

Abstract
The APOBEC family of mammalian cytidine deaminases, such as APOBEC3G (hA3G), has been demonstrated to function as a host viral restriction factor against HIV-1. hA3G has been shown to cause extensive G-to-A mutations in the HIV-1 genome, which may play a role in viral restriction. To investigate the role of G-to-A mutations in HIV-1 pathogenesis, we isolated, amplified, and sequenced HIV-1 sequences (vif, gag, and env) from 29 therapy-naive HIV-1-infected individuals. The levels of G-to-A mutations correlated with the expression levels of hA3G in the vif (rho = 0.438, p = 0.041) and the env regions (rho = 0.392, p = 0.038), but not in the gag region (rho = 0.131, p = 0.582). There is no correlation between viral load and the level of G-to-A mutations in the vif (rho = 0.144, p = 0.522), env (rho = 0.168, p = 0.391), or gag regions (rho = −0.254, p = 0.279). Taken together, these findings suggest that the hA3G-induced G-to-A mutations may not be the mechanism by which hA3G restricts or controls viral replication. Thus, hA3G might be restricting viral growth in infected individuals through a mechanism that is independent of the cytidine deaminase activities of hA3G.
Introduction
HIV-1 infection results in prolonged, continuous viral replication in infected individuals. Viral persistence is not thwarted by the presence of HIV-1-specific immune responses or by innate cellular antiviral defense mechanisms including host restriction factors such as APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G). During the course of the development of acquired immunodeficiency syndrome (AIDS), HIV-1 mutates with high frequency and thus avoids immune response.1 It also avoids the restriction by APOBEC3G (hA3G) by using the viral infectivity factor (VIF) to target hA3G for degradation through the proteosome pathway.2 Interestingly, it has been observed that HIV-1 sequences isolated from infected individuals show a high rate of G-to-A substitutions.3–5 In the past, this mutagenic phenomenon was attributed to the error-prone retroviral reverse transcriptase together with imbalances in the available deoxynucleotide pools in the cell.6 However, recent studies show that hA3G is responsible for G-to-A substitutions in viral genomes.4,7 hA3G causes G-to-A substitutions by rapidly deaminating nascent reverse transcripts to yield U-containing DNA, which later pairs with nucleotide A.
Originally, hA3G was thought to restrict viral replication through G-to-A substitutions by introducing stop codons or triggering a repair mechanism that degraded viral DNA. Recent work by Bishop and colleagues has shown that a strong antiviral effect can be achieved in the absence of detectable G-to-A substitution.8,9 Several lines of evidence now suggest that hA3G can function by a second, editing-independent, antiviral mechanism. Mutated hA3G protein that is unable to function as a cytidine deaminase retains a substantial level of antiviral activity.10 In an in vivo editing assay, alteration to the C-terminal cytidine deaminase domain, such that the protein could no longer mediate the deamination of either HIV-1 cDNA or bacterial DNA, was still packaged into virions and retained significant antiviral activity. This suggests that the antiviral activity of hA3G is not related to the G-to-A substitution but rather to alternative antiviral mechanisms.11
The role of G-to-A mutations mediated by hA3G in HIV-1 pathogenesis is not well understood. A recent study showed that HIV-1-infected patients with hypermutated sequences had lower viral loads when compared to patients with non-hypermutated sequences,12 although APOBEC levels were not measured. Here, we measure hA3G mRNA levels and analyze G-to-A mutations in gag, env, and vif sequences from peripheral blood mononuclear cell (PBMC) samples obtained from HIV-1-infected individuals. We also analyze polymorphism in vif motifs that have been shown to be important for vif expression and its activity against hA3G.
