

Abstract
Early-onset torsion dystonia is the most severe heritable form of dystonia, a human movement disorder that typically starts during a developmental window in early adolescence. Deletion in the DYT1 gene, encoding the torsinA protein, is responsible for this dominantly inherited disorder, which is non-degenerative and exhibits reduced penetrance among carriers. Here, we explore the hypothesis that deficits in torsinA function result in an increased vulnerability to stress associated with protein folding and processing in the endoplasmic reticulum (ER), where torsinA is located. Using an in vivo quantitative readout for the ER stress response, we evaluated the consequences of torsinA mutations in transgenic nematodes expressing variants of human torsinA. This analysis revealed that, normally, torsinA serves a protective function to maintain a homeostatic threshold against ER stress. Furthermore, we show that the buffering capacity of torsinA is greatly diminished by the DYT1-associated deletion or mutations that prevent its translocation to the ER, block ATPase activity, or increase the levels of torsinA in the nuclear envelope versus ER. Combinations of transgenic Caenorhabditis elegans designed to mimic clinically relevant genetic modifiers of disease susceptibility also exhibit a direct functional correlation to changes in the ER stress response. Furthermore, using mouse embryonic fibroblasts (MEFs) from torsinA knockout mice, we demonstrated that loss of endogenous torsinA results in enhanced sensitivity to ER stress. This study extends our understanding of molecular mechanisms underlying dystonia, and establishes a new functional paradigm to evaluate therapeutic strategies to compensate for reduced torsinA activity in the ER as a means to restore homeostatic balance and neuronal function.
INTRODUCTION
The capacity of cells to carry out their various functions is wholly dependent upon efficient protein synthesis, processing, trafficking and degradation. Given the concentration of protein in an average cell, approximately 300 mg/ml (a number close to the density of packed protein crystals at ∼400 mg/cm3), it is readily apparent that vital mechanisms are required to successfully manage this congested milieu (1,2). The efficiency with which the endoplasmic reticulum (ER) in neurons manages protein trafficking has functional consequences on neurotransmission, as changes in the levels of neurotransmitter transporters, receptors, and accessory proteins, can dramatically affect synaptic activity. Thus, deficits in the capacity of the ER to properly handle intracellular stressors, such as misfolded proteins, may disrupt homeostasis and contribute to disease susceptibility and progression in neurologic diseases (3).
A single codon deletion (ΔE302/303), within the gene encoding human torsinA, underlies a dominantly inherited movement disorder called early-onset generalized torsion dystonia (DYT1) (4). DYT1 dystonia is the most severe heritable form of dystonia, where symptoms include sustained muscle contractions and abnormal posturing, which typically appear in childhood (5). Human torsinA is a member of the large, structurally and functionally diverse AAA+ family of ATPase proteins (6). AAA+ proteins use energy released through ATP hydrolysis to remodel their target substrates and are involved in processes that can be broadly sub-classified as having roles in microtubule binding/severing, non-destructive recycling of proteins or protein quality control (7). Torsins are an evolutionarily conserved sub-family of the AAA+ proteins, with homologs across all sequenced invertebrates and mammalian genomes, that appears to have arisen uniquely in metazoans (8). Consistent with a role in protein folding and/or trafficking, evidence from multiple systems indicates that the ER is a native site of torsinA localization (9–11). Notably, torsins are the only AAA+ proteins known to be located inside the ER of eukaryotic cells where they are positioned to influence protein folding and trafficking (12).
Within the brain, torsinA is expressed primarily in neurons, with highest levels in the basal ganglia, cerebellum and cortex (13–15). Overexpression of the dystonia-associated glutamic acid deleted (ΔE302/303) form of torsinA induces the formation of aberrant membranous inclusions and redistribution of the protein to the nuclear envelope (16–18), thereby resulting in a net loss of native torsinA function at the ER. We and others have shown that overexpression of human wild-type (WT) torsinA and related invertebrate orthologs can suppress the accumulation of misfolded proteins (19–21), thereby revealing an activity for torsins as chaperone-like proteins potentially involved in ER-resident quality control mechanisms. Significantly, fibroblasts isolated from DYT1 patients, that contain endogenous levels of both torsinA and torsinA(ΔE), display a deficit in the processing of a reporter protein through the secretory pathway (9,10).
Considering that the pathology of DYT1 dystonia brains does not display evidence of neurodegeneration and that only 30–40% of heterozygous carriers of the torsinA(ΔE) mutation exhibit symptoms, it becomes apparent that disease incidence is a potential consequence of additional factors that cause neurons to exceed an intrinsic homeostatic threshold and enter into neuronal dysfunction. We hypothesize that torsinA normally acts as a pre-emptive regulator of the intracellular stress response in the ER and that deficits in torsinA activity in patients lead to a state of selective vulnerability, that along with other factors, manifest as dystonia.
