

Abstract
The BH3-only protein BIK normally induces apoptotic cell death. Here, we have investigated the role of BCL-2 in BIK-induced cell death using Bcl-2+/+ and Bcl-2−/− mouse embryo fibroblasts. Ectopic expression of BIK in Bcl-2−/− cells resulted in enhanced cell death compared to Bcl-2+/+ cells. In these cells, while caspase-8 was activated, there was no significant activation of caspase-9 and 3. There was no detectable mitochondrial to cytosolic release of cytochrome-c. However, there was significant redistribution of AIF from mitochondria to the nucleus. The extent of BIK-induced cell death was augmented by treatment with the pancaspase inhibitor, zVAD-fmk. The Bcl-2 null cells expressing BIK exhibited autophagic features such as cytosolic vacuoles, punctate distribution of LC3 and enhanced expression of Beclin-1. The survival of BIK-expressing Bcl-2−/− cells was enhanced in the presence of PI3 kinase inhibitors 3-methyladenine and Wortmannin and also by depletion of Atg5 and Beclin-1. Death of BIK-expressing Bcl-2−/− cells treated with zVAD-fmk was increased under caspase-8 depletion. Our results suggest enhanced expression of BIK in the Bcl-2 deficient cells leads to cell death with autophagic features and the extent of such cell death could be increased by inhibition of caspases.
Keywords: BIK, Bcl-2, autophagy, Beclin-1, LC3
Introduction
Programmed cell death is a normal physiological process that maintains tissue homeostasis during development. Apoptosis is the widely studied form of programmed cell death. The BCL-2 family proteins play a central role in regulation of apoptosis by promoting cell survival as well as cell death in animals. BCL-2 and its close relatives such as BCL-xL promote cell survival. Members of a second subfamily of the BCL-2 family proteins (known as BH123 proteins) share three conserved domains with BCL-2 and promote cell death. The proapoptotic proteins BAX and BAK are well-known members of this family. The third subfamily members share only the BH3 domain with other BCL-2 family proteins. The BH3-only proteins promote cell death and function upstream of BAX and BAK (Wei et al., 2001; Zong et al., 2001). The molecular mechanisms by which BH3-only proteins induce cell death via BAX and BAK remain a hotly debated issue.
Recently, studies on other forms of regulated cell death such as autophagic death and necrosis have received deserved attention (Tsujimoto and Shimizu, 2005; Golstein and Kroemer, 2007). Although autophagy has been recognized as a survival mechanism during nutritional deprivation, it is now accepted that the process ultimately leads to cell death that morphologically resemble necrosis. Certain known cytotoxic agents that normally induce apoptosis have been shown to induce autophagic death in cells that are deficient in apoptosis due to the lack of BAX and BAK (Shimizu et al., 2004; Lum et al., 2005). Thus, deficiency in apoptosis appears to divert cells to autophagic cell death. Chemical inhibitors of the PI3 kinase pathway (Shimizu et al., 2004) and ectopic expression of prosurvival proteins such as BCL-2 and vBCL-2 (Pattingre et al., 2005) have been shown to inhibit autophagic cell death.
BIK is the founding member of the BH3-only family proteins (Boyd et al., 1995). A number of reports have shown that ectopic overexpression of BIK results in apoptotic cell death. However, BIK has also been reported to cause nonapoptotic cell death in human malignant glioma (Naumann et al., 2003) and melanoma cells (Oppermann et al., 2005). While investigating the effect of BCL-2 in modulating the activity of BIK, we discovered that Bcl-2 null mouse embryo fibroblasts (MEFs) were highly sensitive to BIK-induced death. In these cells, BIK induced caspase-independent cell death with autophagic features. This form of BIK-induced cell death was enhanced by inhibition of the caspases.
Results
Effect of ectopic BIK overexpression in Bcl-2+/+ and Bcl-2−/− MEFs
To investigate the role of BCL-2 in modulating the activity of BIK, immortalized Bcl-2+/+ and Bcl-2−/− MEFs were infected with an adenovirus vector that expresses BIK (Ad-BIK) or LacZ (Ad-LacZ) at 500 PFU per cell. The effect of BIK expression on cell viability was determined using the MTS assay (Figure 1a). In Bcl-2+/+ cells, there was a modest reduction in viability from about 90% at 24 h infection to about 70% after 72 h. The extent of cell death was enhanced in Bcl-2−/− cells, where the viability ranged from 70% at 24 h infection to about 45% after 72 h. The levels of BIK (including the phosphorylated form) expression were comparable in both cell types (Figure 1b). Visualization of the nuclei stained with DAPI revealed largely intact nuclei, without significant condensation in Bcl-2+/+ cells infected with Ad-BIK or Ad-LacZ. In Bcl-2−/− cells, atypical nuclear morphologies and some nuclear condensation were observed (Figure 1c). These results suggest that BCL-2 protects against BIK-induced cell death (under the levels of ectopic BIK expression observed) while BCL-2 deficiency enhances BIK-induced cell death in MEFs.
Figure 1.
