

Abstract
Production of the Gag-Pol polyprotein in human immunodeficiency virus (HIV) requires a -1 ribosomal frameshift, which is directed by a highly conserved RNA stemloop. Building on our discovery of a set of disulfide-containing peptides that bind this RNA, we describe medicinal chemistry efforts designed to begin to understand the structure-activity relationships and RNA sequence-selectivity relationships associated with these compounds. Additionally, we have prepared analogues incorporating an olefin or saturated hydrocarbon bioisostere of the disulfide moiety, as a first step towards enhancing biostability. The olefin-containing compounds exhibit affinity comparable to the lead disulfide, and importantly have no discernable toxicity when incubated with human fibroblasts at concentrations up to 1 millimolar.
Introduction
The design and synthesis of compounds able to selectively bind specific RNA sequences with high affinity represents a signature challenge in chemistry.1 Unlike DNA, which to a great extent has been rendered an “open book” for molecular recognition,2 the problem of sequence-selective RNA recognition remains largely unsolved. In addition to the fundamental importance of this challenge (a subset of the more general challenge of function-oriented synthesis3), RNA targets of potential therapeutic value are being discovered at an increasing rate. For example, a notable pathogen against which RNA-targeted therapeutics can potentially have an impact is the human immunodeficiency virus (HIV). The causative agent of AIDS, HIV continues to be a major threat to human life despite intensive ongoing research. While modern antiretroviral drugs have dramatically increased the life spans of those infected with the virus, a number of factors drive the need for new anti-HIV drugs.4 Key among these include the complexity of current antiretroviral regimens, and the emergence of drug resistance.5,6,7 Screening and directed design approaches to the development of small molecules interfering with HIV at the RNA level have primarily focused on REV8 and TAR,9 and those efforts have yielded a number of important insights into HIV biology, as well as strategies for RNA recognition. Recently, however, another RNA has garnered interest as a potential therapeutic target. This RNA’s importance derives from the fact that the structural and enzymatic proteins of HIV are encoded in overlapping gag and pol open reading frames, respectively.10 Since pol is always translated as a Gag-Pol fusion protein, the ribosome is required to slip one nucleotide backwards to produce the desired polyprotein. This −1 frameshift event occurs with a frequency of 5–10%,11 and disruption of this percentage has been demonstrated to severely decrease viral replication and infectivity.12
Two cis-acting elements in viral mRNA are responsible for the −1 ribosomal frameshift: a heptameric (UUUUUUA) slippery sequence where the frameshift takes place, and a downstream stimulatory signal.13 Modification of the stimulatory sequence (either via natural variation or laboratory mutation) in ways affecting frameshifting efficiency translates to a decrease in viral replication.14 Thus, targeting this structure with small molecules is a potential strategy for inhibiting viral replication.15,16 Efforts by several laboratories, including three NMR structural analyses,17 indicate that the stimulatory RNA in HIV-1 forms a stem-loop structure. This in turn may be divided into an upper stem-loop and lower stem, separated by a purine bulge (Figure 1). It has been hypothesized that frameshift efficiency is dependent on the mechanical stability of the downstream RNA regulatory element, with higher frameshifting rates correlating with a greater force required to unfold the downstream RNA.18 Recent studies by the Visscher and Fourmy groups suggest that the lower stem and bulge regions of the HIV-1 FSS readily unfold, while unfolding of the upper stemloop requires more force.19 Thus, further stabilization (or, alternatively, destabilization) of the upper stemloop may alter frameshift efficiency, suggesting that this sequence may constitute a useful target for drug discovery. We have previously reported the discovery of a compound (1) able to selectively bind the HIV-1 frameshift-stimulating RNA upper stem-loop (hereafter abbreviated “HIV-1 FSS”).20 Compound 1 was identified via the synthesis and in situ screening of an 11,325 member resin-bound dynamic combinatorial library (RBDCL) designed based on a core structure inspired by nucleic acid-binding, bisintercalating natural products.21 This molecule binds the HIV-1 FSS with a dissociation constant (KD) of 4.1 ± 2.4 μM as measured by surface plasmon resonance (SPR), and serves as an important lead for the development of high affinity ligands for the HIV-1 FSS. Other recent reports of compounds targeting frameshifting in HIV include a description by Butcher and Tor of the interaction of guanidinoneomycin with the HIV-1 FSS upper stemloop,22 and a recent report of several molecules targeting the bulge region.23 This latter study suggests that the question of the “best” region of the FSS to target should not yet be regarded as settled. Heveker and colleagues have described peptides able to interfere with frameshifting in a two-reporter bacterial system, but it is uncertain whether this interference involves a direct interaction with the FSS.24
Figure 1.

(Left) Secondary structure of the HIV-1 frameshift-inducing RNA stem-loop. The sequence shown is that of the HIV group M subtype D.25 The upper stem-loop sequence (or “HIV-1 FSS”) used as the primary target for RBDCC screening and for the experiments described herein is boxed. The slippery sequence where the −1 frameshift occurs is underlined. (Right) Lead molecule 1, previously identified via RBDCC.
This paper details our efforts to expand our understanding of the structural factors governing recognition of the HIV-1 FSS by 1. We approached the problem via solution-phase binding analysis, and by the directed synthesis of several analogs of 1 to explore structure-activity relationships with a particular focus on modifications to the quinoline heterocycle. As part of the structure-activity analysis, we also report the first steps towards enhancing the biostability of 1, via the synthesis and binding analysis of analogues of 1 incorporating hydrocarbon replacements (bioisosteres) for the disulfide.
Analysis of the binding selectivity of 1
Previously reported SPR measurements on 1 were conducted with the compound tethered to the SPR chip via one of its amino groups, and involved a relatively limited set of RNA sequences. As a first step towards increasing our understanding of the interaction of 1 and related compounds with the HIV-1 FSS, we carried out a series of solution-phase fluorescence titration measurements. In these experiments, the fluorescence of 5′ Cy3-labeled RNA was monitored as a function of added compound.26,27
The RNA and DNA sequences employed, and results of titrations (alongside previously reported SPR values, where available) are shown in Table 1; selected titration data and associated curve fits are shown in Figure 2. All data were fit to a 1:1 binding model incorporating ligand depletion. Binding to the HIV-1 FSS derived from HIV group M subtype D (entry 1) was found to be 0.35 ± 0.11 μM in solution, roughly an order of magnitude tighter than the binding constant obtained previously by SPR. This is not altogether surprising, as immobilization of 1 on the SPR chip both reduces the degrees of freedom available to the compound, and its overall charge. The measured binding affinity of 1 for the HIV-1 FSS did not change in the presence of an excess of unrelated competing RNAs (either yeast tRNA, entry 2, or total yeast RNA, entry 3), although the total change in fluorescence decreased. To confirm that this reduction in the total change in fluorescence reflected a nonspecific interaction between the Cy3-labeled HIV-1 FSS and competitor RNAs rather than off-target binding by compound 1, we titrated an identical solution of yeast tRNA into Cy3-labeled HIV-1 FSS. Consistent with our hypothesis, the dilution-corrected Cy3 fluorescence decreased in a manner dependent on the concentration of tRNA (supplementary information page S32). Use of excess yeast tRNA as a measure of selectivity was pioneered by Tor and colleagues;28 following Tor’s terminology, where “specificity” is defined as the ratio of the KD to the sequence of interest over the KD observed in the presence of a large excess of competitor RNAs, our results suggest a specificity ratio of approximately 1. As expected, the measured binding constant could be altered by addition of unlabeled (competing) HIV-1 FSS (entry 4).
Table 1.
Binding constants (KD, micromolar) for 1 and selected oligonucleotides. “Solution” measurements were carried out by titrating unlabeled compound into Cy3-labeled RNA and measuring changes in the fluorescence spectrum, while SPR measurements were performed by immobilizing the compound on the sensor chip and flowing increasing concentrations of unlabeled RNA.
| Entry | Sequence | SPR | Solution | |
|---|---|---|---|---|
| 1 | 4.1±2.4 | 0.35 ± 0.11 | 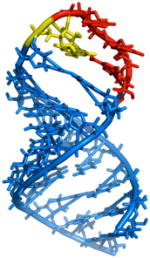 |
|
| 2 | As entry 1, + 40x total yeast tRNA | ND | 0.20 ± 0.03 | |
| 3 | As entry 1, + 20x total yeast RNA | ND | 0.57 | |
| 4 | As entry 1, + 4x unlabeled HIV-1 FSS | ND | 0.65 ± 0.13 | |
| 5 | ND | 0.45 ± 0.04 | ||
| 6 | ND | 0.33 ± 0.12 | ||
| 7 | ND | 0.76 ± 0.06 | ||
| 8 | >90 | 1.10 ± 0.1 | ||
| 9 |
|
No binding | No binding | |
| 10 |  |
No binding | No binding | |
| 11 | 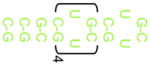 |
ND | >40 |
ND = Not Determined. Right: Butcher, et al. NMR structure of the HIV-1 FSS RNA (PDB code 1PJY)17a color-coded by binding constant change in altered sequences (blue = least; red = most)
Figure 2.

