

Abstract
The discordance in results of independent genome-wide association studies (GWAS) indicates the potential for Type I and Type II errors. We assessed the repeatibility of current Affymetrix technologies that support GWAS. Reasonable reproducibility was observed for both raw intensity and the genotypes/copy number variants. We also assessed consistencies between different SNP arrays and between genotype calling algorithms. We observed that the inconsistency in genotypes was generally small at the specimen level. To further examine whether the differences from genotyping and genotype calling are possible sources of variation in GWAS results, an association analysis was applied to compare the associated SNPs. We observed that the inconsistency in genotypes not only propagated to the association analysis, but was amplified in the associated SNPs. Our studies show that inconsistencies between SNP arrays and between genotype calling algorithms are potential sources for the lack of reproducibility in GWAS results.
Keywords: repeatability, association, genotype, calling algorithm, intensity, copy number
Introduction
Genome-wide association studies (GWAS) aim to identify genetic variants across the human genome that might be associated with phenotypic traits. The flourishing of GWAS1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 makes the technology a promising field of research. However, replication studies show that only a small portion of associated loci in the initial GWAS can be replicated, even within the same populations. For example, in replication studies of GWAS for type 2 diabetes mellitus, Zeggini et al.5 replicated associations for only 10 out of 77 SNP-based loci tested, Scott et al.6 10 out of 80, Easton et al.8 8 out of 57, and Steinthorsdottir et al.16 2 out of 47. Moreover, lists of associated SNPs identified in different GWAS for a disease, such as type 2 diabetes mellitus, can be quite different. Though the differences might be explained by population structure, they might also be due to technical biases, or both.
Given the complexity of GWAS, multiple sources of Type I (false positive) and Type II (false negative) errors exist. GWAS are based on the common trait-common variant hypothesis, which implies that the genetic architecture of complex traits consists of a number of common alleles, each conferring a small increase in risk to the individual.31 Therefore, the likelihood of detecting an individual SNP association is usually small and requires a large sample size to achieve adequate statistical power to detect true associations. The selection of participants for GWAS is an additional potential source of variability because of inaccurate participant ascertainment, biased selection of cases or controls, and population stratification. Case–control misclassification can reduce study power and result in spurious associations.32 Non-genetic covariates (for example smoking33 and obesity3), when confounded with outcome, also generate Type I errors. Population stratification inflates the Type I error rate around variants that are informative about the population substructure,34 but its influence is a matter of debate.35, 36 Statistical tools have been developed to correct for population stratification34, 37, 38 and are now incorporated into GWAS analyses. An emerging standard in GWAS analysis is to filter low-quality arrays and SNPs before statistical testing as genotyping errors, especially if distributed differentially between cases and controls, can generate spurious associations.39 Further complexities emerge because of the need for multiple testing corrections. Methods used in GWAS include Bonferroni correction, false discovery rate,40 and false positive report probability,41 all of which have a different impact on evaluating the significance of associations.
In addition, attention to accurate genotyping is needed.42 Efforts to detect, prevent, and eradicate sources of technical errors and biases in genotyping are important for improving the quality and gaining confidence in GWAS results. This study was designed to evaluate aspects of technical robustness of genotyping.
Accurate and reproducible genotype calls are paramount, as biases in genotypic measurements can lead to an inflation of false associations. Large variances in genotypic measurements diminish the accuracy of calls and may inflate the Type II error rate. Different SNP array technologies exhibit different biases and variance characteristics because of probe and protocol differences. In addition, within the same technology platform, there are different genotypic calling algorithms developed and used. To our knowledge, there are currently no thorough evaluations of the replication consistency (repeatability and reproducibility) between genotype calls obtained using different calling algorithms or between different SNP arrays. Furthermore, it is important to assess how differences in genotype calls (because of technical reasons: algorithm or array) impact the downstream association analyses.
This study addresses several fundamental questions in GWAS: (1) Are current genotyping technologies robust? (2) Can consistent genotypes be obtained when different SNP arrays are used? (3) What is the likelihood that different calling algorithms generate different calls given identical raw intensity data? (4) Do differences in genotype calls impact downstream association analysis and generate discordant results?