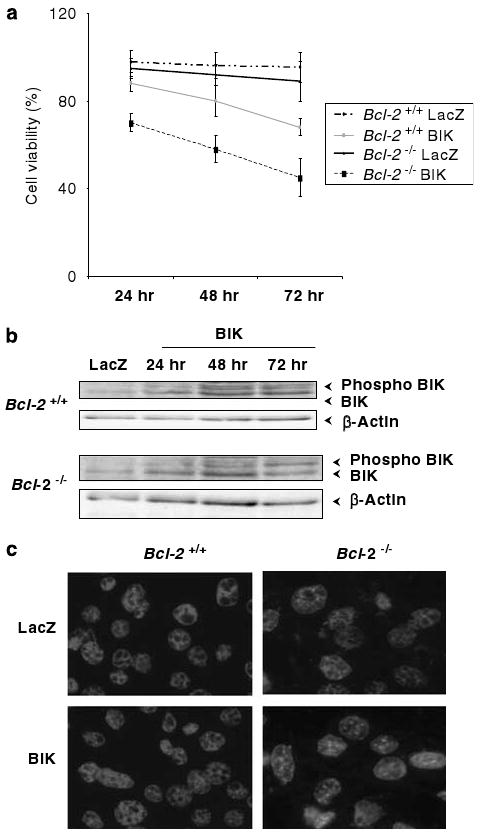
Effect of Bcl-2 on BIK-mediated cell death. (a) Viability of Bcl-2+/+ and Bcl-2−/− cells infected with Ad-LacZ or Ad-BIK. Cells were seeded in 96-well plates and cell viability assay was performed using the MTS assay (Promega, Madison, WI, USA); n = 3. (b) Western-blot analysis of unphosphorylated and phosphorylated forms of BIK. (c) DAPI-staining patterns of infected cells. The cells were stained 48 h of infection, photographed under × 60 magnification.
Nonapoptotic cell death induced by BIK in Bcl-2 null cells
We examined whether cell death induced by BIK in Bcl-2 null cells exhibited the common biochemical signatures associated with apoptotic cell death. Expression of BIK induced significant activation of caspase-8 in both Bcl-2+/+ and Bcl-2−/− cells (Figure 2a). In contrast, there was no detectable activation of either caspases-9 or -3 in both Bcl-2+/+ and Bcl-2−/− cells infected with Ad-BIK. Similarly, BIK expression also did not cause detectable accumulation of cytochrome-c in the cytosol. Consistent with this result, there was no significant change in the levels of mitochondrial cytochrome-c (Figure 2b, left panel). The indirect immunofluorescence analysis using antibodies directed against cytochrome-c also revealed clear perinuclear punctate mitochondrial fluorescence in both cell types (Figure 2b, right panel). In contrast to the results with cytochrome-c, there was significant loss of mitochondrial apoptosis-inducing factor (AIF) with concomitant increase in nuclear accumulation in BIK-expressing Bcl-2−/− cells compared to LacZ-expressing cells (Figure 2c). In Bcl-2+/+ cells, there was no significant loss of mitochondrial AIF as a result of BIK or LacZ expression. Consistent with the western-blot data, the immunofluorescence analysis also revealed significant accumulation of AIF in the nuclei of Bcl-2−/− cells infected with Ad-BIK and not in Bcl-2+/+ cells (Figure 2c, right panel). These results suggest that BIK-induced cell death of Bcl-2 null MEFs does not conform to the features such as activation of caspases hyp9 and hyp3 and mobilization of cytochrome-c from the mitochondria to the cytosol, which are normally associated with apoptosis. However, there was significant nuclear accumulation of AIF in these cells. These data are consistent with the interpretation that BIK induces nonapoptotic type of cell death in the absence of Bcl-2 in MEFs.
Figure 2.
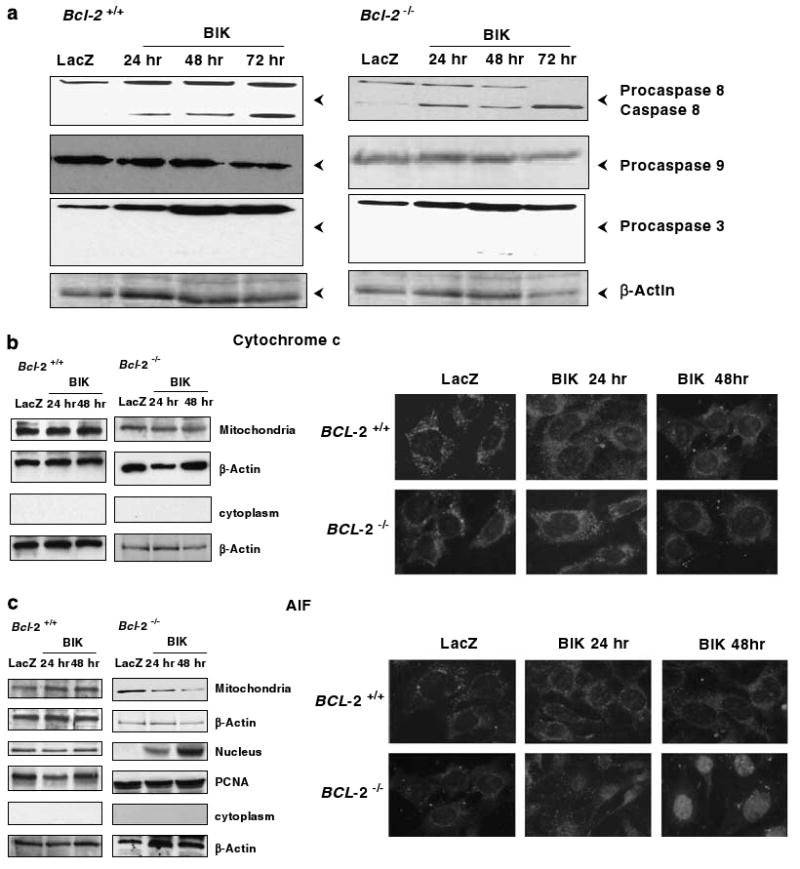
Effect of BIK on expression of apoptosis markers. (a) Western-blot analysis of caspases. The blots were probed with antibodies that recognize both procaspases and processed forms of caspase-8, 9 and 3. The status in Ad-LacZ infected cells at 72 h and in Ad-BIK infected cells at 24, 48 and 72 h is shown. β-Actin was used as the loading control. (b) Mitochondrial and cytoplasmic distribution of cytochrome-c. (c) Mitochondrial and nuclear distribution of apoptosis-inducing factor (AIF). Both western-blot analyses of subcellular fractions and indirect immunofluorescence analysis are shown. In panel (c), PCNA is shown as the control for the nuclear fraction.