Selected fluorescence titration data (dilution-corrected fluorescence change as a function of ligand concentration) and curve fits for the experiments summarized in Table 1.
Single mutation (entry 5) or complete sequence flipping (entry 6) of the stem did not produce an experimentally significant change in the binding constant. In contrast, the introduction of single (entry 7) or multiple (entry 8) mutations into the loop region caused a 2-fold and 4-fold reduction in the binding, respectively. No saturable binding was observed to a DNA homolog of the HIV-1 FSS (entry 9) or to unrelated RNA hairpins (entries 10 and 11; note that these sequences have been examined in previous studies in our laboratory29 and are known to be folded under the conditions of the titrations). Taken together these results confirm that compound 1 binds the target HIV-1 FSS with high affinity and good selectivity. Alterations to the tetraloop had the greatest impact on binding.
Structure-Activity Relationships of the 2-ethylquinoline 3-carboxamide moiety
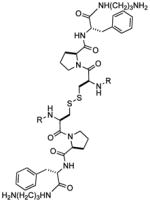
With a more detailed understanding of the solution-phase binding selectivity of 1 in hand, we next turned toward modifications of the compound itself in order to examine the structural features governing binding. Based on prior reports of the ability of quinolines to act as general (non sequence-selective) RNA intercalators,1b one would hypothesize intercalation as the primary function of the 2-ethyl quinoline 3-carboxamide moieties of 1. To test that hypothesis, compounds 2–7 were synthesized on solid-phase resin beads using methods previously described for the synthesis of 1. Binding constants were measured by fluorescence titration (Table 2). Changing the 2-ethyl group to methyl (2) or proton (3) yielded experimentally insignificant (2) or only modest (3) reduction in affinity for the HIV-1 FSS. This is consistent with molecular mechanics calculations indicating that there is little difference in the conformational landscape of these three compounds. Decreasing the available pi-surface, exemplified in the 2-methyl-3-carboxypyridine compound (4) and uncapped peptide (5), completely ablated binding. This stark difference in affinity between 1 and 5 was confirmed by a filter-binding assay (Supplementary Information, Figure S7). The introduction of a dioxolane ring (6) resulted in a compound with roughly four-fold lower affinity than 1, while replacement of the quinoline with an anthraquinone yielded a molecule (7) with affinity equivalent to 1.
Table 2.
binding constants (KD) for heterocycle analogs of 1 binding to the HIV-1 FSS RNA, as measured by fluorescence titration. All values are an average ± standard deviation of 3 replicate titrations. N.B. = No Binding. Compounds 1–7 all likely carry a charge of +2.
| Compound | R | MW | KD (μM) | |
|---|---|---|---|---|
 |
1 | 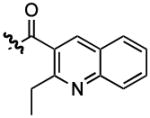 |
1206 | 0.35 ± 0.11 |
| 2 | 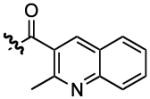 |
1178 | 0.43 ± 0.05 | |
| 3 | 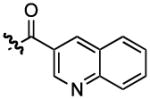 |
1150 | 0.65 ± 0.04 | |
| 4 | 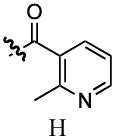 |
1078 | No Binding | |
| 5 | 840 | No Binding | ||
| 6 | 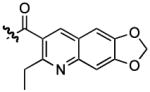 |
1295 | 1.42 ± 0.17 | |
| 7 | 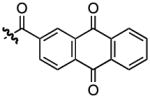 |
1308 | 0.23 ± 0.03 | |
Observation that 2-methyl pyridine derivative 4 is unable to bind supports an intercalative binding mechanism for 1, as it is well known (at least in a DNA-binding context) that pyridines have insufficient pi-surface area to support intercalation. As discussed below, the (Cys-Pro-Phe)2 peptide is not a passive player in binding, however, despite the lack of RNA-binding affinity displayed by 4 and 5. Further efforts are underway to explore the ability of other heterocycles with larger pi-surface area to enhance affinity.
Replacement of the disulfide with non-labile bioisosteres
The ability of the disulfide to undergo exchange with other thiols in solution30 was a critically important design feature of the RBDCL used to identify 1. However, its susceptibility to reduction or exchange in a biological environment would present an obvious obstacle to investigation of this class of molecules in a cellular context. Reduction is particularly a concern in the cytosol, where the ratio of reduced: oxidized glutathione (GSH:GSSG) ranges from 30:1 to 100:1.31 Previous work has shown that in most “biologically active” peptides, the disulfide linkage serves only a structural role, and replacing it with thioether (-S-CH2-) or all carbon (olefin, -CH2-CH2-) bioisosteres generally enhances biostability of the molecule without affecting its function.32 A particularly striking example of the interchangeability of disulfide and olefin linkages was reported in 2000 by Nicolaou and coworkers, who found similar selectivities in the receptor-accelerated synthesis of disulfide- and olefin-linked vancomycin dimers.33 Saturated hydrocarbon analogs of disulfides have also been reported, for example in the enkaphalins.32d Therefore, in order to build towards compounds suitable for cellular studies, we next synthesized dicarba (olefin and hydrocarbon) analogs of compound 1.
Ruthenium-catalyzed olefin metathesis is an attractive and powerful tool for the formation of carbon-carbon double bonds.34 Grubbs’ catalysts (1st and 2nd generation) have been previously used in the synthesis of cyclic peptides via ring-closing metathesis.32 35 Likewise, olefin cross-metathesis of amino acid derivatives36 and more complex peptides is well established. Therefore, to substitute for the cysteine amino acid in 1, the olefin was introduced in the form of L-allylglycine37. Following preparation of the metathesis precursor peptide 8, initial efforts focused on carrying out olefin self-metathesis of this compound in solution. However, we were unable to remove all traces of catalyst from the product olefin-containing peptide despite the application of various catalyst removal strategies.38 To avoid this issue, we turned to the use of on-bead cross-metathesis between resin-bound and solution-phase 8. As shown in Scheme 1, cleavage of the resin-bound adduct following cross-metathesis gave a mixture of geometrical isomers (9 and 10) in a 2:3 ratio and 64% yield. The mixture was then separated by preparative HPLC to give the purified E and Z isomers. Preparation of the saturated analog 11 was accomplished by on-bead hydrogenation of metathesis adduct (Wilkinson’s catalyst; 40 psi H2), followed by TFA-mediated cleavage.
Scheme 1.

Synthesis of olefin and saturated hydrocarbon analogs of 1.
As measured by fluorescence titration, both 9 and 10 bound to the HIV-1 FSS with an affinity comparable to that of compound 1. Thus, bioisosteric replacement of the disulfide linkage in 1 with an olefin does not interfere with RNA recognition (Table 3). Olefin isomers 9 and 10 show only a 2-fold difference in their binding affinity, suggesting that the linker is sufficiently flexible to access a favorable conformation for binding regardless of olefin geometry. The four-fold decreased affinity of the saturated analog (11) relative to 1 may either be a reflection of the increased flexibility of the compound relative to the olefin bioisostere, or potentially a result of its increased hydrophobicity. As with compound 1, the binding affinity of dicarba analogs to the stem-loop was unchanged in the presence of excess yeast tRNA, evidence of selectivity for binding to the HIV-1 RNA stem-loop. Interestingly, monomeric compound 8 also bound to the HIV-1 RNA stem-loop with similar affinity to that of both the parent compound (1) as well as analogs 9 and 10, but binding was completely ablated by the presence of excess yeast tRNA. This is similar to the behavior shown by 2-ethylquinoline 3-carboxylic acid, which binds RNA nonspecifically. Thus, both the full-length peptide and quinoline are required for selective binding to the HIV-1 FSS.
Table 3.