To answer these questions, technical robustness was assessed by genotyping six subjects four times using Affymetrix Genome-Wide Human SNP 6.0 array (Affy6), the consistencies in the genotype calls between algorithms DM,43 BLRMM,44 and Birdseed45 and between Affy6 and Affymetrix GeneChip Human Mapping 500K array set (Affy500K) were examined using the 270 samples from the HapMap,46 and the impact on the association analyses was evaluated.
Materials and methods
DNA samples
DNA samples for the three HapMap subjects (NA10385, NA12448, and NA12449, labeled as N10385, N12248, and N12249 in our study) are from a trio and were obtained from the HapMap consortium.
The NCTR DNA samples (labeled as N13, N59, and N8) are from three anonymous human liver specimens from the US Cooperative Human Tissue Network that were used for human genomic DNA extraction, and these liver tissue samples were confirmed by pathological analysis to be obtained from normal donors.
Genotyping
Four replicates of the six DNA samples were genotyped using Affy6 according to the standard protocol from Affymetrix. On a 96-well plate, DNA samples are placed in 24 wells. Each well contains 2.0–2.5 μg of DNA at a concentration of ∼100 ng μl–1. The 24 DNA samples were placed in three columns of the 96-well plate (samples are randomized on the plate, with their layout given in Supplementary Figure 7) for genotyping with Affy6.
HapMap data
The raw data (CEL files) for Affy500K for the 270 HapMap samples were downloaded from the International HapMap project website (http://www.hapmap.org/downloads/raw_data/affy500k/). The raw data (CEL files) from the Affy6 for the 270 HapMap samples were obtained from Affymetrix.
Genotype calling and copy number variant calling
The quality of raw data was assessed using the program apt-geno-qc in the Affymetrix Power Tools (APT) before genotype calling. Genotype calling was conducted using apt-probeset-genotype in APT. All the parameters were set to the default values recommended by Affymetrix. In earlier work, we assessed calling batch effect and found that uniform and large batch sizes with homogenous samples should be used to make genotype calls for GWAS.47 Therefore, for our genotyping data, all raw data of the 24 samples were called in one batch. For HapMap data of Affy500K, three batches were used to make genotype calls: each used 90 samples from one of the three population groups.
Copy number variant (CNV) were called using the program apt-canary in the APT.
Comparing raw data
The raw intensity data at probe level used for the comparisons were extracted from the CEL files using the program apt-cel-extract in the APT. Thereafter, the pair-wise Pearson's correlation coefficients were calculated using the program corr in the statistical tool box for MatLab.
Comparing CNV results
The CNV calling results were compared by calculating the pair-wise concordance between the samples using the formula:
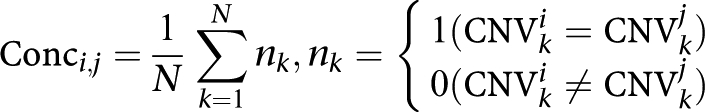 |
where N indicates total CNV regions, CNVki is the copy number called on CNV region k for sample i, and CNVkj is the copy number called on CNV region k for sample j.
Comparing genotype calling results
The pair-wise concordances of genotypes between samples were calculated using the formula:
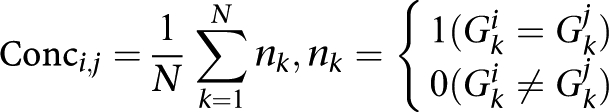 |
where N indicates total SNPs, Gki is the genotype called on SNP k for sample i, and Gkj is the genotype called on SNP k for sample j.
Association analysis
Before association analysis, quality control (QC) of the calling results was conducted to remove SNPs and samples of low quality using minor allele frequency, call rates per SNP and per sample, and testing for departure from Hardy–Weinberg equilibrium. In an association analysis, a 2 × 2 contingency table (allelic association) and a 2 × 3 contingency table (genotypic association) were generated for each SNP and tested for association using χ2 test.
Statistical analysis
To evaluate statistical significance of the difference in missing call rates between all pair-wise comparisons, paired t-tests were performed to test the hypothesis that two matched (or paired) samples/SNPs in the genotype calling results x and y come from distributions with equal means. The difference between arrays or between calling algorithms is assumed to come from a normal distribution with unknown variance. The significance level of α=0.05 was used in all of the tests.