Here, we report the application of the nematode model organism, Caenorhabditis elegans, as an animal system to investigate the capacity of torsinA, and structural variants of this protein, to mediate the onset of misfolded protein stress at the ER. Using a suite of transgenic nematode strains in conjunction with a quantitative in vivo fluorescent readout for the ER stress response, as well as correlative studies using mammalian fibroblasts that are either torsinA WT or null, we report that WT torsinA supports intracellular homeostasis and attenuates the consequences of protein misfolding stress. In contrast, mutant torsinA(ΔE) does not function in this capacity, and further exacerbates intracellular stress. The results of this study enhance our understanding of the mechanism by which torsinA functions and provide insights into the molecular basis of dystonia that can be potentially exploited for targeted therapeutic development.
RESULTS
Regulation of ER stress response by torsinA
Tight regulation of protein processing and assembly at the ER is a mechanism by which cells maintain homeostatic balance. The proper recognition, targeting and translocation of proteins through the ER are all essential aspects of successful trafficking and consequent functionality of secreted and membrane proteins. Our understanding of cellular quality control comes largely from studies investigating the consequences of the misfolded protein stress response in cultured cells and model organisms, which have illuminated numerous proteins involved in ER stress (22–24).
Previous studies showing that torsinA prevents accumulation of misfolded proteins and is also an ER resident protein led us to consider that torsinA may function in modulating the threshold to ER stress. C. elegans is well-suited to evaluating this hypothesis, as levels of ER stress can be readily monitored in transgenic animals using a fluorescent reporter consisting of the promoter from the hsp-4 gene (the worm homolog of the ER chaperone BiP) fused to GFP (25). While hsp-4 expression is ubiquitous, its levels are most strongly responsive to stress in the worm intestine. In the presence of ER stressors, such as tunicamycin (an inhibitor of protein glycosylation), transcription of hsp-4 is induced and GFP is highly expressed (Fig. 1A and B, top panel), thereby providing a direct and quantitative measure of in vivo stress response.
Figure 1.

TorsinA-mediated effects on the ER stress response in C. elegans. (A) Bar charts of GFP intensity from the stress reporter hsp-4::GFP alone or with WT torsinA or torsinA(ΔE) measured consistently in the region shown in (B). Data presented has been normalized to untreated hsp-4::GFP samples and is calculated as the mean ± SD of three experiments, where 30 animals were analyzed per replicate. *P < 0.05; NS, non-significant. Strains were treated with increasing concentrations of tunicamycin for 5 h (or DMSO as control). WT torsinA attenuated the response to ER stress observed at all concentrations of tunicamycin tested, whereas mutant torsinA did not. (B) Fluorescent micrographs of tunicamycin-treated (5 µg/ml; 5 h) C. elegans strains carrying the ER stress reporter hsp-4::GFP alone (upper panel), with a transgene of intestinally expressed WT torsinA (middle panel) or with mutant torsinA(ΔE) (lower panel). The white boxes indicate the 100 × 100 µm region just below the pharynx measured within all animals, which is magnified to the right of each animal. This is the region within animals that consistently exhibits the highest levels of fluorescence intensity. Scale bar = 100 µm. (C) Western blot showing torsinA expression in C. elegans strains from (B). Equal amounts of protein extract were loaded in each lane; actin was used as a loading control. The normalized intensity values for the various torsinA lines are shown below the blot image. (D) Stained agarose gel depicting the products of semi-quantitative RT–PCR reactions following treatment with 7 µg/ml tunicamycin (or solvent control). WT torsinA reduced the spliced xbp-1 transcript under both basal and tunicamycin-treated conditions, whereas torsinA(ΔE) increased the levels of stress-spliced xbp-1. For comparison, a loading control, ama-1, was used in all samples.
To determine the impact of torsinA on the ER stress response, transgenic nematodes were generated expressing the cDNA-encoding human torsinA under the ges-1 intestinal promoter by microinjection of worms containing the integrated hsp-4::GFP reporter. Effects of human torsinA are not complicated by the presence of endogenous torsin-like proteins, since the only post-embryonic C. elegans homolog, TOR-2, is expressed in the vulva and a small subset of head and tail neurons (26). The presence of WT torsinA dramatically reduced the response to ER stress observed in these animals (Fig. 1B, middle panel). To quantitate this reduction, the GFP intensity was consistently measured in the same region of the intestine (Fig. 1B) and these pixel values normalized to the hsp-4::GFP-alone control. Strikingly, the presence of WT human torsinA reduced the induction of the stress response by 35% as expressed across a range of tunicamycin concentrations (Fig. 1A). Additionally, the basal, uninduced levels of hsp-4::GFP were significantly suppressed ∼40%) in the presence of torsinA (Fig. 1A).
To determine the effect of the mutant torsin(ΔE) protein on the ER stress response, we generated transgenic nematodes that co-expressed torsinA(ΔE) in a genetic background with the hsp-4::GFP reporter. In contrast to the dramatic suppression exhibited by WT torsinA, levels of ER stress detected by the reporter were significantly higher in the presence of mutant torsinA(ΔE) (Fig. 1A and B, bottom panel). The relative increase in GFP expression ∼60%) in the presence of dystonia-associated torsinA(ΔE) was substantial in the absence of an exogenous ER stressor (Fig. 1A). Thus, torsinA(ΔE) expression is sufficient to induce the ER stress response and might be saturating the ER stress system in the animals because the addition of an exogenous stressor, such as tunicamycin, does not significantly enhance ER stress in these worms when compared with hsp-4 controls (Fig. 1A). Expression of torsinA was verified by western blotting (Fig. 1C). Furthermore, to ensure effects observed via the GFP reporter were reflective of a correlative impact on HSP-4 protein levels, we also confirmed torsinA-mediated changes using a human BiP antibody (Supplementary Material, Fig. S1).