Binding affinity of dicarba analogs to the HIV-1 FSS RNA (4 × 10−7 M) in comparison to 1 as measured by fluorescence titrations in 1X PBS buffer (pH 7.2) at 25 °C. The reported KD values are an average of two separate titration experiments in each case.
| Compound | MW | Charge | Binding affinity (KD) (μM) | Binding affinity (KD) in the presence of 20x yeast tRNA (μM) |
|---|---|---|---|---|
| 1 | 2 | 0.35 ± 0.11 | 0.20± 0.03 | |
| 2-ethylquinoline 3- carboxylic acid | 201 | −1 | 0.29 ± 0.03 | No binding |
| 8 | 598 | 1 | 0.47 ± 0.04 | No binding |
| 9 | 1168 | 2 | 0.33 ± 0.02 | 0.39 ± 0.02 |
| 10 | 1168 | 2 | 0.18 ± 0.02 | 0.26 ± 0.04 |
| 11 | 1170 | 2 | 1.27 ± 0.11 | 1.41 ± 0.20 |
Study of Binding Kinetics using SPR
While the equilibrium dissociation constant (KD) is obviously an important measure of ligand quality in vitro, kinetic parameters (and in particular, the dissociation rate koff) have been proposed as more effective predictors of in vivo selectivity.39 Slower values for koff mean longer target-bound residence times40 and higher target selectivity. In contrast, increases in koff have been correlated with decreases in inhibitor activity.41 Therefore, to assess the kinetics of binding by dicarba analogs, we turned to surface plasmon resonance.
For binding studies using SPR, biotinylated HIV-1 FSS RNA was immobilized on a Biacore CM5 SPR sensor chip coated with streptavidin. Compounds 1 and 9–11 were then flowed over the HIV-1 FSS RNA functionalized SPR chips in the running buffer (1X PBS buffer equipped with 5 mM MgCl2 and 0.005 % Tween-20). Representative sensorgrams are provided in supplementary information, while kinetic and equilibrium binding constants are summarized in Table 4. The binding constant (KD = 7.88 μM) obtained for 1 (Table 4) in this experimental setup (where HIV-1 FSS RNA is immobilized onto the chip, or the reverse of our previously reported method) is in conformity with binding constant (KD = 4.1 ± 2.40 μM) measured with the earlier experiments. It is interesting to note that immobilization of either binding partner for SPR appears to cause an order-of- magnitude loss in the affinity of these compounds relative to solution-phase measurements. Likewise, measured dissociation constants for 9–11 were at least 10-fold weaker in this format than in the fully solution phase measurements.
Table 4.
Evaluation of binding kinetics using SPR. The kinetic parameters kon and koff were obtained from a 1:1 global fit to six sensorgrams generated by injecting different concentrations of each ligand. The dissociation constant (KD) for neomycin was obtained from a steady-state analysis of seven sensorgrams; obtaining kinetic constants for this compound was not possible.
| Compound | Dissociation rate (koff) s−1 | Association rate (kon) M−1s−1 | Dissociation Constant (KD) μM | Chi2 |
|---|---|---|---|---|
| 1 | 1.41 × 10−2 | 1.79 × 103 | 7.88 | 1.07 |
| 9 | 1.28 × 10−2 | 1.15 × 103 | 11.20 | 1.85 |
| 10 | 7.5 × 10−3 | 1.62 ×103 | 4.66 | 2.73 |
| 11 | 1.3 ×10−2 | 1.05 × 103 | 12.30 | 1.09 |
| Neomycin | ND | ND | 2.6 | 0.18 |
The kinetics data obtained from SPR studies (Table 4) show that the koff of compounds 1 and 9-11 lie in the range of 10−2 to 10−3 s−1, corresponding to a dissociative half-life (t1/2 = 0.693/koff) of 50 to 90 seconds. Dissociative half-life is a parameter that determines the residence time of a ligand at the target site, which in turn governs target selectivity. Indeed, it has been hypothesized that the poor selectivity shown by some aminoglycosides is due at least in part to their relatively high koff (significant amount of dissociation in 30 s).42 To test this in the context of the HIV-1 FSS sequence, we examined the binding of neomycin by SPR. While steady state methods could be used to derive a binding constant (2.6 μM), both kon and koff were too rapid to determine accurately from the data (Figure S13). This is consistent with the hypothesized relationship between the off rate and sequence selectivity, as unlike our compounds neomycin is known to bind with similar affinity to a diverse range of RNAs.1 Hence, these data support the assertion that compounds of the structural class defined by 1, 9, 10, and 11 are promising candidates for further development as selective RNA binding ligands.
While the analyses described thus far suggest compounds 9 - 11 possess significant selectivity for target RNA sequences, a more stringent test is to examine their compatibility with cells, since a lack of selectivity in RNA-binding ability can manifest as toxicity.43 We therefore determined the tolerance of human fibroblasts to compound 10 using the widely employed MTT assay,44 which serves as a reporter for mitochondrial activity. We were gratified to observe that compound 10 caused no statistically significant change in cell viability at concentrations up to 1.0 millimolar. In contrast, mitomycin C, a DNA cross-linking agent widely used in chemotherapy, caused significant cell death at all concentrations tested (Figure 3).
Figure 3.

MTT assay for compound 10 (blue) vs. Mitomycin C (red). Results suggest that compound 10 is non-toxic to human fibroblasts at concentrations up to 1.0 millimolar.
In conclusion, we have employed directed analog synthesis to improve our understanding of the interaction of RBDCC-derived compounds with the HIV-1 FSS RNA, a critical regulatory element of HIV replication. The picture that emerges based on the data obtained to date is that the 2-ethyl 3-carboxyquinoline moieties of 1 and dicarba analogs 9 - 11 are the primary source of affinity, most likely via intercalation, while selectivity of binding resides in the peptide (Figure 4). Replacement of the disulfide moiety with an olefin bioisostere does not diminish activity, a discovery that is an important first step towards the production of compounds suitable for cellular assays of frameshifting. Finally, compounds are non-toxic to cells at relevant concentrations. Neither compound 1 nor analogs 9 - 11 are sufficiently conformationally rigid to permit generation of meaningful hypotheses with regard to their RNA-bound conformation in the absence of experimental data; thus, X-ray or NMR structural analysis will be an important next step towards understanding the behavior of these compounds. Efforts are underway to employ this newfound understanding in the design and synthesis of new compounds with further improved affinity, selectivity, and biostability. In particular, as substantial portions of 9 and 10 are still peptidic, conversion to peptidomimetic structures is a primary current focus. It is also not yet clear whether the full peptide is required for selectivity, and therefore deletion studies will be important to test that question. Completion of these experiments will set the stage for in vitro studies designed to test the ability of such RNA-binding compounds to interfere with frameshifting in HIV.
Figure 4.

Schematic of binding results for analogs described herein: selective, high-affinity binding to the HIV-1 FSS requires both peptide and heterocyclic portions of lead compounds. Affinity is largely determined by the presence of the quinoline moiety, while selectivity derives from the peptide. Binding appears insensitive to olefin geometry.
Experimental Section
Materials and General Methods
Commercially available reagents were obtained from Aldrich Chemical Co. (St. Louis, MO), Fluka Chemical Corp. (Milwaukee, WI), and TCI America (Portland, OR) and used as received unless otherwise noted. Water used for reactions and aqueous work-up was double distilled. Reagent grade solvents were used for all non-aqueous extractions. Reactions were monitored by analytical thin-layer chromatography using EM silica gel 60 F-254 pre-coated glass plates (0.25 mm). Compounds were visualized on the TLC plates with a UV lamp (λ = 254 nm) and staining with I2/SiO2. Synthesized compounds were purified using flash chromatography on EM silica gel 60 (230–400) mesh, or alternatively via preparative reverse-phase HPLC.
Analysis
1H NMR spectra were recorded at 25 °C on either a Bruker Avance 400 (400 MHz) or a Bruker Avance 500 (500 MHz) instrument. Chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane and referenced to the residual protium signal in the NMR solvent (CDCl3, δ = 7.26). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), integration, and coupling constant (J) in Hertz (Hz). 13C NMR spectra were recorded at 25 °C on a Bruker Avance 400 (100 MHz) or Bruker Avance 500 (125 MHz). Chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane and referenced to carbon resonances in the NMR solvent. High resolution mass spectra (HRMS) were acquired at the University of Buffalo mass spectrometry facility, Buffalo, New York, or at the mass spectrometry facility of the University of California, Riverside.