Results
Robustness of genotyping
The experiment that assessed the robustness of genotyping used Affy6. DNA samples of three HapMap subjects and three NCTR subjects were genotyped, each with four replicates. The Birdseed-v1 in APT (1.10.0) was used to make genotype calls. CNV were determined using the apt-canary program in APT. To assess reproducibility across laboratories, the raw data of the three HapMap subjects from Affymetrix were included in our comparisons. The results are depicted in Figure 1 (data in Supplementary Table 1).
Figure 1.

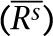 of correlation coefficients or concordances of genotypes or CNV (Rs) between technical replicates (BTR) within each of the six DNA samples, calculated as
of correlation coefficients or concordances of genotypes or CNV (Rs) between technical replicates (BTR) within each of the six DNA samples, calculated as 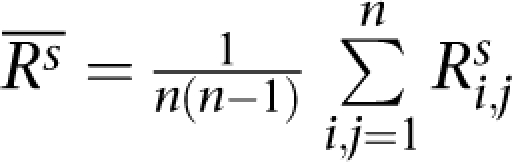 where, n=4, i≠j, s = one of the six samples (N10385, N12248, N12249, N13, N59, and N8, as shown from left to right in the figure). The up-triangles of the second-left panel are the means
where, n=4, i≠j, s = one of the six samples (N10385, N12248, N12249, N13, N59, and N8, as shown from left to right in the figure). The up-triangles of the second-left panel are the means 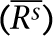 of correlation coefficients or concordances of genotypes or CNV (Rs) between our experimental data and the data from Affymetrix (BEH) for each of the three HapMap samples that were calculated as
of correlation coefficients or concordances of genotypes or CNV (Rs) between our experimental data and the data from Affymetrix (BEH) for each of the three HapMap samples that were calculated as 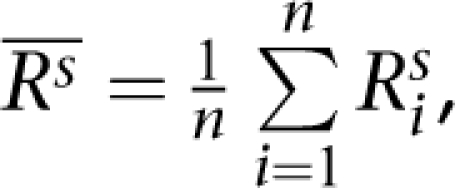 where, n = 4 and s=one of the HapMap samples (N10385, N12248, and N12249, from left to right). The diamonds of the third-left panel are the means (R̄) of correlation coefficients or concordances of genotypes or CNV (R) between parents (N12248 and N12249) and son (N10385) (BPS) that were calculated as
where, n = 4 and s=one of the HapMap samples (N10385, N12248, and N12249, from left to right). The diamonds of the third-left panel are the means (R̄) of correlation coefficients or concordances of genotypes or CNV (R) between parents (N12248 and N12249) and son (N10385) (BPS) that were calculated as 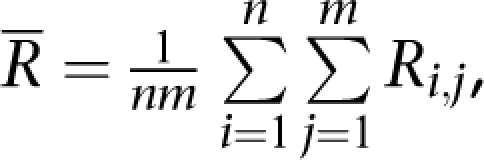 where, n=5 (four replicates from our experiment and one from Affymetrix for N10385) and m=10 (four replicates from our experiment and one from Affymetrix for each of N12248 and N12249). The down-triangles of the right panel are the means (R̄) of correlation coefficients or concordances of genotypes or CNV (R) between not-related samples (BNS) that were calculated as
where, n=5 (four replicates from our experiment and one from Affymetrix for N10385) and m=10 (four replicates from our experiment and one from Affymetrix for each of N12248 and N12249). The down-triangles of the right panel are the means (R̄) of correlation coefficients or concordances of genotypes or CNV (R) between not-related samples (BNS) that were calculated as  |
The QC scores of the 24 CEL files were in the range of 88.6–98.3% (Supplementary Figure 1), similar to the HapMap data from Affymetrix (88.2–99.1%, Supplementary Figure 5c) and compatible with Affymetrix guidelines, indicating that data were of acceptable quality for the comparative study.
The consistency of log2-scaled intensity data were examined using Pearson's correlation. Each pair-wise comparison is summarized in Supplementary Figure 2. The average correlation (Figure 1) between technical replicates (BTR) for five subjects was 0.9514. One subject (N13) had noticeably lower average correlation (0.9231), with one of its replicates determined to be an outlier (lower quality). For the HapMap samples, the average correlation between experiments and Affymetrix data (BEH) was 0.9403, slightly lower than the value corresponding to BTR (0.9515). The average correlation between not-related samples (BNS) was much lower (0.7576). The average correlation between parent and son (BPS) was 0.8456.