It is possible that the reduced GFP levels seen in animals with torsinA could be the result of blockage of the unfolded protein response (UPR), a conserved compensatory mechanism triggered by the presence of misfolded proteins in the ER. The UPR alleviates the protein load by both temporarily reducing the production of proteins (via PERK-mediated translation inhibition), increasing levels of chaperone proteins (via transcription mediated by XBP-1 and ATF6) and increasing the exit and degradation of misfolded proteins via the ERAD pathway (XBP-1-mediated) (3). Blockage of the UPR prevents the induction of hsp-4::GFP (27) and if WT torsinA were to block the UPR, then the lower levels of GFP observed would not actually be indicative of lower stress levels. However, since animals with WT torsinA are still able to induce GFP when exposed to tunicamycin (Fig. 1A), a functional blockage of the UPR by torsinA is an unlikely explanation for the observed reduction in stress response. In contrast, another integral UPR-related protein, IRE-1, which when mutated causes a blockage in the UPR, was unable to induce GFP expression (27) (Supplementary Material, Fig. S2) in response to tunicamycin. Furthermore, animals require the UPR to survive stress-inducing treatments (27), such as dithiothreitol (DTT, a strong reducing agent). Mutant xbp-1 animals cannot trigger the UPR and, importantly, their survival is sensitive to exogenous stressors (28). Indeed, exposure to DTT resulted in approximately 15–20% death in either the hsp-4::GFP only, WT torsinA or mutant torsinA(ΔE)-containing animals, whereas 53% of the xbp-1 animals died (Fig. 2A). Although there was no statistically significant difference in survival between WT torsinA and hsp-4::GFP worms, there was an increasing trend toward survival, which would correlate well with a reduction in ER stress, when compared with a direct downregulation of the UPR by WT torsinA which, if anything, would lead to a decrease in survival following exposure to an exogenous stressor such as DTT. These collective data imply that the reduction in the ER stress response observed in the presence of WT torsinA activity is independent of a blockage of the UPR and is more likely a consequence of torsinA chaperone function, which has been recently demonstrated in vitro using biochemical methods (29).
Figure 2.

TorsinA does not suppress the UPR, while mutant torsinA(ΔE) increases ER stress via UPR factor xbp-1. (A) Bar chart comparing survival of hsp-4::GFP alone, or with WT or ΔE torsinA expression compared with xbp-1 mutant animals, with or without 3 mm DTT. These data are presented as a mean percentage of survival ±SD of three experiments, where 30 animals were analyzed per replicate. (B) Bar chart of GFP intensity from the stress reporter hsp-4::GFP alone (−) or with WT or ΔE torsinA measured consistently in the region shown in Fig. 1B. Data presented have been normalized to untreated hsp-4::GFP samples and is the mean ± SD of three experiments, where 30 animals were analyzed per replicate. *P < 0.05; NS, non-significant. Strains were fed either control or xbp-1 RNAi bacteria. The low level of stress observed with WT torsinA was the same with or without xbp-1(RNAi). Knockdown of xbp-1 eliminated the stress from torsinA(ΔE) demonstrating that this stress is mediated through the UPR. (C) Stained agarose gel showing the products of a semi-quantitative RT–PCR reaction. Control (ama-1) or stress-specific xbp-1 alternative splice product was amplified with samples from (B), demonstrating that levels of xbp-1 were reduced in RNAi animals.
In addition to monitoring the response to ER stress with the hsp-4::GFP reporter, we also assessed this in a secondary assay by detecting levels of the stress-specific spliced isoform of xbp-1. Upon the detection of ER stress, the xbp-1 transcript is alternatively spliced to produce an active version of xbp-1 mRNA (25,27). Semi-quantitative reverse transcriptase–polymerase chain reaction (RT–PCR) was used to assess the levels of spliced (i.e. stressed) xbp-1 mRNA in transgenic animals exposed to tunicamycin (Fig. 1D). As anticipated, spliced xbp-1 levels increased in response to tunicamycin in the absence of torsinA, whereas animals expressing WT torsinA maintained xbp-1 mRNA at the lower, unstressed level, even in the presence of stressor. In contrast, the expression levels of spliced xbp-1 were increased in the presence of torsinA(ΔE), with or without tunicamycin (Fig. 1D).