Binding analysis
Fluorescence titrations were performed on a Cary Eclipse Fluorescence spectrophotometer using a 10 mm path-length semi-micro quartz fluorescence cell with 400 μL sample holding capacity. The 5′-Cy3 labeled HIV-1 FSS RNA was purchased from Integrated DNA Technologies, Inc. Autoclaved 1X Phosphate-Buffered Saline (PBS) (4.3 mM Na2HPO4, 1.47 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl; pH = 7.2) was used as a buffer to dissolve the RNA and the compounds to be tested. A solution of HIV-1 FSS RNA in 1X PBS buffer (400 μL, 400 nM or 500 nM) was heated to 65 °C for 4 min and 2 μL of a MgCl2 solution (1 M) was added to it. The solution was then allowed to cool slowly to RT in the presence of MgCl2 in order to ensure that the RNA assumes its secondary stem-loop structure. This solution of 5′-Cy3 labeled HIV-1 FSS RNA was taken in the cell and excited with a wavelength of 550 nm and a 2.5 nm slitwidth. The emission spectrum was collected from 555 to 600 nm wavelength range at a PMT voltage of 715 V and a 5 nm slitwidth. The compound in 1X PBS buffer was added to the cell in either 2 or 4 μL increments from the stock solution (either 20, 50 or 200 μM) leading to a concentration range starting from 100 nM to 10 μM. After each addition, the solution was allowed to equilibrate for a minimum of 10 minutes, and fluorescence emission spectra were taken. Equilibrium was determined to be established after obtaining three similar fluorescence spectra taken at 1-minute intervals. The decrease in fluorescence of 5′-Cy3 labeled HIV-1 FSS RNA was then noted at 564.1 nm for each added concentration of the compound. The fluorescence units (FU) were then dilution corrected, and the FU after each addition was subtracted from the FU at zero compound concentration to give ΔFU. This ΔFU was plotted against compound concentration using Origin 7 (OriginLab corporation). The data were fit to a 1-site binding model accounting for ligand depletion.45 In this analysis, the free ligand concentration term is substituted with total ligand concentration minus the bound ligand concentration (LT-B), as:
Solving the equation for B (real solution for the quadratic equation),
Where,
B = bound ligand:receptor complex concentration.
LT= total ligand concentration
KD = dissociation constant
RT = total receptor concentration
Titrations were carried out with each compound at least 2 times. Buffer control titrations were performed by titrating similar amounts of PBS pH 7.2 into the Cy-3 RNA, and confirmed a concentration dependant linear fluorescence change matching what is theoretically expected. For the competition experiments with yeast total tRNA, the competing RNA was added to the 5′-Cy3 labeled HIV-1 FSS RNA at a concentration of 8 or 16 μM. This mixture of RNA was then titrated with the compounds in a similar manner described above. Titrations into other labeled sequences (entries 5 through 11, Table 1) were carried out analogously to the procedure employed for the HIV-1 FSS RNA. Surface plasmon resonance (SPR) experiments were conducted using a Biacore-X instrument (GE Healthcare); procedures are described in the main text, and in greater detail in the Supplementary Information.
Synthesis of analogs 2–7
Compounds 2–7 were synthesized on resin using methods previously described for compound 1. Following completion of the synthesis, compounds were cleaved from resin, ether precipitated, and purified by reverse phase preparative HPLC using water-acetonitrile gradient with 0.1 % TFA. Purified compounds were verified as being > 95% purity by analytical HPLC.
Compound 2: FTIR (neat)
3065.52, 3018.87, 2953.30, 2920.99, 2915.69, 1664.45, 1643.24, 1634.56, 1538.12, 1433.38, 1312.95, 1296.08, 1241.50, 1198.68, 1173.60, 1125.87, 1021.72, 952.29 cm−1. 1H NMR (400 MHz, CD3OD): δ 8.6 (s, 1H), 8.58 (s, 1H), 8.06 (d, 1H, J = 8.3), 8.03-7.83 (m, 6H), 7.71-7.61 (t, 1H, J = 7.34, J = 7.83), 7.30-7.7.12 (m, 10H), 7.09 (d, 4H, J = 6.85), 5.33 (q, 1H, J = 4.88), 4.5-4.3 (m, 4H), 4.01-3.91 (m, 1H), 3.91-3.78 (m, 2H), 3.59-3.50 (m, 1H), 3.5 (dd, 1, J = 9.23, J = 4.88), 3.40-3.30 (m, 1H), 3.27-2.96 (m, 2H), 2.90-2.70 (m, 10H), 2.71-2.65 (t, 2H, J = 1.95, J = 2.93), 2.66-2.56 (s, 3H), 2.22-2.04 (m, 4H), 2.05-1.91 (m, 4H), 1.92-1.79 (m, 4H), 1.80-1.67 (m, 6H), 1.66-1.46 (m, 4H). 13C NMR (100 MHz, CD3OD): δ 172.5, 169.6, 167.7, 161.45, 161.1, 156.4, 143.9, 136.9, 132.6, 129, 128.8, 128.2, 127.8, 127.7, 126.5, 125.9, 124.3, 60.7, 56.6, 55.1, 51.1, 39, 36.9, 36.6, 35.5, 28.9, 27.1, 24.5, 20.9. HRMS m/z calculated for C62H75N12O8S2 [M+H] +: 1179.5267; found: 1179.5299
Compound 3: FTIR (neat)
3068.12, 2960.53, 2940.76, 2450.23, 2270.34, 1651.43, 1634.08, 1538.12, 1435.90, 1199.16, 1127.32 cm−1. 1H NMR (400 MHz, CD3OD): δ 9.08 (s, 1H), 8.68 (d, J = 18.2 Hz, 1H), 8.06–7.75 (m, 6H), 7.62 (dd, J = 17.0, 10.1 Hz, 2H), 7.32–7.07 (m, 10H), 5.35–5.21 (m, 2H), 4.53–4.32 (m, 3H), 4.23 (d, J = 8.4 Hz, 1H), 4.07 – 3.91 (m, 2H), 3.80 (s, 2H), 3.55 – 2.89 (m, 22H), 2.76 (dd, J = 14.5, 7.2 Hz, 4H), 2.10 (dd, J = 19.8, 8.1 Hz, 2H), 2.00 – 1.63 (m, 9H), 1.67 – 1.35 (m, 2H). 13C NMR (100 MHz, CD3OD): δ 172.6, 170.0, 161.2, 147.7, 137.2, 136.8, 132.0, 128.8, 126.9, 126.5, 60.7, 59.3, 55.1, 54.1, 50.1, 39.1, 39.8, 36.5, 35.4, 28.8, 27.1, 25.6. HRMS m/z calculated for C60H71N12O8S2 [M+H]+: 1151.4954; found: 1151.4931.