Genotype concordances were calculated for all pair-wise comparisons (Supplementary Figure 2) and averaged for BTR, BEH, BPS, and BNS (Figure 1). The average concordance for BTR was 0.9886, excluding N13 (0.9799), indicating a high repeatability. The average concordance for BEH was 0.9883, showing a high reproducibility across laboratories. As expected, the average concordance for BNS (0.6177) was low, and for BPS (0.7290) moderate.
Except for one replicate of N13 with a significantly lower heterozygous rate, the call rates and heterozygous rates were very similar for comparisons between replicates and between these experiments and Affymetrix data (Supplementary Figure 3). The lower heterozygous rate for the replicate of N13 is consistent with its lower average intensity correlation and genotype concordance. After removal of this replicate, the average intensity correlation (0.9568) and genotype concordance (0.9899) for N13 were similar to the other subjects.
Technical robustness was also evaluated by calculating CNV concordances for all pair-wise comparisons (Supplementary Figure 4), and averaging them for BTR, BEH, BPS, and BNS (Figure 1). The average concordance for BTR was 0.9804, except for N13 (0.9414), indicating a reasonable CNV repeatability. For the HapMap samples, the average concordance for BEH was 0.9605, similar to the corresponding BTR (0.9784), showing reasonable robustness across laboratories. As expected, the average concordance for BNS (0.8662) was low, and for BPS (0.8978) moderate.
In spite of the apparent overall reproducibility, an outlier was detected only after replicate measurements were completed. The outlier would have not otherwise been detected, as the array met the guidelines for Affymetrix genotyping array quality.
Inconsistencies between SNP arrays
To examine whether genotype calls from different SNP arrays are consistent, genotypes of SNPs interrogated in common in both Affy500K and Affy6 were compared using the 270 HapMap samples.46
The QC scores for Affy500K (Supplementary Figures 5a and b) and Affy6 (Supplementary Figure 5c) data met Affymetrix guidelines. Therefore, all CEL files were used.
After quantile normalization, genotypes were called using the same calling algorithm, Birdseed, with the same parameter settings. Thereafter, the 482 215 common SNPs were used for the comparisons (Figure 2a).
Figure 2.

Overview of the procedures for evaluating consistency between SNP arrays (a). Both data sets of the 270 HapMap samples from Affy500K and Affy6 were genotype called using algorithm Birdseed. The 482 251 SNPs interrogated in both arrays were used in the downstream association analysis. The same QC process was applied to both sets of genotypes before the same statistical tests for associations (see Materials and methods). Overview of the procedures for evaluating consistency between genotype calling algorithms (b). The raw data (CEL files) of Affy500K of the 270 HapMap samples were genotype called using algorithms DM, BRLMM, and Birdseed. The same QC process and the same statistical tests were used for the three sets of genotypes. In the association analysis, both allelic and genotypic association tests were conducted. Three different case–control frameworks were used: each of the three population groups (European, African, and Asia) was set as ‘case' with the other two as ‘control' (see Materials and methods).
The missing call rates per SNP (Figure 3a) and per sample (Figure 3b) were compared between Affy500K (x axis) and Affy6 (y axis). Many SNPs and samples are not consistent, some of which show large differences between the two arrays. Moreover, the missing call rates from Affy6 are slightly lower than those from the Affy500K. The P-values (Supplementary Table 2) of paired two-sample t-tests for comparing the missing call rates per SNP and per sample were <0.05, indicating that the difference of missing call rates is statistically significant.
Figure 3.

Comparison of genotype calls between SNP arrays. The missing call rates per SNP (a) and per sample (b) between arrays Affy500K and Affy6 were plotted. The red diagonal lines indicate the locations of SNPs (a) and samples (b) when their missing call rates are exactly same between these two arrays. The concordances of homozygote calls (AA), heterozygote calls (AB), and variant homozygote calls (BB) between Affy500K and Affy6 were given by the bars in the left panel of (c) (left y axis). Each blue bar represents a ratio (n500K−6g/n500Kg, g=AA or AB or BB) of the number of the specific genotypes from both Affy500K and Affy6 (n500K−6g) and the total of corresponding genotypes from Affy500K (n500Kg). Each red bar is for n500K−6g/n6g where n6g is the total from Affy6. The distribution of discordant successful genotype calls between Affy500K and Affy6 are depicted in the right panel of (c) (right y axis). The value of green bar and magenta bar are for Affy500K and Affy6 that were calculated using ng500Kg6/ng500K and ng500Kg6/ng6, respectively, where n500Kg6 is the number of genotypes assigned to g500K from Affy500K, but to g6 from Affy6; ng500K, the number of genotypes assigned to g500K from Affy500K; and ng6, the number of genotypes assigned to g6 from Affy6 (g500K, g6=AA or AB or BB; g500K≠g6).