The UPR protein, xbp-1, participates in torsinA action on the ER stress response
As discussed above, the UPR is triggered in cells under ER stress. If torsinA is acting through the stress pathways, as opposed to a more direct (and trivial) action on the hsp-4::GFP reporter itself, then UPR signaling should be involved. As expected, interference RNA (RNAi)-mediated knockdown of xbp-1 in untreated animals lowers the basal expression of hsp-4::GFP (Fig. 2B). Notably, the markedly elevated hsp-4::GFP levels observed in mutant torsinA(ΔE) or WT/ΔE-expressing animals were eliminated when xbp-1 was reduced by RNAi (Fig. 2B and Supplementary Material, S3A). This indicates that the stress response observed in the presence of torsinA(ΔE) or WT/ΔE is mediated through the UPR pathway. To confirm that the RNAi was specifically targeting xbp-1, semi-quantitative RT–PCR was performed in these strains with and without RNAi treatment (Fig. 2C and Supplementary Material, S3B). RNAi-treated animals showed a reduction in xbp-1 levels, whereas the control (ama-1) stayed consistent. These results (Figs 1D and 2B) show that the cellular action of torsin functions through its effect on the UPR. Specifically, WT torsinA lowers stress by a mechanism that reduces the activated, alternatively spliced form of xbp-1 mRNA, whereas mutant torsinA acts to increase splicing of xbp-1 mRNA, thereby increasing the ER stress response.
Together, these results indicate that, in its WT form, torsinA functions potently to combat the onset of the ER stress response. Furthermore, these data demonstrate that mutant torsinA(ΔE) itself induces the ER stress response and thereby results in a reduction of the normal capacity to attenuate stress at the ER, thus contributing to the overall homeostatic burden.
torsinA/torsinA(ΔE) animals exhibit diminished capacity and variability in attenuating ER stress response
Patients with DYT1 dystonia are heterozygous for the torsinA(ΔE) mutation and the disorder is inherited dominantly (4). To mimic the heterozygous state, worms were generated carrying transgenes encoding both WT and mutant torsinA(ΔE). Examination of ER stress in the absence of exogenous stressor revealed that hsp-4::GFP fluorescence in WT/ΔE animals was elevated (Fig. 3A) and that the stress response occurred through the UPR, similar to mutant torsinA(ΔE) alone (Supplementary Material, Fig. S2). Furthermore, WT/ΔE transgenic animals exposed to tunicamycin showed high ER stress response levels statistically equivalent to what was observed with torsinA(ΔE) alone (Fig. 3A and B). We further evaluated these transgenic animals using an alternate ER stressor, DTT. As seen with the tunicamycin treatment, WT torsinA was protective against DTT-induced stress, whereas ΔE and WT/ΔE torsinA animals were both more susceptible to stress and also were not significantly different from each other in this response (Fig. 3A). Expression of torsinA constructs was verified by western blotting (Fig. 1C). These data suggest that the presence of the mutant gene product is sufficient to mask the protective activity of the normal gene product in vivo, perhaps indicative of the dominant nature of inheritance of DYT1 dystonia.
Figure 3.

ER stress is not attenuated in heterozygous WT torsinA/torsinA(ΔE) animals. (A) Bar charts of GFP intensity from the stress reporter hsp-4::GFP alone (−), with WT, ΔE or WT/ΔE torsinA, measured consistently in the region shown in Figs 1B or 3B. Data presented have been normalized to untreated hsp-4::GFP samples and is the mean ± SD of three experiments, where 30 animals were analyzed per replicate. *P < 0.05; NS, non-significant. Strains were treated with 5 µg/ml tunicamycin for 5 h or 3 mm DTT for 4 h. (B) Fluorescent micrograph of a tunicamycin-treated C. elegans nematode expressing hsp-4::GFP with a combination of WT/ΔE torsinA. The white box indicates the 100 × 100 µm region measured within all animals, which is magnified to the right of the animal. Scale bar = 100 µm. (C) GFP intensity distribution for 100 animals per strain were categorized according to pixel intensity [very low (<1000 a.u.), low (1000–2000 a.u.), medium (2000–3000 a.u.) and high (>3000 a.u.)]. Notably, most worms expressing WT torsinA have very low pixel intensity while most worms expressing ΔE have medium or high pixel intensities. Examples of pixel intensity differences are displayed in Supplementary Material, Fig. S4.
Interestingly, we observed greater animal-to-animal variability in managing the basal ER stress response in both the torsinA(ΔE) and strains co-expressing both mutant and WT torsinA together than in control and WT torsinA strains (Figs 1A and 3A). To precisely examine this variability, GFP intensities were quantified within individual nematodes in the populations of the various isogenic strains (Fig. 3C). For each transgenic line, 100 animals were categorized as exhibiting either a very low (<1000 arbitrary units [a.u.]), low (1000–2000 a.u.), medium (2000–3000 a.u.) or high (>3000 a.u.) stress response in the absence of stressor (see Supplementary Material, Fig. S4 for images of each category). While a third of the hsp-4::GFP animals had very low levels of GFP (34%), a majority of the WT torsinA-expressing animals had predominantly very low levels of GFP (73%); this is consistent with the ability of WT torsinA to minimize the ER stress response. There was a substantially greater number of mid- to high-level GFP animals within these populations (53 and 28%, respectively). Notably, the mutant torsinA(ΔE) and the heterozygous WT/ΔE strains still had a few animals with very low levels of GFP (8% and 15%, respectively). Thus, when torsinA(ΔE) is expressed, even in genetically isogenic animals, the degree of stress observed is variable and stochastic. Importantly, the tendency away from homeostasis and toward greater stress levels within a population is substantially attenuated by WT torsinA activity (Fig. 3C).