Compound 4: FTIR (neat)
3150.13, 3100.92, 2950.45, 2834.56, 1651.92, 1635.04, 1553.55, 1539.09, 1516.91, 1435.90, 1419.03, 1199.16, 1173.60, 1127.80, 1021.24, 951.81 cm−1. 1H NMR (400 MHz, CD3OD); δ 8.48-8.3 (m 1H), 8.38 (d, 1H, J = 4.89), 7.75-7.67 (m, 2H), 7.66 (d, 1H, J= 7.83), 7.38 (dd, 2H, J = 4.40, J = 4.40), 7.35-7.15 (m, 12H), 7.05 (dd, 2H, J = 1.96, J = 1.46), 5.22-5.16 (m, 2H), 4.91 (d, 1H, J = 3.91), 4.81 (d, 1H, J = 3.91), 4.42 (q, 2H, J = 6.84, J = 1.95, J = 6.35), 4.34 (q, 2H, J = 4.89, J = 3.42, J = 4.89), 3.94 (m, 2H), 3.75 (m, 2H), 3.62 (d, 1H, J = 2.44), 3.26-3.17 (m, 4H), 2.86-2.75 (m, 4H), 2.64 (s, 3H). 13C NMR (100 MHz, CD3OD); δ 18.59, 24.45, 24.38, 27.05, 28.78, 35.45, 36.64, 38.98, 50.57, 55.11, 60.89, 125.86, 126.51, 128.16, 128.82, 134.63, 136.96, 140.43, 145.59, 147.31, 166.74, 170.23, 172.50. HRMS m/z calculated for C54H71N12O8S2 [M+H]+: 1079.4954; found: 1079.4957
Compound 5: FTIR (neat)
3068.39, 3029.00, 2995.31, 2967.15, 2950.40, 2932.08, 2929.67, 1655.29, 1650.95, 1645.171575.25, 1524.14, 1488.46, 1444.10, 1435.42, 1388.17, 1350.08, 1320.18, 1199.64, 1176.50, 1131.65, 1001.95, 959.05. 1H NMR (400 MHz, CD3OD); δ 7.35-7.27 (m, 10H), 4.64-4.49 (tt, 6H, J = 3.66, J = 5.79, J = 5.49, J = 6.71), 3.86-3.80, (m, 2H), 3.79-3.70 (m, 2H, 3.55-3.49 (tt, J = 4.57, J = 7.62, J = 5.19, J = 5.79, J = 8.55) 3.28-3.09 (m, 10H), 2.90-2.71 (m, 6H), 2.29-2.24 (m, 2H), 2.14-2.06 (m, 7H), 1.99-1.73 (m, 5H). 13C NMR (125 MHz, CD3OD); δ 13C NMR (126 MHz, MeOD) δ 173.33, 172.97, 166.88, 162.37, 137.67, 129.94, 129.10, 129.03, 127.48, 61.31, 56.19, 51.57, 49.00, 48.83, 48.66, 48.49, 48.32, 48.15, 47.98, 39.91, 38.19, 37.79, 37.49, 36.37, 29.92, 27.99, 25.60. HRMS m/z calculated for C40H61N10O6S2 [M+H]+: 841.4211 found: 841.4208
Compound 6: FTIR (neat)
3021.78, 2985.12, 2966.80, 2920.51, 1668.31, 1651.92, 1634.56, 1538.61, 1468.69, 1464.35, 1298.00, 1265.70, 1231.46, 1198.88, 1173.60, 1050.34 cm−1. 1H NMR (400 MHz, CD3OD); δ 8.46 (d, 1H, J = 16.62), 8.37 (s, 2H), 7.29-7.20 (m, 18H), 7.06 (d, 6H, J = 7.83), 6.22 (d, 4H, J = 4.40), 5.54 (s, 2H), 5.28 (q, 1H, J = 4.48, J = 4.40, J= 4.48), 4.44, (m, 2H), 3.89 (q, 1H, J = 7.34, J = 6.85), 3.89 (q, 1H, J = 7.34, 6.85), 3.79 (q, 3H, J = 8.80, J = 6.35), 3.43 (dd, 1H, J = 8.91, J = 8.91), 3.29-2.95 (m, 10H), 2.74 (q, 2H, J = 6.85, J = 6.85), 2.68, (d, 1H, J = 1.96), 2.64 (q, 2H), 2.06 (m, 2H), 1.94 (q, 2H, J = 6.35, J= 6.84, J = 8.31), 1.72 (t, 3H, J = 6.84), 1.27 (t, 3H, J = 7.82). 13C NMR (100 MHz, CD3OD); δ 172.5, 171.12, 169.52, 167.55, 157.77, 149.46, 143.96, 136.89, 128.89, 129.02, 125.86, 103.39,102.95, 60.59, 56.63, 55.142, 38.99, 36.61, 35.466, 28.92, 27.05, 24.51, 13.45. HRMS m/z calculated for C66H79N12O12S2 [M+H]+: 1295.5376; found: 1295.5350
Compound 7: FTIR (neat)
3100.23, 3050.10, 2950.66, 1690.89, 168.31, 1668.28, 1652.40, 1635.04, 1567.89, 1234.99, 1200.61, 1174.09, 1129.24 cm−1. 1H NMR (400 MHz, CD3OD); δ 8.64 (d, 2H, J = 1.96), 8.33-8.18 (m, 9H), 7.85 (m, 4H), 7.32-5.15 (m, 14H), 5.16 (q, 2H, J = 6.36, J = 2.44, J = 5.87), 4.46 (m, 4H), 3.95 (q, 2H, J = 9.78, J = 7.34, J = 6.85), 3.78 (m 2H), 3.27 (m, 4H), 3.24 (d, 4H), 3.20-3.08 (m, 6H), 3.04-2.95 (m, 4H), 2.7 (m, 5H), 2.6 (d, 4H, J = 4.40), 2.15 (m, 2H), 2.01-1.85 (m, 2H), 1.69 (t, 4H, J = 6.84), 1.29 (d, 4H, J = 3.913). 13C NMR (100 MHz, CD3OD); δ 182.07, 182.02, 172.53, 170.42, 166.74, 138.36, 136.89, 135.20, 134.19, 133,40, 133.23, 132.50, 128.80, 128.72, 28.16, 127.02, 126.70, 126.54, 125.81, 60.84, 55.09, 51.96, 40.41, 38.98, 36.83, 36.61, 35.45, 28.89, 28.76, 27.07, 24.44. HRMS m/z calculated for C70H73N10O12S2 [M+H]+: 1309.4845; found: 1309.4823
Synthesis of dicarba analogs
Resin bound 8 was synthesized using standard Fmoc methodology for peptide synthesis. Wang resin (1.0 g, 100–200 mesh, 1 mmol/g loading) was activated with 1,1′-carbonyl-di-imidazole (DIC, 3.3 g, 10 mmol) in 12 mL of DMF, for 12 h on a LabQuake™ rotator. The resin was then washed three times each with DMF, CH2Cl2 and DMF again, followed by reaction with 1,3-diaminopropane (0.72 mL, 10 mmol) in DMF (12 mL), for another 12 h. The wash cycle was then repeated. The coupling of the first amino acid was carried out by adding Fmoc-L-Phe-OH (1.16 g, 3 mmol), HBTU (1.14 g, 3 mmol) and DIPEA (0.85 mL, 5 mmol) in 12 mL of DMF to the resin, and rotating the reaction mixture for 1 h. Following a wash cycle, Fmoc deprotection was accomplished using 20% piperidine in CH2Cl2 for 0.5 h followed again by the wash cycle. Similarly, Fmoc-L-Pro-OH, Fmoc-L-allylglycine and 2-ethyl-quinoline-3-carboxylic acid were coupled to synthesize resin-bound monomeric compound (8).
The resin was then split into two equal parts of 0.50 g. One part was treated with 30% TFA/1% TEA in CH2Cl2 for 0.5 h to obtain a cleaved product 8 (0.23 g) to be used as the solution olefin component for the metathesis reaction. The other part of the resin was dried in a dessicator under vacuum for 12 h, then allowed to swell in dry CH2Cl2 (12 mL) for 20 min. The resin was washed with CH2Cl2 (3 × 10 mL) followed by 0.8 M LiCl in DMF (10 mL) for 10 min. The resin was then washed with DMF (10 mL) and the 0.8 M LiCl wash was repeated for two more times. Finally, the resin was washed with dry, degassed 1,2-dichloroethane (10 mL) and then suspended in the same solvent (5 mL). To this suspension were added Grubbs’ 2nd generation catalyst (0.14 g, 0.17 mmol) in 5 mL of 1,2-dichloroethane and the cleaved 8 (0.25 g, 0.42 mmol) in 10 mL of a 1:4 mixture of CH2Cl2 and 1,2 dichloroethane. The reaction mixture was refluxed for 24 hours. The reaction was then cooled to room temperature and additional Grubbs’ 2nd generation catalyst (0.07 g, 0.09 mmol) was added to the reaction mixture and refluxed for another 24 hours. After repeating this cycle a third time, the reaction mixture was cooled to room temperature and transferred to a standard solid-phase reaction vessel with filtering. The resin was then washed with CH2Cl2 (3 × 10), DMF (3 × 10 mL) and suspended in 10 mL of DMF. DMSO (0.2 mL) was added to the suspension and rotated for 12 h, in order to remove colored ruthenium impurities.46 The resin was then washed and the product cleaved off the resin with 25% TFA in CH2Cl2 (10 mL) for 0.5 h to obtain a crude mixture of olefins 9 and 10 (0.25 g, 64%). The isomers were then separated using preparative RP-HPLC (isocratic elution, 30% acetonitrile in water with 0.1% TFA).
Compound 8: FTIR (neat)
3249, 2934, 2817, 1672, 1659, 1643, 1634, 1529, 1517, 1201, 1177, 1128, 1026 cm−1. 1H NMR (400 MHz, CDCl3): δ 8.90 (s, 1H), 8.30 (t, 1H, J = 8.8 Hz), 8.09 (d, 1H, J = 8.4Hz), 8.03 (t, 1H, J = 7.8 Hz), 7.82 (t, 1H, 7.8 Hz), 7.71-7.69 (m, 3H), 7.31-7.27 (m, 3H), 7.12 (d, 2H, 6.8Hz), 5.88-5.84 (m, 1H), 5.34-5.24 (m, 2H), 4.49-4.88 (m, 1H), 4.59-4.5 (m, 1H), 4.32-4.28 (m, 1H), 3.98 (s, 1H), 3.68-3.62 (m, 1H), 3.39-3.21 (m, 4H), 3.11-3.10 (m, 2H), 2.94-2.90 (m, 2H), 2.46-2.42 (m, 2H), 2.22-2.13 (m, 1H), 2.07-2.01 (m, 1H), 1.96-1.90 (m, 1H), 1.85-1.80 (m, 3H), 1.35 (t, 3H, J = 7.4Hz), 1.25 (s, 2H). 13C NMR (100 MHz, CDCl3): 172.2, 171.9, 171.4, 166.2, 161.4, 136.4, 132.6, 129.7, 129.0, 128.6, 128.4, 127.1, 126.1, 119.3, 61.4, 54.5, 51.4, 47.9, 37.0, 36.7, 35.9, 35.8, 28.7, 27.4, 26.8, 24.9, 15.2, 14.2. HRMS m/z calculated for C34H42N6O4 [M+H]+: 599.3346; found: 599.3357.