Three possible genotypes (homozygote: AA; heterozygote: AB; and variant homozygote: BB) are provided for each call. The concordance of each paired calls between Affy500k and Affy6 was analyzed (Supplementary Table 3). The analysis revealed 267 608 (0.21%) genotype differences between the two arrays. Further comparison regarding the nature of the differences (Figure 3c) shows that concordance of homozygous calls (AA and BB) was higher than the concordance of heterozygous calls (AB). Moreover, discordant genotypes between heterozygote and homozygote were more prevalent than those between two homozygous types.
Inconsistencies between calling algorithms
Genotype concordances were determined between three algorithms (DM, BRLMM, and Birdseed) that were released along with three recent generations of Affymetrix arrays (Figure 2b). Affy500K raw data for the 270 HapMap samples were called using the three algorithms. Thereafter, the calls were compared to determine consistency between algorithms.
The missing call rates per SNP (Figure 4a) and per sample (Figure 4b) were compared. Many SNPs and samples had different missing call rates between the three algorithms. Furthermore, the missing call rates of the single-chip-based algorithm DM were higher compared with the multiple-chip-based algorithms BRLMM and Birdseed (caused by the default cutoff used in this study, see Discussion), whereas differences between BRLMM and Birdseed were much smaller. The P-values (Supplementary Table 2) of paired two-sample t-tests when comparing missing call rates per SNP and per sample were <0.05, indicating that the algorithms have significantly different missing call rates.
Figure 4.

Comparison of genotype calls between calling algorithms. The missing call rates per SNP (a) and per sample (b) between algorithms Birdseed, BRLMM, and DM were plotted. The red diagonal lines indicate the locations of SNPs (a) and samples (b) when their missing call rates are exactly same between two of these three algorithms. The concordances of homozygote calls (AA), heterozygote calls (AB), and variant homozygote calls (BB) between BRLMM and Birdseed (c), between DM and Birdseed (d), and between DM and BRLMM (e) were shown by the bars in the left panels (left y axes). The blue bars represent ratios (nA1–A2g/nA1g, g=AA or AB or BB) of the numbers of specific genotypes by both algorithms (nA1–A2g) to the totals of corresponding genotypes from the first algorithm A1 (nA1g) (A1=BRLMM (c) and DM (d, e)). The red bars are for ngA1–A2/nA2g, where nA2g are totals from the second algorithms (A2=Birdseed (c, d) and BRLMM (e)). The discordant successful genotype calls between two algorithms are depicted in the right panels of (c, d, e) (right y axes). The values of green bars and magenta bars are for the first algorithms and the second algorithms that were calculated using ngA1gA2/ngA1 and ngA1gA2/ngA2, respectively, where ngA1gA2 is the number of genotypes assigned to gA1 from the first algorithm, but to gA2 from the second algorithm; nA1, the number of genotypes assigned to gA1 from the first algorithm; and ngA2, the number of genotypes assigned to gA2 from the second algorithm (gA1, gA2=AA or AB or BB; gA1≠gA2).
The consistencies of successful calls between the three algorithms were calculated as concordances given in Supplementary Table 3. A total of 538 774 genotypes (0.41%) differed between DM and Birdseed; 200 592 genotypes (0.15%) between DM and BRLMM; and 285 788 genotypes (0.21%) between Birdseed and BRLMM. The concordance of the successful calls between BRLMM and Birdseed stratified on three genotypes that are given in Figure 4c. The concordance for homozygous calls was higher than for heterozygous calls for both BRLMM and Birdseed. Moreover, discordance between heterozygote and homozygote was higher than between the two homozygous types. Comparisons between DM and Birdseed and between DM and BRLMM are depicted in Figures 4d and e, respectively, with similar trends to the comparison between BRLMM and Birdseed prevailing, such as homozygous calls being more concordant than heterozygous calls.