Effect of zVAD-fmk on BIK-induced cell death
BIK has been known to induce cell death in a caspase-independent fashion in human melanoma cells (Oppermann et al., 2005). The results presented in Figure 2 and the above report prompted us to examine the effect of the broad-spectrum caspase inhibitor zVAD-fmk in BIK-induced death of Bcl-2 null MEFs. Treatment of Bcl-2+/+ cells infected with Ad-BIK or Ad-LacZ with zVAD-fmk enhanced the cell viability modestly (Figure 3a). To our surprise, we observed a dose-dependent increase in death of BIK-expressing Bcl-2−/− cells that were treated with 10 or 25 μM concentrations of zVAD-fmk (Figure 3b). DAPI staining of Bcl-2−/− cells infected with Ad-BIK and treated with zVAD-fmk showed bright spots throughout the nuclei and the nuclei appeared highly condensed. Cells infected with Ad-LacZ and treated with zVAD-fmk showed intact nuclear morphology (Figure 3c). These results suggest that inhibition of caspases (by treatment with zVAD-fmk) in the absence of BCL-2 enhances BIK-induced cell death in MEFs whereas such a treatment inhibited cell death in the presence of BCL-2.
Figure 3.
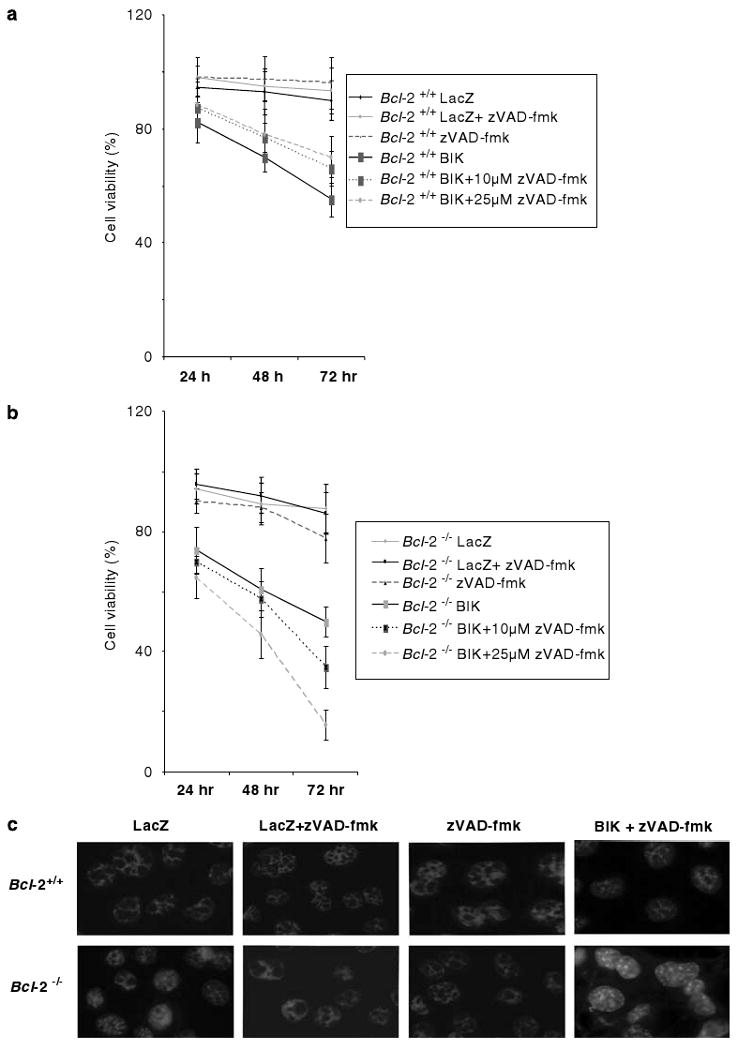
Effect of zVAD-fmk on BIK-induced cell death. Viability of Bcl-2+/+ (a) and Bcl-2−/− (b) cells infected with Ad-LacZ or Ad-BIK in the presence or absence of zVAD-fmk; n = 4. (c) DAPI-staining patterns of cells expressing LacZ or BIK (±zVAD-fmk, 10 μM).