Materials and Methods
Sample acquisition
PBMC samples used in this study were obtained from a cohort of female sex workers in Dakar, Senegal, that our laboratory has been following since 1985. Epidemiologic and clinical aspects of this cohort have previously been described elsewhere.13 At enrollment all subjects gave informed consent and participated in protocols approved by the Counseil National de Lutte Contre le Sida Comite Ethique et Juridique and the Harvard School of Public Health Human Subjects Committee. CD4+ T cell counts were determined, serum samples were tested for HIV-1-specific antibodies, and viral load measurements were done as previously described.14 All subjects enrolled in this study were antiretroviral therapy naive and had CD4+ T cell counts above 200/μl at the time of sample acquisition.
DNA extraction and amplification
Cryopreserved PBMC samples were thawed and rested overnight in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum and 1% antibiotics to ensure sample viability and to remove dead cells. The following day, DNA was extracted (blood and cell culture DNA mini kit; QIAGEN), and DNA concentrations were determined by an optical density reading at 260 nm. Approximately 1 μg of DNA was used for polymerase chain reaction (PCR) amplification. The vif gene was amplified in a nested PCR using first-round primers nzV1f (5′-GGGTACAGTGCAGGGGAAAG-3′) and nzV1r (5′-CTGCCATAGGAGATGCCTAAG-3′) and second round primers nzV2f (5′-CTCTGGAAAGGTGAAGGGGC-3′) and nzV2r (5′-TGTTGRCACCCAATTCTGAAAATG-3′). A fragment of the GAG region was amplified using heminested PCR using first-round primers P108 (5′-GACTAGCGGAGGCTAGAA-3′) and P109 (5′-AGGGGTCGTTGCCAAAGA-3′) and the second-round primers P91 (5′-CACCTATCCCAGTAGGAGAAATC-3′) and P109 (5′-AGGGGTCGTTGCCAAAGA-3′). Each PCR reaction contained 1 μl of DNA in a volume of 50 μl with 10 × PCR buffer, 2 mM MgSO4, 0.8 mM deoxynucleoside triphosphate mix, 100 nM of each forward and reverse primer, and 1 U of Taq polymerase, under the following conditions: 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 3 min; and a final extension for at 72°C for 10 min. The PCR products were then purified using microcon (utracel YM-100, Millipore) according to the manufacturer's protocol. Purified products were sequenced using the second-round primers and the two internal primers: primers nzVsf (5′-AAAGCCACCTTTGCCTAGTGT-3′) and nzVsr (5′-TCTTCTGGGGTTGTTCCATCT-3′) for vif sequences and primers G100 (5′-TAGAAGAAATGATGACAG-3′) and G25 (5′-ATTGCTTCAGCCAAAACTCTTGC-3′) for gag sequences. The env region was amplified and sequenced as described.15
Quantification of hA3G mRNA
We used the TaqMan assay to quantify hA3G mRNA transcripts in PBMCs from infected individuals. The reactions were performed in a 96-well Optical Reaction Plate (Applied Biosystems). Primer and probes for hA3G and β-actin were ready-made assay-on-demand (TaqMan; Applied Biosystems). Each reaction contained 2.25 μl H2O, 12.5 μl TaqMan Universal PCR Master Mix (Applied Biosystems), 1.25 μl primers and probes, and 10 μl cDNA. Each reaction was performed in duplicate. The reactions were run on the ABI PRISM 7000 Sequence Detection System (Applied Biosystems). The amplification program was 50°C for 2 min, 95°C for 10 min, followed by 45 cycles of 95°C for 15 s and 60°C for 1 min. The levels of hA3G were quantified using standard curves generated using plasmids; pTrc99A with hA3G (NM_021822) insert. The quantity of β-actin was determined by a standard curve generated by human genomic DNA. The results were analyzed (Sequence Detection Software; Applied Biosystems), and hA3G mRNA expression was normalized to the expression of the housekeeping gene, β-actin (NM_001101). The levels of hA3G mRNA transcripts were reported as median copies of hA3G per 100 copies of β-actin.