Localization of WT torsinA to the ER is required for attenuation of stress response
The localization of torsinA to the ER has been considered essential for protein activity, however the absence of functional assays by which to evaluate torsinA have previously precluded studies to investigate this assumption in vivo. Therefore, we evaluated the consequences of structural changes in torsinA that would hypothetically alter its ATPase activity, translocation into the ER and distribution within the ER and monitored their impact on attenuating the ER stress response.
The first 40 amino acids of torsinA encode a signal sequence and a hydrophobic membrane-associated domain. Deletion of these motifs causes torsinA to localize in the cytoplasm (30). To determine if torsinA can retain function in reducing the stress response without proper entry and localization in the ER, we generated distinct strains of transgenic nematodes expressing either the WT, mutant (ΔE) or both WT/ΔE torsinA gene products, all containing a deletion of the N-terminal 1–40 amino acids (Δ1–40). Examination of animals expressing an N-terminal truncation of WT torsinA indicated that deletion of these motifs resulted in the loss of torsinA-dependent reduction in tunicamycin-induced stress (Fig. 4A). Stress response levels in either mutant (ΔE) or WT/ΔE torsinA animals with the truncation remained consistently high (Fig. 4A). Likewise, a series of hydrophobic amino acids (24–40) within this N-terminal region is predicted to localize torsinA within the ER lumen (30), but does not necessarily allow the protein to associate with the membrane. Animals expressing this internal deletion within the WT torsinA N-terminus were also not able to combat ER stress in response to tunicamycin (Fig. 4B). Finally, two point mutations within the ATPase domain were assessed. One mutation, E171Q, prevents ATP hydrolysis and causes torsinA to preferentially localize within the NE (16,17,31). The second mutation, K108A, cannot bind ATP but remains localized to the ER (16,17). Neither of these mutated proteins were able to reduce response to stressors (Fig. 4C). In all cases, these proteins were expressed at similar levels to non-mutated forms of torsinA, but Δ1–40 and Δ24–40 proteins were slightly smaller in size than WT torsinA, correlating to amino acid deletions and a lack of glycosylation (Fig. 4D). Taken together, these data indicate that the ability of torsinA to maintain a homeostatic threshold against stress is dependent upon its proper localization within the ER and that the ATPase domain is required for this activity (29).
Figure 4.

Only correctly localized WT torsinA attenuates ER stress. (A–C) Bar charts of GFP intensity from stress reporter hsp-4::GFP alone or with WT or ΔE or WT/ΔE torsinA measured consistently in the region shown in Fig. 1B. Data presented have been normalized to untreated hsp-4::GFP samples and is the mean ± SD of three experiments, where 30 animals were analyzed per replicate. *P < 0.05; NS, non-significant. Animals were treated with 5 µg/ml tunicamycin for 5 h. (A) Amino acids 1–40 are required for WT torsinA to reduce ER stress in response to tunicamycin. The deleted amino acids, which contain the signal sequence and hydrophobic membrane localization sequences, were removed from WT, torsinA(ΔE) and WT/ΔE. None of the mutated forms reduced ER stress response in comparison with full-length WT torsinA. (B) The hydrophobic membrane localization sequence (amino acids 24–40) within WT torsinA is required to combat tunicamycin-induced ER stress. (C) Two previously characterized mutations in the ATPase region of torsinA were expressed in C. elegans and found to be required for activity. Specifically, the E171Q mutation, which prevents ATP hydrolysis and is localized to the NE, and the K108A mutation, which cannot bind ATP and is localized to the ER, did not prevent ER stress against tunicamycin. (D) Extracts from transgenic animals carrying hsp-4::GFP alone or torsinA mutants from parts (A–C) were isolated and analyzed by western blotting. Equal amounts of protein extract were loaded in each lane and actin was used as a loading control.
Modulation of ER stress response by torsinA correlates with phenotype of DYT1 patient-associated polymorphisms
Prior genetic analyses of DYT1 dystonia patients have discerned that disease penetrance is correlated with a specific polymorphism, D216H, in the coding sequence of torsinA (32). The H216 allele, when carried in trans to the ΔE allele, is increased in frequency in non-manifesting DYT1 carriers and decreased in affected DYT1 dystonia patients (32) (Fig. 5A). In a manner similar to torsinA(ΔE), overexpression of the H216 allele alone shows a tendency to form inclusions in cell culture, when these forms of torsinA are overexpressed together, inclusions are reduced (33), consistent with a protective compensatory interaction of H216 torsinA with torsinA(ΔE). To evaluate the consequences of this disease-modifying polymorphism in a functional context, we constructed multiple lines of transgenic nematodes to investigate its impact on inducing or preventing the response to ER stress in C. elegans. The results of this analysis indicated that while animals expressing the H216 allele alone showed an increased stress response, combined expression of the H216 torsinA variant in trans with either WT or torsinA(ΔE) containing the usual D216 residue, led to a striking reduction of the stress response, returning protection to the level exhibited by WT torsinA alone (Fig. 5B). The trans combinations of WT/216H, as well as ΔE/216H, were tested with and without tunicamycin, and stress response levels were significantly reduced back to WT torsinA levels in all cases, as long as the 216H protein was expressed (Fig. 5B). This observed restoration of WT torsinA function in attenuating the response to ER stress by the trans configuration was in contrast to that seen when WT D216 torsinA was placed in trans with torsinA(ΔE) (Fig. 3A). Expression of torsinA was verified by western blotting (Fig. 5C). While such ectopic expression data across species must always be considered within context, these results collectively demonstrate that the effect of torsinA and its polymorphic variants on ER stress appears to correlate with the disease penetrance observed in human populations.