Additionally, we also examined the effect of treatment of Bcl-2−/− cells with zVAD-fmk on mobilization of mitochondrial death factors, cytochrome-c and AIF by western blot and immunofluorescence analysis (Supplementary Figure S2). Cells treated with zVAD-fmk revealed a pattern similar to that of untreated cells (as in Figures 2b and c), suggesting that the enhanced cell death induced by zVAD-fmk in Bcl-2−/− cells might not be attributable to mobilization of cytochrome-c from mitochondria. However, in the absence or presence of zVAD-fmk, there was selective mobilization of mitochondrial AIF to nucleus.
Autophagic features of BIK-induced death in Bcl-2 null cells
As BIK induced an atypical form of cell death, we examined the ultra structure of MEFs expressing BIK by transmission electron microscopy (EM) (Figure 4). In Bcl-2 null cells infected with Ad-BIK, cytosolic vacuoles resembling autophagic vacuoles were very clearly visible in about one third of the cells examined (Figure 4, lower panel, middle image). However, when BIK-expressing cells were treated with zVAD-fmk, more extensive cytosolic vacuoles were seen in more than half of the number of cells examined. In contrast, Bcl-2+/+ cells expressing BIK, with or without zVAD-fmk treatment, no such vacuoles were readily detectable.
Figure 4.
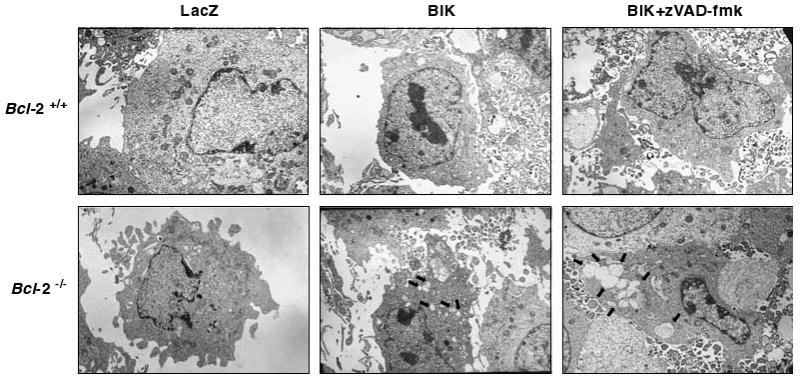
Electron micrographs of Bcl-2+/+ and Bcl-2−/− cells expressing LacZ or BIK. Thin sections of cells infected with Ad vectors (±zVAD-fmk) were analysed by scanning EM. The arrows in the lower panels point to cytosolic vacuoles.
To substantiate that the vacuoles observed in Bcl-2 null cells were the consequences of autophagy, we examined the cytosolic redistribution of the autophagy marker, LC3. The cytosolic protein LC3 is the mammalian homolog of the yeast gene Atg8 which is recruited to the autophagosomal membranes during autophagy (Kabeya et al., 2000). A chimeric LC3-EGFP reporter is widely used for detection of autophagy. Bcl-2 proficient and Bcl-2 null MEF cells were transiently transfected with plasmids expressing the LC3-EGFP and BIK, and the distribution of EGFP fluorescence was determined. In Bcl-2 proficient cells, most EGFP-positive cells showed diffused fluorescence throughout the cells (Figure 5a, upper panel). In contrast, about one fourth of the EGFP positive Bcl-2 null cells revealed punctate fluorescence (Figure 5a, lower panel). The Bcl-2 null cells cotransfected with BIK and LC3-GFP and then treated with zVAD-fmk, large punctate granules of LC3 protein was seen. The punctate fluorescence was observed in about 25–30% of Bcl-2−/− cells infected with Ad-BIK and after treatment with of z-VAD-fmk about 65–70% of cells exhibited such fluorescence (Figure 5c). In contrast, the level of punctate distribution of LC3 was significantly reduced in cells transfected with BIKΔBH3, suggesting that the BH3 domain might contribute to the nonapoptotic cell death, at least partially. We also examined the distribution of the endogenous LC3 by immunofluorescence analysis. In agreement with transient transfection studies, in Bcl-2 null cells infected with Ad-BIK, LC3-specific fluorescence was observed in punctate structures (Figure 5d, lower panel). These results suggest that LC3 might be associated with autophagosomes in Bcl-2−/− as a consequence of BIK expression and such association might be enhanced by treatment with zVAD-fmk.
Figure 5.