Sequence analysis and calculations
Sequences were aligned using CLUSTAL X software (version 1.81)16; manual adjustments were made using McClade software (version 4; Sinauer).17 The subtype identity of each sequence was established using phylogenetic trees generated by PAUP (version 4; Sinauer). To estimate GA substitutions, we used a previously described method12; briefly, individual HIV-1 proviral DNA sequences were aligned against the consensus sequence of its subtype. We used HYPERMUT18 to examine GA substitutions, where GA, GC, GG, and GT dinucleotides in the consensus sequence were observed as AA, AC, AG, and AT, respectively, in the sample sequence. To determine GA substitutions for each sequence, we estimated the level of general GA substitutions in each sequence using the following formula12:
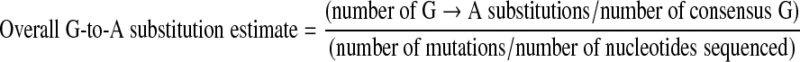 |
The formula accounts for the proportion of all GA substitutions adjusted for the proportion of G nucleotides in the consensus sequence.
Analysis of APOBEC3G target motifs
To investigate the contribution of APOBEC3G (3G) to G-to-A substitution, we examined the dinucleotide sequence contexts of GG substitutions using the following formula:
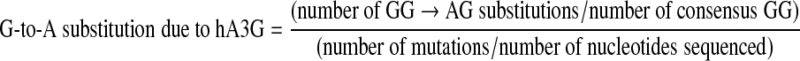 |
The formula accounts for the proportion of all G-to-A substitutions that occurred in the GG context adjusted for sequence length.
Vif polymorphism
To investigate the association between vif amino acid polymorphisms and G-to-A mutations, we performed a putative translation of vif DNA sequences (amino acids 1 to 193). We analyzed the VIF protein sequence focusing on motifs that have been shown to be important in interactions with the hA3G protein as well as regions that are important for VIF's stability and function including degradation of hA3G. Polymorphisms in the following motifs—DRRMR, YRHHY, EWRKKR, PPLP, and SLQXLA—were analyzed to determine if there were any associations with viral load or level of G-to-A substitutions.
Results
G-to-A mutations in vif, env, and gag
We analyzed G-to-A mutations in vif, env, and gag sequences amplified from HIV-1-infected individuals (Table 1). Each of the three regions of the HIV-1 genome examined had a different level of G-to-A substitutions; of the analyzed sequences, the G-to-A mutation in the vif represented 22% of all mutations, while in env and gag regions it was 15% and 18%, respectively. Since hA3G is known to specifically target the single-stranded polynucleotide GG-motif,19 we determined the amount of G-to-A substitutions caused by hA3G. In the vif, the GG-to-AG substitution was 33% of all G-to-A substitutions while the GA-to-AA substitution comprised 47%. In the env region, GA-to-AA substitutions were 33% of all G-to-A substitutions and GG-to-AG substitutions were 42%. In the gag region, GG-to-AG substitutions accounted for 34% of all G-to-A substitutions and GA-to-AA substitutions accounted for 36% (Fig. 1).
Table 1.
Characteristics of Sequences Amplified from HIV-1-Infected Individualsa
| Vif | Env | Gag | |
|---|---|---|---|
| Number of sequence | 25 | 28 | 22 |
| Nucleotide sequenced | 592 (580–590) | 346 (335–346) | 721 (488–740) |
| G-to-A mutations | 12 (9–50) | 15.5 (7–32) | 65 (6–96) |
| General G-to-A mutations score | 0.85 (0.21–1.12) | 0.62 (0.38–1.139) | 0.54 (0.37–0.89) |
| hA3G-specific G-to-A mutations score | 1.23 (0.15–0.1.54) | 1.05 (0.40–1.17) | 1.08 (0.34–1.89) |
| hA3G mRNA level | 1.54 (0.28–2.73) | 1.39 (0.70–4.40) | 1.37 (0.55–2.55) |
| Median CD4+ cells count | 547 (300–1096) | 547 (300–1096) | 547 (300–1096) |
| Median viral load | 4.02 (1.7–4.88) | 4.02 (1.7–5.25) | 4.4 (0.10–5.25) |
Their viral load and CD4 count values are provided. Values are expressed as median (range).