Figure 5.

Polymorphic D216H allele of torsinA effects stress differently when alone or in trans with ΔE mutant. (A) Schematic drawing summarizing human genetic data obtained from Risch et al. (32), illustrating that non-manifesting DYT1 carriers have an increase in a polymorphism that is protective in trans. The uppermost drawing depicts the torsinA protein with the approximate location of the D216H polymorphism, as well as the ΔE deletion. The lower pair of drawings demonstrates that, within an individual, when the 216H allele is in trans to the ΔE allele, there is a decrease in affected DYT1 dystonia patients. (B) Bar chart of GFP intensity from the stress reporter, hsp-4::GFP, alone (−) or with WT torsinA or torsinA(ΔE) (each of these torsinA constructs contains and expresses the normal D216 amino acid) or with torsinA H216 alone or in a trans combination with the WT or ΔE torsinA. Data presented have been normalized to untreated hsp-4::GFP samples and is the mean ± SD of three experiments, where 30 animals were analyzed per replicate. *P < 0.05; NS, non-significant. C. elegans expressing the torsinA H216 mutation alone displays high levels of ER stress comparable to torsinA(ΔE) while combined expression of H216 torsinA in trans with WT or torsinA(ΔE) restored ER stress to levels observed from WT torsinA alone. (C) Extracts from transgenic animals carrying hsp-4::GFP alone or torsinA D216H mutants from (A) were isolated and analyzed by western blotting. Equal amounts of protein were loaded in each lane; actin was used as a loading control.
Endogenous torsinA is required to manage ER stress response in mammalian cells
To further extend the hypothesis that torsinA functions to maintain a homeostatic threshold against ER stress in mammals, we used MEFs to evaluate the stress response with and without endogenous torsinA. MEF cells, isolated from either WT or homozygous torsinA knockout mouse embryos, were treated with either DTT (Fig. 6A and B) or tunicamycin (Fig. 6C and D) to induce protein misfolding in the ER. The degree of stress within the ER was quantitated by measuring BiP (GRP78) protein expression (34). In the absence of torsinA and stressor, BiP levels were intrinsically high, and became higher when exposed to the stressor (Fig. 6). In contrast, the presence of torsinA in WT MEFs significantly lowered levels of the basal stress response and also showed a reduced sensitivity to the onset of incrementally induced stress when exposed to increasing concentrations of these drugs when compared with torsinA-null MEFs (Fig. 6). These MEF data, with endogenous torsinA levels, complement findings in C. elegans, whereby the presence of WT torsinA substantially reduces HSP-4/BiP levels (Supplementary Material, Fig. S1).
Figure 6.
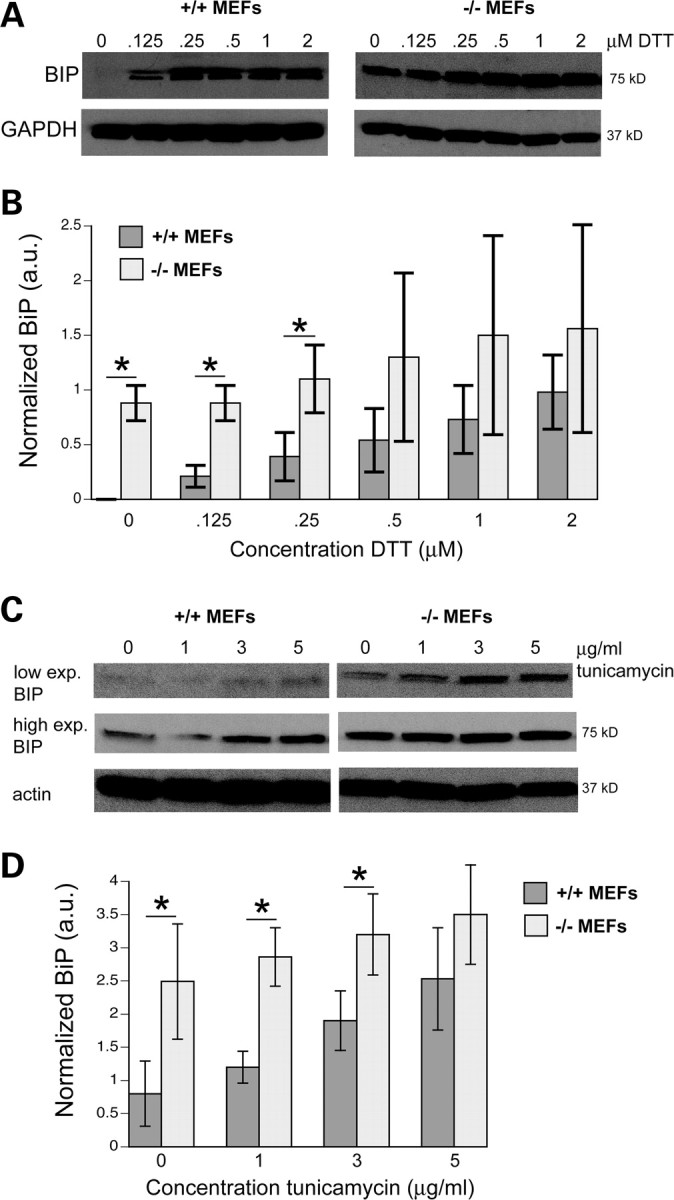
Absence of endogenous torsinA increases ER stress response in torsinA−/− MEFs. (A) Western blot analysis of whole-cell lysates of torsinA+/+ and torsinA−/− MEFs treated with 0, 0.125, 0.25, 0.5, 1 and 2 µm of DTT for 4 h. Cells were harvested 20 h after treatment and 40 µg of each lysate was resolved by electrophoresis in 10% SDS–polyacrylamide. In order to evaluate the level of ER stress induction, the expression of BiP (GRP78), a stress chaperone and GAPDH (used as loading control) were measured by western blot and densitometry analysis using Quantity one 4.6 (BioRad). This experiment was performed in triplicate and the western blot figure shows a representative blot. (B) Graph of values of western blots from DTT-treated samples conducted in triplicate showing BiP levels significantly higher for torsinA−/− MEFs at 0 (P<0.001), 0.125 (P < 0.007) and 0.25 µM DTT (P < 0.04) when compared with torsinA+/+ MEFs after normalizing to GAPDH levels (using a two-tailed Student's t-test). (C) Western blot analysis of whole-cell lysates of torsinA+/+ and torsinA−/− MEFs treated with 0, 1, 3, 5 µg/ml of tunicamycin for 5 h. Blotting was performed as described in (A) except that β-actin was used as a loading control. This