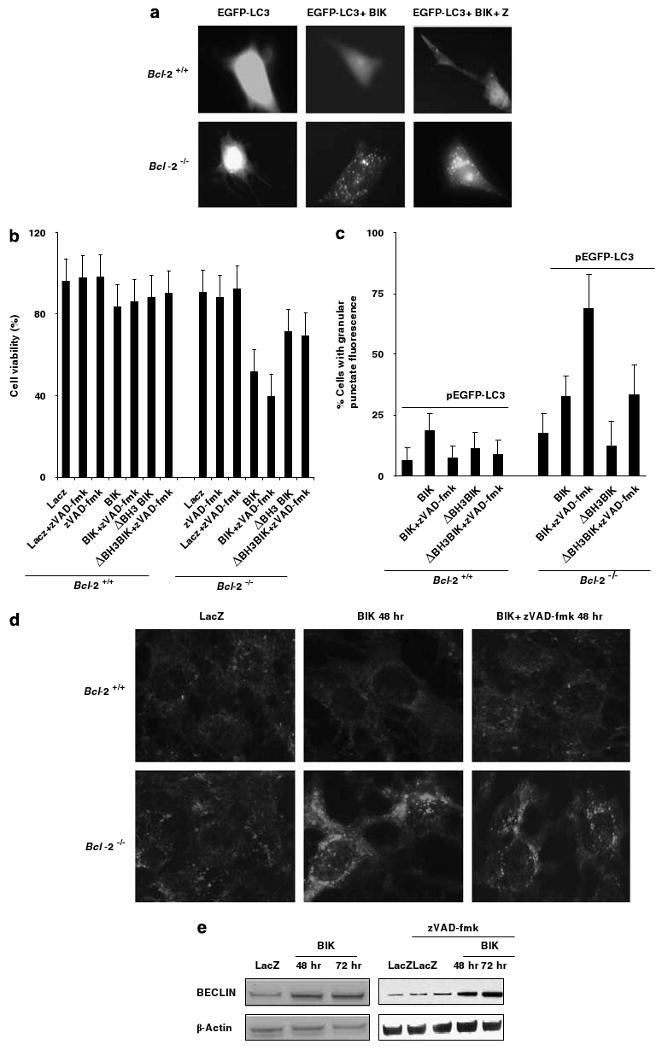
Autophagic features of BIK-induced cell death. (a) Distribution of LC3 in cells transfected with BIK (±zVAD-fmk). (b) Role of BIK BH3 domain on cell death. Cells were infected with Ad-LacZ or Ad-BIK or Ad-BIKΔBH3 (±zVAD-fmk) and viabilities were determined after 48 h; n = 3. (c) Quantification of cells showing punctate distribution of LC3; n = 3. At least 150 cells per sample were examined at random. (d) Immunofluorescence analysis of LC3 distribution. (e) Expression of Beclin-1 in BIK-expressing Bcl-2−/− cells treated with zVAD-fmk.
Increased levels of Beclin-1 expression are generally associated with autophagy (Levine and Klionsky, 2004). We examined the effect of BIK on expression of Beclin-1 (Figure 5e). Bcl-2 null cells were infected with Ad-LacZ or Ad-BIK and levels of Beclin-1 was determined 48 or 72 h after infection. The levels of expression of Beclin-1 were clearly increased in cells infected with Ad-BIK compared to cells infected with Ad-LacZ (Figure 5e, left panel). Similarly, there was increased level of Beclin-1 in BIK-expressing cells that were treated with zVAD-fmk (Figure 5e, right panel). Thus, these results further support the conclusion that BIK expression in Bcl-2 null MEFs might induce autophagy and this effect might be enhanced by zVAD-fmk.
Effect of autophagy inhibitors on BIK-induced cell death
Since autophagy and autophagic death are regulated by PI3 kinases (Tsujimoto and Shimizu, 2005), inhibitors such as 3-methyladenine (3-MA) and Wortmannin (WM) against these kinases are commonly used to inhibit these processes. We determined the effects of 3-MA and WM on BIK-induced death in Bcl-2−/− MEFs. BIK expression was mediated either by transfection of pcDNA-BIK or by infection with Ad-BIK. Viability of BIK-expressing cells that were either mock treated or treated with zVAD-fmk in presence or absence of 3-MA or WM was determined. Treatment with 3-MA (Figure 6a) or WM (Figure 6b) increased viability of BIK expressing cells with or without zVAD-fmk treatment. The extent of inhibition was more in cells treated with zVAD-fmk. Our results suggest that BIK-induced autophagy-like death of Bcl-2−/− cells is modulated by PI3 kinases.
Figure 6.
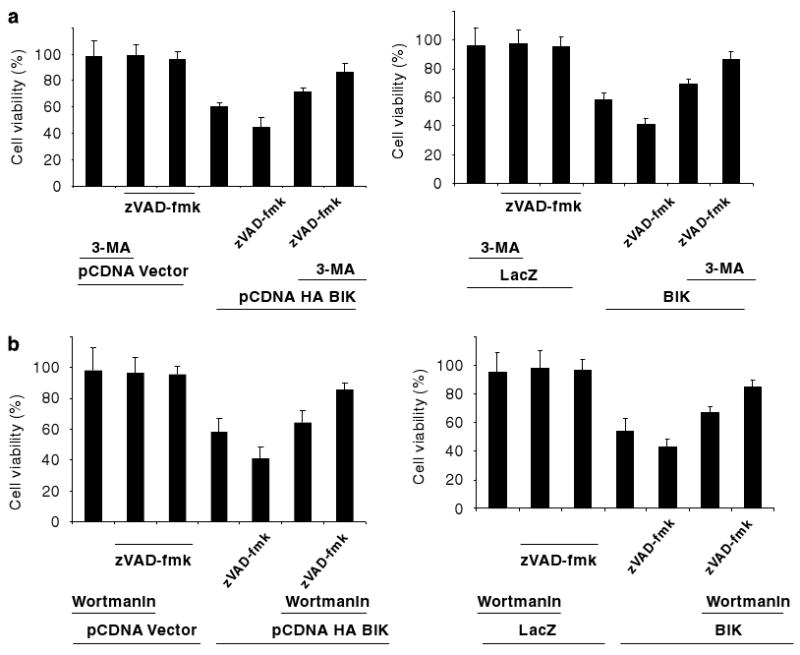
Effect of PI3 kinase inhibitors on BIK-induced cell death. (a) Effect of 3-Methyladenine (3-MA). (b) Effect of Wortmannin. Bcl-2−/− cells were either transfected with pcDNA3-BIK or empty vector (left panels) or infected with Ad-LacZ or Ad-BIK (right panels). These cells (±zVAD-fmk) were treated with 1mM 3-methyladenine or 0.2 μml−1 Wortmannin and the viability was determined; n = 4.