FIG. 1.
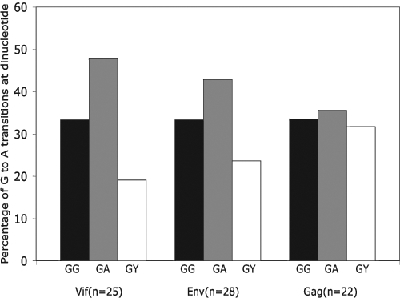
Histogram depicting the percentage of GG-to-AG, GA-to-AA, and GY-to-AY mutations in the vif, env, and gag sequences. Mutation occurring at GG, GA, and GY dinucleotide contexts are indicated by black, gray, and white, respectively.
Since hA3G is thought to restrict viral replication through the introduction of G-to-A substitutions,4,20,21 we sought to investigate the relationship between accumulation of G-to-A substitutions and viral load. G-to-A substitutions in the vif region were not correlated with viral load (rho = 0.144, p = 0.522). In the env region the G-to-A substitutions were also not correlated with viral load (rho = 0.168, p = 0.381) and the same trend was observed in the gag region where no correlation was found between the levels of G-to-A transition and levels of viral load (rho = −0.254, p = 0.279).
Since the levels of hA3G expression have been shown to correlate with the levels of the active forms (low molecular mass) of hA3G,22,23 we investigated the relationship between the levels of hA3G mRNA and G-to-A substitutions. There was a statistically significant correlation between the levels of hA3G and number of G-to-A substitutions in the vif (rho = 0.438, p = 0.041) and env regions (rho = 0.392, p = 0.038), but not in the gag region (rho = 0.131, p = 0.582) (Fig. 2).
FIG. 2.

Correlations of the levels of G-to-A mutations [in vif (a), env (b), and gag (c)] and the amount of hA3G mRNA. Correlation coefficients (rho) and p values were calculated using Spearman's rank correlation; p values <0.05 were considered significant.
Association of Vif amino acid polymorphism, viral load, and G-to-A substitutions
Since HIV-1 Vif interacts with hA3G, we sought to establish the effect of polymorphism in specific Vif motifs that have been demonstrated to be important for Vif function, especially in targeting hA3G for degradation. First, we examined Y40 RHHY44 and D14RMR17, regions that have recently been shown to be important for the binding of Vif to hA3G.24 We analyzed polymorphisms in the D14RMR17 motif and found no association between polymorphisms at any position in the motif and patient viral load or number of G-to-A substitutions. Next, we examined Y40 RHHY44 motif polymorphisms; the amino acid change at position 42 was not significantly associated with viral load or G-to-A substitutions (Fisher's exact test = 0.099 and 0.143, respectively; Table 2).
Table 2.
Effect of Polymorphisms in Important Vif Motifs on Viral Load and G-to-A Mutationsa
| Motif | Polymorphism | Polymorphic | Nonpolymorphic | G-to-A mutations (p-value) | Viral load (p-value) |
|---|---|---|---|---|---|
| D14RMR17 | None | ||||
| Y40RHHY44 | H42 | 3 | 21 | 0.143 | 0.099 |
| E88WRKKR93 | W89 | 2 | 22 | 0.446 | 0.036 |
| P161PLP164 | P162 | 3 | 21 | 0.145 | 0.521 |
| SLQXLA | None |
Vif was putatively translated and polymorphisms identified by comparison to concensus peptide sequence. The association polymorphism with viral load and G-to-A mutation was tested by Fisher's exact test; p < 0.05 was considered statistically significant.