Compound 9: FTIR (neat)
2957.1, 2920.2, 2853.5, 1673.1, 1667.8, 1457.1, 1377.0, 1201.1, 1179.3, 1134.1 cm−1. 1H NMR (500 MHz, CD3OD): δ 8.63 (s, 2H), 8.08 (d, 2H, J = 8.0 Hz), 8.04 (d, 2H, J = 9.0 Hz), 7.89 (t, 2H, J = 8Hz), 7.68 (t, 2H, J = 8.0 Hz), 7.21-7.11 (m, 10H), 5.78 (m, 2H), 4.48-4.42 (m, 2H), 4.36 (t, 2H, J = 7.5 Hz), 3.92-3.88 (m, 2H), 3.66-3.60 (m, 4H), 3.55-3.51 (m, 1H), 3.17-3.10 (m, 4H), 3.02-2.90 (m, 4H), 2.81 (s, 1H), 2.17-2.62 (m, 6H), 2.54-2.52 (m, 1H), 2.15-2.08 (m, 2H), 1.91-1.88 (m, 4H), 1.83-1.79 (m, 2H), 1.68-1.63 (m, 5H), 1.34-1.21 (m, 8H), 0.87-0.80 (m, 2H). 13C NMR (100 MHz, CD3OD): 173.9, 171.9, 168.8, 162.3, 138.0, 134.2, 130.7, 130.2, 130.1, 129.9, 129.8, 129.5, 129.4, 129.3, 127.8, 127.4, 61.5, 56.5, 53.3, 38.4, 37.9, 36.7, 35.0, 29.1, 28.4, 25.9, 14.5. HRMS m/z calculated for C66H80N12O8 [M+H]+: 1169.6300; found: 1169.6296
Compound 10: FTIR (neat)
2957.6, 2920.0, 2900.7, 2851.1, 1664.3, 1634.3, 1557.4, 1538.6, 1201.1, 1180.3, 1133.1 cm−1. 1H NMR (500 MHz, CD3OD): δ 8.67 (s, 2H), 8.11-8.05 (m, 4H), 7.98-7.93 (m, 2H), 7.77-7.71 (m, 2H), 7.25-7.17 (m, 10H), 5.82-5.79 (m, 2H), 4.97-4.94 (m, 1H), 4.48-4.45 (m, 2H), 3.97-3.92 (m, 2H), 3.72-3.65 (m, 4H), 3.21-3.16 (m, 8H), 3.05-3.20 (m, 4H), 2.76-2.69 (m, 6H), 2.18-2.12 (m, 2H), 1.97-1.93 (m, 3H), 1.87-1.83 (m, 2H), 1.72-1.69 (m, 4H), 1.39-1.28 (m, 10H). 13C NMR (100 MHz, CD3OD): δ 172.6, 172.5, 170.3, 167.7, 161.0, 136.8, 132.8, 129.5, 128.7, 128.5, 128.1, 127.9, 127.6, 126.5, 126.1, 124.2, 69.8, 60.3, 55.2, 51.9, 37.1, 36.6, 35.4, 29.0, 27.6, 24.6, 13.2. HRMS m/z calculated for C66H80N12O8 [M+H]+: 1169.6300; found: 1169.6297
Compound 11
Hydrogenation was performed on resin. The resin-bound olefin metathesis product (resin: 0.25 g, 0.25 mmol) was suspended in 6 mL of 10 % MeOH in CH2Cl2 in a 16 mL glass vial. Wilkinson’s catalyst (0.06 g, 0.06 mmol) was added to this suspension, and the mixture was agitated on a Parr hydrogenator under 40 psi H2 gas pressure at RT for 5 h. The resin was then washed with 10 % MeOH in CH2Cl2 (3 × 10 mL), MeOH (3 × 10 mL), and CH2Cl2 (3 × 10 mL). The resin-bound reduced product was then cleaved using 30 % TFA/1 % TES in CH2Cl2 (6 mL). The cleaved product was collected and the solvent was removed in vacuo. The crude product was then precipitated using cold ether, centrifuged (5000 × g, 5 minutes), and the ether was decanted to obtain pure 11 in 60 % yield (0.08 g).
FTIR (neat)
3300.3, 2959.5, 2910.2, 1675.4, 1652.4, 1641.2, 1621.5, 1535.7, 1199.2, 1190.5, 1177.0, 1124.9 cm−1. 1H NMR (500 MHz, CD3OD): δ 8.67 (s, 2H), 8.10-8.05 (m, 4H), 7.95-7.91 (m, 2H), 7.74-7.69 (m, 2H), 7.24-7.20 (m, 10H), 4.50-4.40 (m, 4H), 4.20 (dd, 2H, J1=7.5 Hz, J2 = 2.5 Hz), 4.03-3.98 (m, 2H), 3.82-3.64 (m, 3H), 3.48 (q, 2H, J = 17.5 Hz), 3.27-3.14 (m, 10H), 3.05-3.02 (m, 4H), 2.98 (bs, 2H), 2.78-2.67 (m, 4H), 2.22-2.09 (m, 3H), 1.73-1.65 (m, 4H), 1.39-1.28 (m, 6H), 0.99-0.89 (m, 4H), 0.59 (m, 2H). δ 172.6, 172.5, 161.2, 161.0, 138.3, 136.8, 131.9, 129.6, 128.9, 128.1, 127.3, 126.4, 125.9, 125.3, 60.2, 55.2, 51.8, 36.9, 36.5, 35.3, 28.9, 28.3, 27.0, 25.1, 24.6, 13.2. HRMS m/z calculated for C66H82N12O8Na [M+Na]+: 1193.6271; found: 1193.6243
Cell culture and in vitro cytotoxicity assay
Human fibroblast cells were cultured in Dulbecco’s modified eagle medium (DMEM), containing 5 % fetal bovine serum (Invitrogen) and 5 % pen-strep. Cell cultures were incubated in a humidified atmosphere containing 5 % CO2 at 37 °C. The viability of human fibroblasts in the presence of compound 10 was tested using the 3-(4,5-dimethylthiazole-2yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were plated in a 96 well sterile plate at concentrations of 3.2 × 104 cells per well in a volume of 100 μL culture media. The cells were then allowed to grow to approximately 80 % confluence by incubating at 37 °C for 48 hours. The old media was removed and replaced with 100 μL of different concentrations (up to 1 mM) of compound 10 or mitomycin C (as a control) in DMEM. Cells were exposed to compounds for period of 24 hours, after which the media were removed. Next, MTT was added to each of the wells and incubated for 4 hours. Isopropyl alcohol was added to the cells after removal of the MTT medium followed by absorbance measurement at 600 nm. Absorbance values were obtained on a Modulus microplate reader (Turner Biosystems).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge financial support of the NIH/NIAID P30AI078498, via the University of Rochester Developmental Center for AIDS Research (DCFAR), and NIH T32AR007472 (training grant support to PCG). We thank Prof. Thomas Foster for the use of his fluorimeter, and Prof. Lisa DeLouise for guidance with the MTT assays.
Footnotes
Supplementary Material Available: Spectral data for compounds 2 - 11, selected fluorescence titration data for compounds 2 - 11, SPR data and controls for compounds 9 - 11, and Dynamic Light Scattering data for compounds 9 - 11. (39 pp.)
Abbreviations: HIV-1 FSS: Frameshift-stimulating RNA sequence of HIV-1; SPR: Surface Plasmon Resonance; RBDCC: Resin-Bound Dynamic Combinatorial Chemistry; RBDCL: Resin-Bound Dynamic Combinatorial Library; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
References
- 1.For reviews, see: Tor Y. Targeting RNA with small molecules. ChemBioChem. 2003;4:998–1007. doi: 10.1002/cbic.200300680.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546.