Role of Atg5 and Beclin-1 in BIK-mediated cell
Death In addition to the kinase inhibitors, we also determined the effect of knock down of two of the autophagy effectors, Atg5 and Beclin-1 on BIK-induced cell death. Transfection of MEF cells (Bcl-2−/−) with siRNA against Atg5 or Beclin-1 resulted in significant knock down of the respective proteins compared to cells transfected with control siRNA (Figure 7a). The transfected cells were infected with Ad-LacZ or Ad-BIK. The infected cells were either mock-treated or treated with zVAD-fmk as in Figure 6 and cell viability was determined 48 h after infection (Figure 7b). Knock down of Atg5 as well as Beclin-1 enhanced the viability of (20–30%) BIK-expressing cells that were treated with zVAD-fmk (compare left most three bars in Figure 7b). Between knock down of Atg5 and Beclin-1, knock down of Atg5 resulted in more enhancement of viability. Modest enhancement in viability was also observed in cells expressing BIK in the absence of zVAD-fmk. Thus, the results from knock-down studies provide additional support for our conclusion that BIK-induces cell death with autophagic features in BCL-2 null cells.
Figure 7.
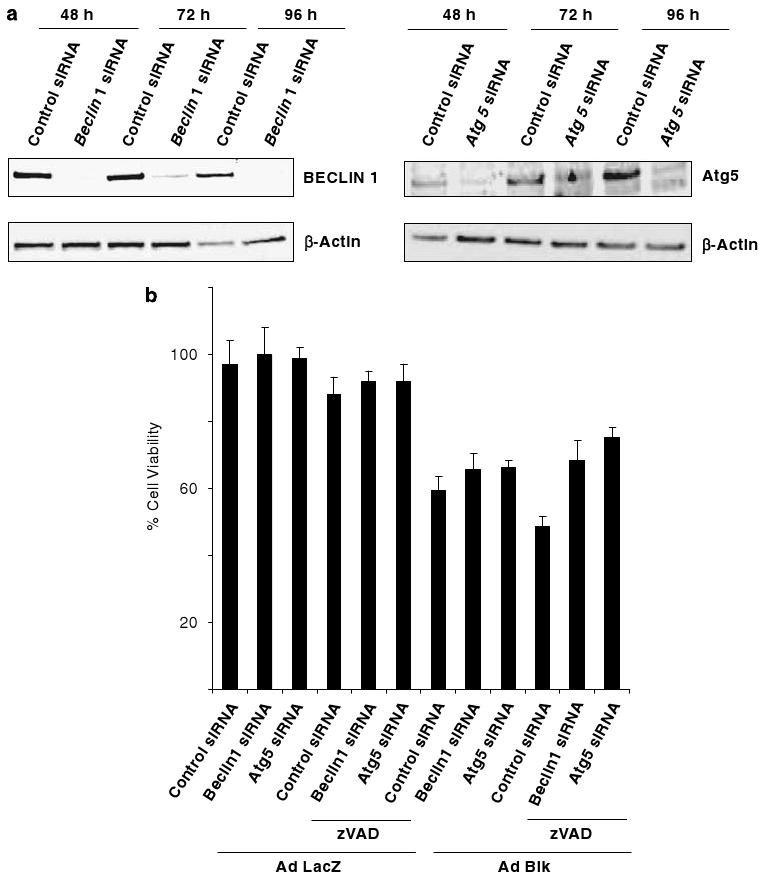
Effect of Atg5 and Beclin-1 on BIK-induced cell death. (a) Western-blot analysis of siRNA-mediated depletion of Atg5 and Beclin-1. Bcl-2−/− cells were transfected with siRNAs and the levels of expression of Atg5 and Beclin-1 were determined by western-blot analysis. (b). Cell viability in siRNA-transfected cells. The cells depleted for Atg5 or Beclin-1 were infected with Ad-LacZ or Ad-BIK (±zVAD-fmk) and cell viability was determined; n = 3.
Discussion
The results presented here show that the BH3-only protein BIK induces a type of cell death with features ascribed to autophagy and autophagic cell death in Bcl-2−/−MEF cells. Treatment with zVAD-fmk enhanced this type of cell death. BIK-induced cell death (±zVAD-fmk) was inhibited by 3-MA and WM which are general inhibitors of autophagy (Tsujimoto and Shimizu, 2005). Previous reports have implicated certain other BH3-only proteins in autophagic cell death. Glioma cells treated with ceramide (Daido et al., 2004) or arsenic trioxide (Kanzawa et al., 2005) expressed elevated levels of a BH3-only protein BNIP3 and underwent nonapoptotic cell death with certain autophagic features (punctate distribution of LC3 and cytosolic vacuoles). Ectopic expression of another BH3-only protein, HSpin1 was reported to induce caspase-independent cell death with autophagic (cytosolic vacuoles) features (Yanagisawa et al., 2003).