experiment was performed in quadruplicate and the western blot figure shows a representative blot. The BiP signal after low (top panels) and high exposures (middle panels) is shown. The bottom panels show actin levels. (D) Graph of values of western blots from tunicamycin-treated samples conducted in quadruplicate showing BiP levels significantly higher for torsinA−/− MEFs at 0 (P < 0.009), 1 (P < 0.001) and 3 µg/ml tunicamycin (P < 0.005) when compared with torsinA+/+ MEFs after normalizing to actin levels (using a two-tailed Student's t-test). We quantified using the high exposures because the BiP signal in the presence of torsinA+/+ was so strongly attenuated in the lower exposure blot.
These data reveal that the capacity for torsinA to act as a functional buffer to the induction of the ER stress response is conserved across species and is not an artifact of atypical expression in the nematode model. More importantly, these results confirm an intracellular requirement for torsinA to maintain homeostasis and demonstrate that endogenous levels of torsinA act inherently to maintain the intracellular threshold to stress at the ER in mammalian cells.
DISCUSSION
While the precise causes underlying the clinical manifestations of dystonia remain unresolved, the means by which the torsinA(ΔE) mutation translates into neuronal dysfunction at low penetrance is suggestive less of an absolute consequence and more of an imbalance in proper cellular dynamics. A proposed role for torsinA in the cellular stress response has been previously investigated (26,35–37). While these reports indicated a cytoprotective role for torsinA, studies to date have not found a change in torsinA expression in response to ER stress (16,35,38). Our data do not contradict these prior reports, but in an important distinction, represent the delineation of the ability of torsinA to prevent the onset of the ER stress response, and thereby to protect cells. Thus, we propose a conceptually different role for this protein that contends that torsinA activity serves natively to increase the overall cellular threshold to which misfolded proteins or other stressors may induce dysfunction. In this model, decreases in the buffering capacity of torsinA in the ER, owing to the presence of mutant torsinA, predispose patient cells to a state of vulnerability where dystonia may result from an inability to combat secondary environmental or genetic modifiers.
Although the decreased function of torsinA in the ER may represent a critical molecular deficit, it is equally important to consider the mislocalization of the mutant protein to the nuclear envelope as either a causative or contributory event in disease onset (16,17,39). Moreover, these aspects need not be considered mutually exclusive etiological effectors. A mutant-associated shift in the intracellular locale and/or substrate interaction for torsinA might result in downstream effects that impact homeostasis and proper neuronal activity. Consistent with this, we show in a cellular background lacking endogenous torsin proteins, the C. elegans intestine, that expression of mutant human torsinA(ΔE) resulted in enhanced ER stress in vivo, while WT torsinA was protective (Figs 1 and 3). Recent studies have also indicated that torsinA mutants exhibit differential cellular stability and are degraded by proteasomal and lysosomal pathways in neurons (40,41). Thus, in addition to resulting in a potential imbalance of torsinA oligmerization, with a concomitant effect on function, premature loss of torsinA function as a consequence of degradation may also contribute to an enhanced vulnerability to stress. Conversely, very high levels of either WT or mutant torsinA(ΔE) expressed from the murine prion promoter result in neuronal dysfunction and phenotypic abnormalities (42). Importantly, our comparative analyses of MEFs from normal versus torsinA knockout mouse embryos clearly show that a change in endogenous torsinA levels directly correlates with a diminished capacity to manage ER stress.