Propagation of array inconsistency to associated SNPs
The objective of a GWAS is to identify genetic markers associated with a phenotype. It is critical to assess how inconsistencies between different SNP arrays propagate to the associated SNPs identified in the downstream association analysis. To mimic case–control GWAS, three association analyses were conducted for genotypes obtained from Affy6 and Affy500K data for the 270 HapMap samples (Figure 2a). Each of the three population groups (EU: European; AS: Asian; and AF: African) were set in turn as the cases, whereas the other two groups were set as the controls. Associations were analyzed to identify SNPs that can differentiate cases from controls. The significantly associated SNPs were compared using Venn diagrams.
Comparisons of significantly associated SNPs obtained from allelic and genotypic tests (on 482 251 common SNPs) between the two arrays are given in Figures 5a and b, respectively. For all case–control frameworks and both allelic and genotypic tests, the inconsistency in genotypes between arrays influenced the downstream association analyses, resulting in differently associated SNPs. For example, using allelic testing, 4926 SNPs were significant only for the Affy500K using Europeans as case. It is unclear whether these differences are due to Type I errors using Affy500K or Type II errors using Affy6. Alternatively, the variation in associated SNPs could be due to the exclusion of SNPs during QC steps.
Figure 5.

Comparisons of the lists of associated SNPs from Affy500K and Affy6 for assessing propagations of the inconsistency in genotypes between the two arrays to associated SNPs. The significantly associated SNPs identified using allelic association test (a) and genotypic association test (b) from the 482 251 common SNPs were compared between Affy500K (numbers in the black circles) and Affy6 (numbers in the red circles). Numbers in green are the associated SNPs from both arrays, numbers in black are the SNPs only significant from Affy500K, and numbers in red are the SNPs only significant from Affy6. EU∼Others: the association analyses results for European versus others; AF∼Others: for African versus others; AS∼Others: for Asian versus others. The discordant association SNPs caused by QC (solid circles) and by association statistical tests (empty circles) in percentage were plotted in (c). Red points are for SNPs significant from Affy500K, but not from Affy6, the blue points are for SNPs significant from Affy6, but not from Affy500K. The x axis indicates the ‘case' populations for the association analyses.
For associated SNPs not common to both arrays, observed differences in downstream association analysis were examined to see whether they were due to failing to pass QC or to conflicting results for statistical testing. The results depicted in Figure 5c show that most associated SNPs missed with Affy500K were excluded at the QC step. Differences in statistical testing were the major cause for the associated SNPs missed in Affy6.
Propagation of calling algorithm inconsistency to associated SNPs
To assess propagation of inconsistencies in genotypes between calling algorithms to the associated SNPs, associations were analyzed using genotypes obtained from algorithms DM, BRLMM, and Birdseed for Affy500K data for the 270 HapMap samples. The associated SNPs were compared using Venn diagrams (Figures 6a and b) for the allelic and genotypic tests, respectively. The inconsistencies in genotypes between the three algorithms propagated into the downstream association analyses. For example: only 1593, 1349, and 1873 SNPs were significantly associated (genotypic test, European as case) using DM, BRLMM, and Birdseed algorithms, respectively. Again, possible Type I or Type II errors as well as QC exclusion differences contribute to the variability in the associated SNPs.
Figure 6.

Comparisons of the lists of associated SNPs between calling algorithms DM, BRLMM, and Birdseed for assessing propagations of the inconsistence in genotypes to association SNPs. The significantly associated SNPs identified using allelic association test (a) and genotypic association test (b) were compared between algorithms DM (numbers in the black circles), BRLMM (numbers in the blue circles), and Birdseed (numbers in the red circles). Numbers in brown represent the associated SNPs shared by all three algorithms, numbers in green represent the associated SNPs shared by only two algorithms, and the numbers in other colors are the associated SNPs identified by only the corresponding algorithms. EU∼Others: the association analyses results for European versus others; AF∼Others: for African versus others; AS∼Others: for Asian versus others. The discordant associated SNPs (missed from DM: black; BRLMM: red; Birdseed: blue) caused by QC (solid shapes) and by association statistical tests (empty shapes) in percentage were plotted in (c) (SNPs were significant from one algorithm, but not significant from the other two algorithms) and (d) (SNPs were significant from two algorithms, but not significant from the other one algorithm). The x axis indicates the ‘case' populations for the association analyses.