We also examined polymorphisms in two other motifs: E88WRKKR93 and the proline-rich P161PLP164, which have been shown to enhance steady-state levels of Vif and its interaction with tyrosine kinases. In the E88WRKKR93 motif, replacement of the amino acid tryptophan at position 89 was associated with low viral load (Fisher's exact test = 0.036, Table 2). However, there was no association between polymorphism in this motif and G-to-A transition (Fisher's exact test = 0.446). The polymorphism P161PLP164 motif was not associated with either viral load or the G-to-A transition rate (Table 2). The SLQXLA motif that has been shown to be important for Vif function was conserved in all of our sequences.
Discussion
The host restriction factor hA3G is thought to restrict HIV-1 replication through introduction of G-to-A substitutions in HIV-1 sequences.3,20,22 The G-to-A substitutions have been observed in infected individuals,3 but their correlation with hA3G levels and role in HIV-1 replication in vivo is not well understood. In this study, we investigated the relationship between G-to-A substitutions and viral load in HIV-1-in-fected individuals. Our findings indicate that there is no correlation between G-to-A substitutions in HIV-1 sequences and patient plasma viral load, suggesting that accumulation of G-to-A substitutions has no effect on the levels of the viral load in infected patients. This is somewhat paradoxical since the levels of hA3G have been shown to correlate significantly with viral load.25 If this is so, the G-to-A substitution might be expected to similarly correlate with the viral load.3
Although this study has a relatively small sample size, the observation that G-to-A substitutions do not correlate with the viral load is supported by findings from the work of Blankson et al. that analyzed 978 HIV-1 sequences and found that there was no difference in the level of APOBEC-mediated hypermutations in individuals with a high viral load compared to individuals with an undetectable viral load.26 This observation is consistent with recent findings that demonstrate that hA3G can achieve an anti-HIV-1 effect through a mechanism that is distinct from cytidine deamination.10,27 Newman et al. used site-directed mutagenesis of conserved residues to demonstrate that hA3G lacking deaminase activity was able to restrict viral growth.10 A study examining the effect of hA3G on hepatitis B virus (HBV) infection has also suggested that hA3G may exert its antiviral effect in more than one way.5,28 Specifically, the study showed that although hA3G exhibited a strong suppression of HBV growth, the small amount of DNA found associated with viral cores lacked G-to-A substitution, implying that the restriction was through a mechanism other than cytidine deamination.
Our findings suggest that in HIV-1-infected individuals, hA3G may not be exerting its antiviral effect through generation of G-to-A substitutions. Polymorphisms in the vif motif (E88WRKKR93), which has been shown to be crucial for viral infectivity,24,29 affected the levels of viral load but not the levels of G-to-A mutations (Table 2). If hA3G was exerting its antiviral activity through the generation of G-to-A mutations, the motif's polymorphisms would be expected to affect both the levels of G-to-A mutations and viral load. Indeed, a number of recent studies have suggested that hA3G might be targeting a stage of viral cycle other than the reverse transcriptase stage. A study by Yu et al. shows that hA3G interacts with HIV-1 integrase and inhibits proviral DNA formation.30 Another study by Malim et al. has demonstrated that hA3G can inhibit the accumulation of HIV-1 reverse transcription products in the absence of G-to-A mutations.11 These two studies are complemented by Mbisa et al. who show that hA3G decreased the amount of viral DNA that was integrated into the host cell genome and similarly reduced the efficiency with which HIV-1 preintegration complexes (PICs) integrated into a target DNA in vitro.31 Taken together, these results suggest that hA3G might be restricting viral replication by reducing the efficiency of reverse transcripts synthesis and viral integration.