- 2.Dervan PB, Bürli RW. Sequence-specific DNA recognition by polyamides. Curr Opin Chem Biol. 1999;3:688–693. doi: 10.1016/s1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]
- 3.Wender PA, Miller BL. Synthesis at the Molecular Frontier. Nature. 2009;460:197–201. doi: 10.1038/460197a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flexner C. HIV drug development: the next 25 years. Nat Rev Drug Disc. 2007;6:959–966. doi: 10.1038/nrd2336. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Cajas JL, Wainberg MA. Protease inhibitor resistance in HIV-infected patients: molecular and clinical perspectives. Antiviral Res. 2007;76:203–221. doi: 10.1016/j.antiviral.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 6.De Clercq E. HIV resistance to reverse transcriptase inhibitors. Biochem Pharmacol. 1994;47:155–169. doi: 10.1016/0006-2952(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 7.Struble K, Murray J, Cheng B, Gegeny T, Miller V, Gulick R. Antiretroviral therapies for treatment-experienced patients: current status and research challenges. AIDS. 2005;19:747–756. doi: 10.1097/01.aids.0000168968.34810.ca. [DOI] [PubMed] [Google Scholar]
- 8.(a) Zapp ML, Stern S, Green MR. Small molecules that selectively block RNA binding of HIV-1 Rev protein inhibit Rev function and viral production. Cell. 1993;74:969–978. doi: 10.1016/0092-8674(93)90720-b. [DOI] [PubMed] [Google Scholar]; (b) Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, Frankel AD, Williamson JR. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science. 1996;273:1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]; (c) Kirk SR, Luedtke NW, Tor Y. Neomycin-Acridine Conjugate: A Potent Inhibitor of Rev-RRE Binding. J Am Chem Soc. 2000;122:980–981. [Google Scholar]; (d) Hendrix M, Priestley ES, Joyce GF, Wong CH. Direct Observation of Aminoglycoside-RNA Interactions by Surface Plasmon Resonance. J Am Chem Soc. 1997;119:3641–3648. doi: 10.1021/ja964290o. [DOI] [PubMed] [Google Scholar]
- 9.Mei HY, Cui M, Heldsinger A, Lemrow SM, Loo JA, Sannes-Lowery KA, Sharmeen L, Czarnik AW. Inhibitors of protein-RNA complexation that target the RNA: specific recognition of human immunodeficiency virus type 1 TAR RNA by small organic molecules. Biochemistry. 1998;37:14204–14212. doi: 10.1021/bi981308u. [DOI] [PubMed] [Google Scholar]
- 10.(a) Tang H, Kuhen KL, Wong-Staal F. Lentivirus replication and regulation. Annu Rev Genet. 1999;33:133–170. doi: 10.1146/annurev.genet.33.1.133. [DOI] [PubMed] [Google Scholar]; (b) Frankel AD, Young JA. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]; (c) Ludwig V, Krebs A, Stoll M, Dietrich U, Ferner J, Schwalbe H, Scheffer U, Dürner G, Göbel MW. Tripeptides from synthetic amino acids block the Tat-TAR association and slow down HIV spread in cell cultures. Chembiochem. 2007;8:1850–1856. doi: 10.1002/cbic.200700232. [DOI] [PubMed] [Google Scholar]; (d) Wang D, Iera J, Baker H, Hogan P, Ptak R, Yang L, Hartman T, Buckheit RW, Jr, Desjardins A, Yang A, Legault P, Yedavalli V, Jeang K-T, Appella DH. Multivalent binding oligomers inhibit HIV Tat-TAR interaction critical for viral replication. Bioorg Med Chem Lett. 2009;19:6893–6897. doi: 10.1016/j.bmcl.2009.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. 1988;331:280–283. doi: 10.1038/331280a0. [DOI] [PubMed] [Google Scholar]; (b) Parkin NT, Chamorro M, Varmus HE. Human immunodeficiency virus type 1 gag-pol frameshifting is dependent on downstream mRNA secondary structure: demonstration by expression in vivo. J Virol. 1992;66:5147–5151. doi: 10.1128/jvi.66.8.5147-5151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Park J, Morrow CD. Overexpression of the gag-pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J Virol. 1991;65:5111–5117. doi: 10.1128/jvi.65.9.5111-5117.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Karacostas V, Wolffe EJ, Nagashima K, Gonda MA, Moss B. Overexpression of the HIV-1 gag-pol polyprotein results in intracellular activation of HIV-1 protease and inhibition of assembly and budding of virus-like particles. Virol. 1993;193:661–671. doi: 10.1006/viro.1993.1174. [DOI] [PubMed] [Google Scholar]; (c) Hung M, Patel P, Davis S, Green SR. Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J Virol. 1998;72:4819–4824. doi: 10.1128/jvi.72.6.4819-4824.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shehu-Xhilaga M, Crowe SM, Mak J. Maintenance of the Gag/Gag-Pol Ratio Is Important for Human Immunodeficiency Virus Type 1 RNA Dimerization and Viral Infectivity. J Virol. 2001;75:1834–1841. doi: 10.1128/JVI.75.4.1834-1841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Dulude D, Berchiche YA, Gendron K, Brakier-Gingras L, Heveker N. Decreasing the frameshift efficiency translates into an equivalent reduction of the replication of the human immunodeficiency virus type 1. Virol. 2006;345:127–136. doi: 10.1016/j.virol.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 13.Dulude D, Baril M, Brakier-Gingras L. Characterization of the frameshift stimulatory signal controlling a programmed -1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res. 2002;30:5094–5102. doi: 10.1093/nar/gkf657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Telenti A, Martinez R, Munoz M, Bleiber G, Greub G, Sanglard D, Peters S. Analysis of natural variants of the human immunodeficiency virus type 1 gag-pol frameshift stem-loop structure. J Virol. 2002:76, 7868–7873. doi: 10.1128/JVI.76.15.7868-7873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baril M, Dulude D, Gendron K, Lemay G, Brakier-Gingras L. Efficiency of a programmed -1 ribosomal frameshift in the different subtypes of the human immunodeficiency virus type 1 group M. RNA. 2003;9:1246–1253. doi: 10.1261/rna.5113603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hung M, Patel P, Davis S, Green SR. Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J Virol. 1998;72:4819–4824. doi: 10.1128/jvi.72.6.4819-4824.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gareiss PC, Miller BL. Ribosomal Frameshifting: An Emerging Target in HIV. Curr Opin Invest Drugs. 2009;10:121–128. [PubMed] [Google Scholar]
- 17.(a) Staple DW, Butcher SE. Solution structure of the HIV-1 frameshift inducing stem-loop RNA. Nucleic Acids Res. 2003;31:4326–4331. doi: 10.1093/nar/gkg654. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Staple DW, Butcher SE. Solution structure and thermodynamic investigation of the HIV-1 frameshift inducing element. J Mol Biol. 2005;349:1011. doi: 10.1016/j.jmb.2005.03.038. [DOI] [PubMed] [Google Scholar]; (c) Gaudin C, Mazauric MH, Traikia M, Guittet E, Yoshizawa S, Fourmy D. Structure of the RNA signal essential for translational frameshifting in HIV-1. J Mol Biol. 2005;349:1024–1035. doi: 10.1016/j.jmb.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 18.(a) Green L, Kim C-H, Bustamante C, Tinoco I. Characterization of the mechanical unfolding of RNA pseudoknots. J Mol Biol. 2008;375:511–528. doi: 10.1016/j.jmb.2007.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen G, Chang K, Chou M, Bustamante C, Tinoco I. Triplex structures in an RNA pseudoknot enhance mechanical stability and increase efficiency of -1 ribosomal frameshifting. Proc Natl Acad Sci USA. 2009;106:12706–12711. doi: 10.1073/pnas.0905046106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazauric M-H, Seol Y, Yoshizawa S, Visscher K, Fourmy D. Interaction of the HIV-1 frameshift signal with the ribosome. Nucl Acids Res. 2009;37:7654–7664. doi: 10.1093/nar/gkp779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNaughton BR, Gareiss PC, Miller BL. Identification of a selective small-molecule ligand for HIV-1 frameshift-inducing stem-loop RNA from an 11,325 member resin bound dynamic combinatorial library. J Am Chem Soc. 2007;129:11306–11307. doi: 10.1021/ja072114h. [DOI] [PubMed] [Google Scholar]