For SNPs found to be significant only from one algorithm, the SNPs that failed in QC and in statistical tests are given in Figure 6c. Missed SNPs from DM were mainly caused by QC exclusion, whereas missed SNPs from BRLMM and Birdseed were mainly caused by association testing.
For SNPs that were identified as significant only from two algorithms, but not the third, the SNPs that failed in QC and in statistical tests are shown in Figure 6d. QC caused more missed SNPs from DM and Birdseed, whereas association testing caused more missed SNPs from BRLMM.
Discussion
GWAS simultaneously interrogate hundreds of thousands of SNPs and associate genetic variants with health-related traits. In the past 3 years, many loci were identified and replicated.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 However, often GWAS results are not replicated, indicating that each step in GWAS has the potential to introduce Type I and Type II errors. It is important to know the robustness of current genotyping technology. Availability of different SNP arrays and genotype calling algorithms make it vital to be aware of SNP array and calling algorithm inconsistencies and their effect in GWAS.
To evaluate genotyping technical robustness, an experiment was designed and conducted with Affy6 using four technical replicates for six subjects. The results showed that genotyping with Affy6 is generally robust for the raw intensity, genotypes, and CNV. The reproducibility across laboratories with an average concordance of ∼0.99 was observed. However, common diseases being investigated in GWAS are typically influenced by multiple loci, with each locus making a small contribution. Therefore, small errors in any procedure can be amplified in GWAS results, as shown in the results for significant associations under various SNP arrays and genotype calling algorithms. The potential for errors caused by small technical fluctuations of genotyping suggests that technical replicates can increase the reliability of GWAS findings. Furthermore, using technical replicates helps remove low-quality arrays as shown in this study. If no technical replicates were used, one replicate of N13 would not be identified as problematic because its QC looks reasonable. But when comparing with the other three technical replicates, it is obvious that all measures (intensity correlation, concordances of genotypes and CNV, heterozygous rate) show that the data from this array causes problems in genotyping.
This study showed that genotype inconsistency propagates to GWAS results. Sources of errors introduced into genotypes such as experimental design, the type of SNP array, and the genotype calling algorithm have the potential to generate inconsistent associated SNPs, and hence Type I and Type II errors. Furthermore, it was observed that genotype inconsistency not only propagated to the downstream association analysis, but was amplified in the associated SNPs (Supplementary Figure 6).
There were many SNPs (∼15%) identified as significant from BRLMM and Birdseed, but not from DM (Supplementary Figures 6b and 6c). Most of those SNPs had low call rates and were filtered in the QC process and not tested for associations. If a less stringent cutoff was used in DM, it could be expected that some of those SNPs would pass the same QC criterion and the missing rate of associated SNPs from DM would be decreased, but could not be completely eliminated, evidenced by the comparison between BRLMM and Birdseed in which discordant rates of associated SNPs were about three times of the discordant rate of genotypes between the two algorithms (Supplementary Figure 6d).
Genotype discordance was found in both missing calls and successful calls. Our study showed that the propagation of discordant genotypes to the associated SNPs was caused by both sources of discordance (Figures 5 and 6). Our observations suggest that there is room for improvements on both call rate and accuracy of calling algorithms. There is a tradeoff in the source of discordance depending on the chosen cutoff for calling a missing.
An interesting observation was that more associated SNPs were identified in the model using African as case (Figures 5 and 6). In the HapMap samples, it is well known that the Yoruban is more genetically distinct than the Asian and European. However, discordant rates of associated SNPs for the African model were lower than Asian and European models (Figure 6). Therefore, discordance in genotypes might be amplified more in the associated SNPs for weaker traits than for stronger traits. Comparing with the population differences of the HapMap samples used in our study, traits of current GWAS are usually much weaker, and a smaller number of concordant associated SNPs are expected.