To explore the relationship between the G-to-A mutations and the expression of levels of hA3G, we quantified the hA3G mRNA in infected samples. The level of hA3G expression was significantly correlated with the amount of G-to-A mutations in the vif and env regions (Fig. 2). However, in the Gag region the level of hA3G expression was not significantly correlated with G-to-A mutations; this might be due to the fact that the number of Gag sequences analyzed was less than that of Vif or Env. Overall, our results suggest a link between hA3G expression and the levels of G-to-A mutation in infected individuals. Apart from contributing to the diversity of the viral population, the specific role of these mutations in HIV-1 pathogenesis is not clear. Although our data do not show a relationship between G-to-A mutations and viral load, hA3G in infected individuals may function as a “double-edged sword,” inhibiting viral growth on the one hand and generating viral diversity through deaminase activity on the other. The increased viral diversity generated as a result of G-to-A mutations may help the virus to evade the immune response,1 further complicating the role of hA3G in HIV-1 pathogenesis.
In conclusion, our results provide further insight into the hA3G viral restriction function and its role in HIV-1 pathogenesis. Specifically, we have shown that hA3G contributes significantly to the generation of G-to-A mutations in HIV-1-infected individuals and that there is no statistically significant correlation between the levels of G-to-A mutations and plasma viral load. We have also shown that the levels of G-to-A mutations correlate with levels of hA3G mRNA. Taken together, our results suggest that hA3G, contrary to previous reports, might restrict viral growth in a G-to-A mutation-independent manner. Our results underscore the need for further research to elucidate the mechanism of hA3G's antiviral functions so that it could be explored as a potential antiretroviral drug target.
Sequence Data
Sequences have been deposited at GenBank. Vif and gag sequences GenBank accession numbers are EU541976-EU542019. Env sequences GenBank accession numbers are DQ323299-DQ323200, DQ323225-DQ323228, DQ323237, DQ323241, DQ323219, DQ323231, DQ323289, DQ323233, DQ323238, DQ323243, DQ323189, AY646143, AY646144, AY646132, AY646142, AY646135, AY646151, AY646128, AY646145, AF085284, AF526689, AF526813, AF526799, AF020823.
Acknowledgment
This study was supported by the National Institute of Allergy and Infectious Diseases Grant AI6274-04 from the National Institute of Health.
Disclosure Statement
No competing financial interests exist.
References
- 1.Walker BD. Goulder PJ. AIDS. Escape from the immune system. Nature. 2000;407:313–314. doi: 10.1038/35030283. [DOI] [PubMed] [Google Scholar]
- 2.Vartanian JP. Sommer P. Wain-Hobson S. Death and the retrovirus. Trends Mol Med. 2003;9:409–413. doi: 10.1016/j.molmed.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Harris RS. Bishop KN. Sheehy AM. Craig HM. Petersen-Mahrt SK. Watt IN. Neuberger MS. Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 4.Sheehy AM. Gaddis NC. Choi JD. Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 5.Turelli P. Mangeat B. Jost S. Vianin S. Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 6.Julias JG. Pathak VK. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J Virol. 1998;72:7941–7949. doi: 10.1128/jvi.72.10.7941-7949.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lecossier D. Bouchonnet F. Clavel F. Hance AJ. Hyper-mutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen DH. Gummuluru S. Hu J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol. 2007;81:4465–4472. doi: 10.1128/JVI.02510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bishop KN. Holmes RK. Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol. 2006;80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman EN. Holmes RK. Craig HM. Klein KC. Lingappa JR. Malim MH. Sheehy AM. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 11.Holmes RK. Koning FA. Bishop KN. Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 12.Pace C. Keller J. Nolan D. James I. Gaudieri S. Moore C. Mallal S. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J Virol. 2006;80:9259–9269. doi: 10.1128/JVI.00888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanki P. M'Boup S. Marlink R. Travers K. Hsieh CC. Gueye A. Boye C. Sankale JL. Donnelly C. Leisenring W, et al. Prevalence and risk determinants of human immunodeficiency virus type 2 (HIV-2) and human immunodeficiency virus type 1 (HIV-1) in west African female prostitutes. Am J Epidemiol. 1992;136:895–907. doi: 10.1093/aje/136.7.895. [DOI] [PubMed] [Google Scholar]