- 21.(a) Addess KJ, Sinsheimer JS, Feigon J. Solution structure of a complex between [N-MeCys3, N-MeCys7]TANDEM and [d(GATATC)]2. Biochemistry. 1993;32:2498–2508. doi: 10.1021/bi00061a006. [DOI] [PubMed] [Google Scholar]; (b) Addess KJ, Feigon J. Sequence specificity of quinoxaline antibiotics. 1. Solution structure of a 1: 1 complex between triostin A and [d(GACGTC)]2 and comparison with the solution structure of the [N-MeCys3, N-MeCys7]TANDEM-[d(GATATC)]2 complex. Biochemistry. 1994;33:12386–12397. doi: 10.1021/bi00207a005. [DOI] [PubMed] [Google Scholar]
- 22.Staple D, Venditti V, Niccolai N, Elson-Schwab L, Tor Y, Butcher S. Guanidinoneomycin B recognition of an HIV-1 RNA helix. Chembiochem. 2007;9:93–102. doi: 10.1002/cbic.200700251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcheschi RJ, Mouzakis KD, Butcher SE. Selection and characterization of small molecules that bind the HIV-1 frameshift site RNA. ACS Chem Biol. 2009;4:844–854. doi: 10.1021/cb900167m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dulude D, Théberge-Julien G, Brakier-Gringras L, Heveker N. Selection of peptides interfering with a ribosomal frameshift in the human immunodeficiency virus type 1. RNA. 2008;14:981–991. doi: 10.1261/rna.887008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) McCutchan FE. Understanding the genetic diversity of HIV-1. AIDS. 2000;14:S31–S44. [PubMed] [Google Scholar]; (b) Robertson DL, Anderson JP, Bradac JA, Carr JK, Foley B, Funkhouser RK, Gao F, Hahn BH, Kalish ML, Kuiken C, Learn GH, Leitner T, McCutchan F, Somanov S, Peeters M, Pieniazek D, Salminen B, Sharp PM, Wolinsky S, Korber B. HIV-1 nomenclature proposal. Science. 2000;288:55–56. doi: 10.1126/science.288.5463.55d. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Thomas JR, Hergenrother PJ. Deoxystreptamine Dimers bind to RNA Hairpin Loops. J Am Chem Soc. 2004;126:9196–9197. doi: 10.1021/ja048936l. [DOI] [PubMed] [Google Scholar]
- 27.Llano-Sotelo B, Chow CS. RNA-aminoglycoside antibiotic interactions: Fluorescence detection of binding and conformational change. Bioorg Med Chem Lett. 1999;9:213–216. doi: 10.1016/s0960-894x(98)00718-5. [DOI] [PubMed] [Google Scholar]
- 28.Luedtke NW, Liu Q, Tor Y. RNA-ligand interactions: affinity and specificity of aminoglycoside dimers and acridine conjugates to the HIV-1 Rev response element. Biochemistry. 2003;42:11391–11403. doi: 10.1021/bi034766y. [DOI] [PubMed] [Google Scholar]
- 29.Sequence 10: Karan C, Miller BL. RNA-Selective Coordination Complexes Identified via Dynamic Combinatorial Chemistry. J Am Chem Soc. 2001;123:7455–7456. doi: 10.1021/ja010325v.Sequence 11: Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, Miller BL. Dynamic Combinatorial Selection of Small Molecules Capable of Inhibiting the (CUG) Repeat RNA – MBNL1 Interaction in vitro: Discovery of Lead Compounds Targeting Myotonic Dystrophy (DM1) J Am Chem Soc. 2008;130:16254–16261. doi: 10.1021/ja804398y.
- 30.Szajewski RP, Whitesides GM. Rate constants and equilibrium constants for thiol-disulfide interchange reactions involving oxidized glutathione. J Am Chem Soc. 1980;102:2011–2026. [Google Scholar]
- 31.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 32.(a) Fotouhi N, Joshi P, Tilley JW, Rowan K, Schwinge V, Wolitzky B. Cyclic thioether peptide mimetics as VCAM-VLA-4 antagonists. Bioorg Med Chem Lett. 2000;10:1167–1169. doi: 10.1016/s0960-894x(00)00175-x. [DOI] [PubMed] [Google Scholar]; (b) Stymiest JL, Mitchell BF, Wong S, Vederas JC. Synthesis of biologically active dicarba analogues of the peptide hormone oxytocin using ring-closing metathesis. Org Lett. 2003;5:47–49. doi: 10.1021/ol027160v. [DOI] [PubMed] [Google Scholar]; (c) Berezowska I, Chung NN, Lemieux C, Wilkes BC, Schiller PW. Dicarba analogues of the cyclic enkephalin peptides H-Tyr-c[D-Cys-Gly-Phe-D(or L)-Cys]NH(2) retain high opioid activity. J Med Chem. 2007;50:1414–1417. doi: 10.1021/jm061294n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mollica A, Guardiani G, Davis P, Ma S, Porreca F, Lai J, Mannina L, Sobolev AP, Hruby VJ. Synthesis of stable and potent delta/mu opioid peptides: analogues of H-Tyr-c[D-Cys-Gly-Phe-D-Cys]-OH by ring-closing metathesis. J Med Chem. 2007;50:3138–3142. doi: 10.1021/jm061048b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicolaou KC, Hughes R, Cho SY, Winssinger N, Smethurst C, Labischinski H, Endermann R. Target-accelerated combinatorial synthesis and discovery of highly potent antibiotics effective against vancomycin-resistant bacteria. Angew Chem Int Ed. 2000;39:3823–3828. doi: 10.1002/1521-3773(20001103)39:21<3823::AID-ANIE3823>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 34.Hoveyda AH, Zhugralin AR. The remarkable metal-catalysed olefin metathesis reaction. Nature. 2007;250:243–251. doi: 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]
- 35.(a) Wels B, Kruijtzer JA, Garner K, Nijenhuis WA, Gispen WH, Adan RA, Liskamp RM. Synthesis of a novel potent cyclic peptide MC4-ligand by ring-closing metathesis. Bioorg Med Chem. 2005;13:4221–4227. doi: 10.1016/j.bmc.2005.04.027. [DOI] [PubMed] [Google Scholar]; (b) Stymiest JL, Mitchell BF, Wong S, Vederas JC. Synthesis of oxytocin analogues with replacement of sulfur by carbon gives potent antagonists with increased stability. J Org Chem. 2005;70:7799–7809. doi: 10.1021/jo050539l. [DOI] [PubMed] [Google Scholar]
- 36.McNaughton BR, Bucholtz KM, Camaano-Moure A, Miller BL. Self-Selection in Olefin Cross Metathesis: The Effect of Remote Functionality. Org Lett. 2005;7:733–736. doi: 10.1021/ol047361u. [DOI] [PubMed] [Google Scholar]
- 37.(a) Myers AG, Schnider P, Kwon S, Kung DW. Greatly Simplified Procedures for the Synthesis of alpha-Amino Acids by the Direct Alkylation of Pseudoephedrine Glycinamide Hydrate. J Org Chem. 1999;64:3322–3327. doi: 10.1021/jo990341z. [DOI] [PubMed] [Google Scholar]; (b) Ryan SJ, Zhang Y, Kennan AJ. Convenient access to glutamic acid side chain homologues compatible with solid phase peptide synthesis. Org Lett. 2005;7:4765–4767. doi: 10.1021/ol0520222. [DOI] [PubMed] [Google Scholar]
- 38.Galan BR, Kalbarczyk KP, Szczepankiewicz S, Keister JB, Diver ST. A rapid and simple cleanup procedure for metathesis reactions. Org Lett. 2007;9:1203–1206. doi: 10.1021/ol0631399. [DOI] [PubMed] [Google Scholar]
- 39.Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discovery. 2004;3:801–808. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- 40.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 41.Dierynck I, De Wit M, Gustin E, Keuleers I, Vandersmissen J, Hallenberger S, Hertogs K. Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier. J Virol. 2007;81:13845–13851. doi: 10.1128/JVI.01184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hendrix M, Priestley ES, Joyce GF, Wong CH. Direct observation of aminoglycoside-RNA interactions by surface plasmon resonance. J Am Chem Soc. 1997;119:3641–3648. doi: 10.1021/ja964290o. [DOI] [PubMed] [Google Scholar]
- 43.Begg EJ, Barclay ML. Aminoglycosides – 50 years on. Brit J Clin Pharmacol. 1995;39:597–603. [PMC free article] [PubMed] [Google Scholar]
- 44.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 45.(a) Motulsky H, Christopoulos A. Fitting Models to Biological Data Using Linear and Nonlinear Regression. A Practical Guide to Curve Fitting. Oxford University Press; New York: 2004. Analyzing Saturation Binding with Ligand Depletion; pp. 208–210. [Google Scholar]; (b) Fluorescence Polarization Technical Resource Guide. Chapter 7 Invitrogen, Inc; 2006. [Google Scholar]
- 46.Ahn YM, Yang K, Georg GI. A convenient method for the efficient removal of ruthenium byproducts generated during olefin metathesis reactions. Org Lett. 2001;3:1411–1413. doi: 10.1021/ol010045k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.