.