- 14.Sarr AD. Popper S. Thior I. Hamel DJ. Sankale JL. Siby T. Marlink R. Essex M. Mboup S. Kanki P. Relation between HIV-2 proviral load and CD4+ lymphocyte count differs in monotypic and dual HIV infections. J Hum Virol. 1999;2:45–51. [PubMed] [Google Scholar]
- 15.Hamel DJ. Sankale JL. Eisen G. Meloni ST. Mullins C. Gueye-Ndiaye A. Mboup S. Kanki PJ. Twenty years of prospective molecular epidemiology in Senegal: Changes in HIV diversity. AIDS Res Hum Retroviruses. 2007;23:1189–1196. doi: 10.1089/aid.2007.0037. [DOI] [PubMed] [Google Scholar]
- 16.Thompson JD. Gibson TJ. Plewniak F. Jeanmougin F. Higgins DG. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maddison WP. Maddison DR. Interactive analysis of phylogeny and character evolution using the computer program MacClade. Folia Primatol (Basel) 1989;53:190–202. doi: 10.1159/000156416. [DOI] [PubMed] [Google Scholar]
- 18.Rose PP. Korber BT. Detecting hypermutations in viral sequences with an emphasis on G → A hypermutation. Bioinformatics. 2000;16:400–401. doi: 10.1093/bioinformatics/16.4.400. [DOI] [PubMed] [Google Scholar]
- 19.Bishop KN. Holmes RK. Sheehy AM. Davidson NO. Cho SJ. Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 20.Mangeat B. Turelli P. Caron G. Friedli M. Perrin L. Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 21.Harris RS. Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 22.Chiu YL. Soros VB. Kreisberg JF. Stopak K. Yonemoto W. Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- 23.Stopak KS. Chiu YL. Kropp J. Grant RM. Greene WC. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J Biol Chem. 2007;282:3539–3546. doi: 10.1074/jbc.M610138200. [DOI] [PubMed] [Google Scholar]
- 24.Russell RA. Pathak VK. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol. 2007;81:8201–8210. doi: 10.1128/JVI.00395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin X. Brooks A. Chen H. Bennett R. Reichman R. Smith H. APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. J Virol. 2005;79:11513–11516. doi: 10.1128/JVI.79.17.11513-11516.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gandhi SK. Siliciano JD. Bailey JR. Siliciano RF. Blankson JN. Role of APOBEC3G/F-mediated hypermutation in the control of human immunodeficiency virus type 1 in elite suppressors. J Virol. 2008;82:3125–3130. doi: 10.1128/JVI.01533-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shindo K. Takaori-Kondo A. Kobayashi M. Abudu A. Fukunaga K. Uchiyama T. The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. J Biol Chem. 2003;278:44412–44416. doi: 10.1074/jbc.C300376200. [DOI] [PubMed] [Google Scholar]
- 28.Rosler C. Kock J. Malim MH. Blum HE. von Weizsacker F. Comment on “Inhibition of hepatitis B virus replication by APOBEC3G.”. Science. 2004;305:1403. doi: 10.1126/science.1101974. author reply 1403. [DOI] [PubMed] [Google Scholar]
- 29.Fujita M. Sakurai A. Yoshida A. Miyaura M. Koyama AH. Sakai K. Adachi A. Amino acid residues 88 and 89 in the central hydrophilic region of human immunodeficiency virus type 1 Vif are critical for viral infectivity by enhancing the steady-state expression of Vif. J Virol. 2003;77:1626–1632. doi: 10.1128/JVI.77.2.1626-1632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo K. Wang T. Liu B. Tian C. Xiao Z. Kappes J. Yu XF. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J Virol. 2007;81:7238–7248. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mbisa JL. Barr R. Thomas JA. Vandegraaff N. Dorweiler IJ. Svarovskaia ES. Brown WL. Mansky LM. Gorelick RJ. Harris RS, et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007;81:7099–7110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]