Acknowledgments
HH thanks Dr Williams Slikker for his support on this research project. LX thanks NCTR-US FDA for providing the ORISE postdoctoral research fellowship to participate in the project.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website (http://www.nature.com/tpj)
Supplementary Material
References
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2003;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride level. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- Easton DF, Pooley KA, Dunning AM, Pharoah PDP, Thompson J, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium Genome-wide association study of 14 000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raelson JV, Little RD, Ruether A, Fournier H, Paquin B, Eerdewegh PV, et al. Genome-wide association study for Crohn's disease in the Quebec Founder Population identifies multiple validated disease loci. Proc Natl Acad Sci USA. 2007;104:14747–14752. doi: 10.1073/pnas.0706645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, et al. Genome-wide association study identifies a second breast cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, et al. Genome-wide association study of breast cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39:770–775. doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, et al. A genome-wide association study for celiac disease identifies risk variants in the region harbouring IL2 and IL21. Nat Genet. 2007;39:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analysis of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, et al. Genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–988. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007;39:989–994. doi: 10.1038/ng2089. [DOI] [PubMed] [Google Scholar]
- Buch S, Schafmayer C, Völzke H, Becker C, Franke A, von Eller-Eberstein H, et al. A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nat Genet. 2007;39:995–999. doi: 10.1038/ng2101. [DOI] [PubMed] [Google Scholar]
- Winkelmann J, Schormair B, Lichtner P, Ripke S, Xiong L, Jalilzadeh S, et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet. 2007;39:1000–1006. doi: 10.1038/ng2099. [DOI] [PubMed] [Google Scholar]
- Grupe A, Abraham R, Li Y, Rowland C, Hollingworth P, Morgan A, et al. Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum Mol Genet. 2007;16:865–873. doi: 10.1093/hmg/ddm031. [DOI] [PubMed] [Google Scholar]
- Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser M, Liu F, Janssens AC, Rivadeneira F, Lao O, van Duijn K, et al. Three genome-wide association studies and a linkage analysis identify HERC2 as a human iris color gene. Am J Hum Genet. 2008;82:411–423. doi: 10.1016/j.ajhg.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HH, Hu N, Taylor PR, Lee MP. Whole genome-wide association study using Affymetrix SNP Chip: a two-stage sequential selection method to identify genes that increase the risk of developing complex diseases. Methods Mol Med. 2008;141:23–35. doi: 10.1007/978-1-60327-148-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, Garber J, et al. A genome-wide association study provides evidence for a breast cancer risk at 6q22.33. Proc Natl Acad Sci USA. 2008;105:4340–4345. doi: 10.1073/pnas.0800441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of â-thalassemia. Proc Natl Acad Sci USA. 2008;105:1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- Pearson TA, Manolio TA. How to interpret a genome-wide association study. J Am Med Assoc. 2008;82:411–423. doi: 10.1001/jama.299.11.1335. [DOI] [PubMed] [Google Scholar]
- Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal component analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Thomas DC, Witte JS. Point: population stratification: a problem for case-control studies of candidate-gene associations. Cancer Epidemiol Biomarkers Prev. 2002;11:505–512. [PubMed] [Google Scholar]
- Wacholder S, Rothman N, Caporaso N. Counterpoint: bias from population stratification is not a major threat to the validity of conclusions from epidemiological studies of common polymorphisms and caner. Cancer Epidemiol Biomarkers Prev. 2002;11:513–520. [PubMed] [Google Scholar]
- Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;36:598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- Zheng G, Freidlin B, Gastwirth JL. Robust genomic control for association studies. Am J Hum Genet. 2006;78:350–356. doi: 10.1086/500054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskvina V, Craddock N, Holmans P, Owen MJ, O'Donovan MC. Effects of differential genotyping error rate on the type 1 error probability of case-control studies. Hum Hered. 2006;61:55–64. doi: 10.1159/000092553. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96:434–442. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Di X, Matsuzaki H, Webster TA, Hubbell E, Liu G, Dong S, et al. Dynamic model based algorithms for screening and genotyping over 100K SNPs on oligonucleotide microarrays. Bioinformtics. 2005;21:1958–1963. doi: 10.1093/bioinformatics/bti275. [DOI] [PubMed] [Google Scholar]
- See the white paper on BRLMM of Affymetrix: http://www.affymetrix.com/support/technical/whitepapers/brlmm_whitepaper.pdf .
- http://www.affymetrix.com/products/software/specific/birdseed_algorithm.affx .
- The International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–862. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Su Z, Ge W, Shi L, Perkins R, Fang H, et al. Assessing batch effects of genotype calling algorithm BRLMM for the Affymetrix GeneChip Human Mapping 500K Array Set using 270 HapMap samples. BMC Bioinformatics. 2008;9:S17. doi: 10.1186/1471-2105-9-S9-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.