Although autophagy is considered as a defensive mechanism against cell death (Levine and Klionsky, 2004), there is conclusive evidence that it ultimately leads to cellular demise (Shimizu et al., 2004; Yu et al., 2004). BIK-induced cell death appears to meet the critical criteria that have been suggested for distinguishing autophagic death (Tsujimoto and Shimizu, 2005). These criteria include presence of cytoplasmic vacuoles in dying cells, punctate distribution of LC3 (Atg8), dependence on known autophagic effectors and inhibition of cell death by PI3 kinase inhibitors. The mechanism by which BIK induces accelerated nonapoptotic cell death in Bcl-2−/− cells remains to be fully elucidated. We have observed upregulation of Beclin-1, an effector of autophagy and autophagic death. Various cytotoxic stimuli that induced autophagic death have been reported to enhance expression of autophagic genes Atg5 and Beclin-1 through unknown mechanisms. However, it is generally believed that these stimuli may activate various signaling pathways. BIK expression has been reported to cause mobilization of calcium from the endoplasmic reticulum (ER) (Mathai et al., 2005). Elevated cytosolic Ca2+ was reported to induce autophagy which was inhibited by BCL-2 (Hoyer-Hansen et al., 2007). The possibility that BIK may activate calcium-dependent signaling pathways during nonapoptotic cell death remains to be explored.
We have observed enhanced BIK-mediated cell death with autophagic features in Bcl-2 null cells. A previous report showed that downregulation of Bcl-2 in HL-60 cells elicited autophagic-like death (Saeki et al., 2000). Beclin-1-induced autophagic death was also inhibited by ectopic expression of BCL-2 and vBCL-2 (Pattingre et al., 2005). Thus, the lack of Bcl-2 in KO MEF may sensitize the cells for Beclin-1-dependent autophagic death by BIK. Although both BIK (Boyd et al., 1995) and Beclin-1 (Oberstein et al., 2007) are BH3-only proteins, BIK might function as an upstream initiator stimulus. Our results shown in Figures 5b and c, suggest that the BH3 domain of BIK may at least partially contribute to the punctate distribution of LC3 and enhanced cell death in Bcl-2−/− cells treated with z-VAD-fmk. Since BIK contains other determinants, in addition to the BH3 domain, that contribute to its overall cell death activity (Elangovan and Chinnadurai, 1997), other regions of BIK might cooperate with the BH3 domain in inducing cell death with autophagic features.
We have also observed enhanced mobilization of AIF in cells undergoing BIK-mediated nonapoptotic death. It is not known, if the mobilization of AIF is directly linked to BIK-mediated cell death or is a consequence of death. Mitochondrial to nuclear redistribution of AIF has been observed in cells undergoing necrotic-type of cell death (Golstein and Kroemer, 2007). A previous study has reported chromatin condensation in the absence of cytochrome-c release from mitochondria during caspase-independent cell death induced by BIK in human melanoma cells (Oppermann et al., 2005). In light of our present results, it would be interesting to examine if the nonapoptotic cell death induced by BIK in melanoma cells exhibit autophagic features.
BIK-induced cell death was accelerated by zVAD-fmk. Treatment of L929 mouse fibroblast cells with zVAD-fmk induced cell death with characteristic features of autophagy which was found to be dependent on caspase-8 and autophagy genes Atg6 and Atg7 (Yu et al., 2004). Macrophages that were treated with LPS and zVAD-fmk underwent autophagy-like cell death that was inhibited by 3-MA and WM as well as by downregulation of Atg6 (Xu et al., 2006). The mode of action of zVAD-fmk in triggering autophagic cell death remains to be clarified. As zVAD-fmk is a strong inhibitor of caspase-8 probably its mode of action is through its inhibition. We have observed activation of caspase-8 in BIK-expressing cells and enhancement of BIK-induced cell death in the presence of zVAD-fmk. These results suggest that inhibition of caspase-8 by zVAD-fmk might contribute to autophagic-like cell death by BIK. This result is in agreement with a published report (Yu et al., 2006). We have observed that siRNA-mediated depletion of caspase-8 (Supplementary Figure S1) enhanced cell death induced by BIK and zVAD-fmk. These results also support the link between caspase-8 and zVAD-fmk.
Our results suggest an anticancer strategy for certain human cancers. Generally, it is believed that Bik functions as a tumor suppressor since it is deleted in certain cancers (Sturm et al., 2006) and its expression is repressed in many different human cancers (Dai et al., 2006). Paradoxically, Bik expression has also been associated with poor prognosis of non-small cell lung cancer (NSCLC) (Lu et al., 2006). Among the eight prognostic markers associated with high-risk NSCLC, three (Casp8, Bcl-2 and Bik) appear to be relevant to our results. Elevated expression of Bik was also observed in sporadic breast cancers (Garcia et al., 2005). Here, we have shown that zVAD-fmk mediated sensitization for autophagic cell death might be linked to these genes. It could be envisioned that small molecules that mimics the effect of zVAD-fmk might be useful in management of high-risk NSCLC. Manipulation of the autophagic process has been proposed as a therapeutic modality for cancers (Kondo et al., 2005). Anticancer agents, arsenic trioxide (Kanzawa et al., 2005) and ceramide (Daido et al., 2004) have been shown to mediate cell death by upregulation of BNIP3 in malignant glioma cells. It would be interesting to investigate such agents against cancers that overexpress Bik.
Materials and methods
Plasmids, recombinants, cells and reagents
The plasmid EGFP-LC3 was gift from T Yoshimori. Ad-BIK and Ad-LacZ have been described (Theodorakis et al., 2002). Immortalized Bcl-2+/+ and Bcl-2−/− MEFs were a gift from Dr Stanley Korsmeyer (Bassik et al., 2004). The cells were cultured in DMEM supplemented with 10% fetal bovine serum. Antibodies against cytochrome-c, AIF, PCNA, MAPLC3, β-actin, Atg5 and BIK were purchased from SantaCruz Biotechnology (Santa Cruz, CA, USA). Mouse mAb against Beclin-1 and cytochrome-c were purchased from BD Biosciences (San Jose, CA, USA). Antibodies against caspases-8, -9 and -3 were from Cell Signaling Technology (Danvers, MA, USA). siRNA against caspase-8, Beclin-1 and Atg5 were purchased from the smart pool siRNA reagents from Dharmacon (Chicago, IL, USA). WM and 3-MA were purchased from Calbiochem (San Diego, CA, USA) and Sigma (St Louis, MO, USA).
Western blotting, immunofluorescence and EM analysis
Cells were harvested and lysed in RIPA buffer supplemented with the protease inhibitor cocktail (Sigma). Subcellular fractionation was preformed using standard protocols. The total protein content was estimated using the bicinchoninic acid (BCA) protein estimation kit (Pierce Biotechnology, Rockford, IL, USA). The cell lysates were separated by sodium dodecyl sulphate-polyacryalmide gel electrophoresis and analysed by western blotting and the bands were visualized using ECL reagents (Roche, Indianapolis, IN, USA). For immunofluorescence analysis, cells were seeded onto coverslips, and examined as described (Rashmi et al., 2005).
Supplementary Material
Acknowledgments
This work was supported by research grants CA-73803, CA-116262 and CA-33616 from the National Cancer Institute. The MEF cells were kindly provided by late Stanley Korsmeyer.
References
- Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, et al. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene. 1995;11:1921–1928. [PubMed] [Google Scholar]
- Dai Z, Liu S, Marcucci G, Sadee W. 5-Aza-2′-deoxycytidine and depsipeptide synergistically induce expression of BIK (BCL2-interacting killer) Biochem Biophys Res Commun. 2006;351:455–461. doi: 10.1016/j.bbrc.2006.10.055. [DOI] [PubMed] [Google Scholar]
- Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- Elangovan B, Chinnadurai G. Functional dissection of the proapoptotic protein Bik. Heterodimerization with anti-apoptosis proteins is insufficient for induction of cell death. J Biol Chem. 1997;272:24494–24498. doi: 10.1074/jbc.272.39.24494. [DOI] [PubMed] [Google Scholar]
- Garcia N, Salamanca F, Astudillo-de la Vega H, Curiel-Quesada E, Alvarado I, Penaloza R, et al. A molecular analysis by gene expression profiling reveals Bik/NBK overexpression in sporadic breast tumor samples of Mexican females. BMC Cancer. 2005;5:93. doi: 10.1186/1471-2407-5-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2005;24:980–991. doi: 10.1038/sj.onc.1208095. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Lu Y, Lemon W, Liu PY, Yi Y, Morrison C, Yang P, et al. A gene expression signature predicts survival of patients with stage I non-small cell lung cancer. PLoS Med. 2006;3:e467. doi: 10.1371/journal.pmed.0030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Mathai JP, Germain M, Shore GC. BH3-only BIK regulates BAX,BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem. 2005;280:23829–23836. doi: 10.1074/jbc.M500800200. [DOI] [PubMed] [Google Scholar]
- Naumann U, Schmidt F, Wick W, Frank B, Weit S, Gillissen B, et al. Adenoviral natural born killer gene therapy for malignant glioma. Hum Gene Ther. 2003;14:1235–1246. doi: 10.1089/104303403767740777. [DOI] [PubMed] [Google Scholar]
- Oberstein A, Jeffrey P, Shi Y. Crystal structure of the BCL-XL-beclin 1 peptide complex: beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- Oppermann M, Geilen CC, Fecker LF, Gillissen B, Daniel PT, Eberle J. Caspase-independent induction of apoptosis in human melanoma cells by the proapoptotic Bcl-2-related protein Nbk/Bik. Oncogene. 2005;24:7369–7380. doi: 10.1038/sj.onc.1208890. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Rashmi R, Kumar S, Karunagaran D. Human colon cancer cells lacking Bax resist curcumin-induced apoptosis and Bax requirement is dispensable with ectopic expression of Smac or downregulation of Bcl-XL. Carcinogenesis. 2005;26:713–723. doi: 10.1093/carcin/bgi025. [DOI] [PubMed] [Google Scholar]
- Saeki K, Yuo A, Okuma E, Yazaki Y, Susin SA, Kroemer G, et al. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ. 2000;7:1263–1269. doi: 10.1038/sj.cdd.4400759. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a nonapoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S, et al. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 2006;13:619–627. doi: 10.1038/sj.cdd.4401782. [DOI] [PubMed] [Google Scholar]
- Theodorakis P, Lomonosova E, Chinnadurai G. Critical requirement of BAX for manifestation of apoptosis induced by multiple stimuli in human epithelial cancer cells. Cancer Res. 2002;62:3373–3376. [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12(Suppl 2):1528–1534. doi: 10.1038/sj.cdd.4401777. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:19179–19187. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Miyashita T, Nakano Y, Yamamoto D. HSpin1, a transmembrane protein interacting with Bcl-2/Bcl-xL, induces a caspase-independent autophagic cell death. Cell Death Differ. 2003;10:798–807. doi: 10.1038/sj.cdd.4401246. [DOI] [PubMed] [Google Scholar]
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.