Several additional lines of evidence support a role for torsinA in the management of protein misfolding at the ER. It has been demonstrated that torsinA regulates trafficking of the mammalian dopamine transporter (DAT) and other polytopic membrane-bound proteins in mammalian cell cultures (31). We have also shown that torsinA can regulate processing of the C. elegans DAT-1, a membrane protein trafficked through the ER to neuronal membranes, in vivo (26). Interestingly, mutations in ε-sarcoglycan (SGCE), a glycosylated transmembrane protein, also result in an ER-based trafficking defect where mutant, but not WT versions of SGCE were selectively targeted for degradation by torsinA (43). This latter report is significant as it represents a link between two proteins associated with different forms of dystonia, since SGCE is the mutated DYT11 gene product responsible for myoclonus dystonia (44). Collectively, these data suggest a role for torsinA as either directly participating in the folding and assembly of proteins in the ER, and/or trafficking of correctly folded proteins onto the Golgi or misfolded proteins out into the cytoplasm for degradation.
The efficiency by which proteins are processed through the ER is a consequence of a combination of factors including cell-type, translational load, development and aging, among others (45). Through pre-emptive blockage of protein misfolding or co-translational redirection to the cytoplasm for degradation, the burden of misfolded substrates at the ER is reduced, which has an impact on overall cytosolic protein dynamics, including proteosomal degradation (46). This provides an explanation for why torsinA, which we have shown must be retained at the ER for its activity in attenuating stress, also robustly prevents accumulation of different cytoplasmic proteins, such as α-synuclein or polyglutamine-repeat containing proteins, as well as the downstream neurotoxic consequences of their overexpression (18–21,37). Furthermore, overexpression of torp4A, the Drosophila ortholog of torsinA, is neuroprotective and reduction of the gene product results in retinal degeneration (47). It has also been demonstrated that, under conditions of acute ER stress, the translocation of secretory and membrane proteins is quickly and transiently attenuated in a signal sequence-selective manner (46,48). Thus, pre-emptive quality control mechanisms at the ER act to minimize the necessity for UPR induction in response to stress. Changes in torsinA mRNA and protein levels during rodent development have been reported, with torsinA highly expressed during perinatal periods (49–51). Thus, deficits in regulating intracellular stress may also have implications during the postnatal developmental window in which early-onset dystonia patients exhibit symptoms and would be consistent with the concept that age-dependent changes in the homeostatic regulation of the neurons may impact susceptibility in individuals (5).
While the overt differences between humans and nematodes should not be understated, our results are reflective of known patient etiology in several respects. Firstly, the heterozygous state acts dominantly both to cause disease in people (4) and results in a diminished capacity to modulate the ER stress response in C. elegans (Fig. 3). Secondly, only 30–40% of people carrying the torsinA(ΔE) mutation develop the disease. A surprising outcome of our transgenic analyses was the range of stress response levels displayed by populations of genetically invariable, mutant torsinA(ΔE)-expressing worms (Fig. 3C). Thus, the stochastic nature of these data are intriguing in the context of disease penetrance, and may be partly characteristic of the relative variability in disease onset and course in torsinA(ΔE) gene carriers. Thirdly, the differences we observed among transgenic worms using the ER stress response as a readout for torsinA functionality recapitulate elegant human genetic studies, which discerned that interallelic complementation between torsinA(ΔE) and torsinA containing a modifying polymorphism (H216) results in dramatic suppression of symptoms among mutant torsinA(ΔE) gene carriers (33).
The vulnerability of neurons to imbalances in the regulation of protein load, or proteostasis, has been proposed as a mechanism responsible for a variety of disorders (24,52–54). It has recently been shown that selective neuronal vulnerability leading to progressive weakening and paralysis manifested in a mouse model of amyotrophic lateral sclerosis is directly associated with increased ER stress (54). Notably, torsinA and torsin-related proteins have evolved exclusively in metazoans (8); therefore we speculate this family of ATPases emerged in response to a requirement for the management of increased protein load of eukaryotic translation, particularly in neurons. In this regard, reduced function of torsinA in DYT1 dystonia likely represents a deficit in a basic cellular mechanism at the ER, wherein neuronal dysfunction is a consequence of a reduced torsinA-dependent ‘gatekeeping’ activity in attenuating response to stress.
The absence of overt neurodegeneration in dystonia is indicative that the subtle changes in neuronal function that lead to disruption of proper coordination and movement may be reversible. Application of the C. elegans system and related cellular models toward screening of small molecule effectors represents a promising strategy to identify chemical modifiers of torsinA-mediated stress modulation that can advance therapeutic discovery for dystonia (55). Increased understanding of the functional role of torsinA at the ER will serve to illuminate molecular mechanisms underlying dystonia, and perhaps related disorders that are an outcome of homeostatic imbalance.
MATERIALS AND METHODS
Plasmid constructs
Full-length WT and torsinA(ΔE302/303) variants were PCR-amplified using primers 5′-CTAGCTAGCATGAAGCTGGGCCGGGCCGTG-3′ and 5′-GGGGTACCTCAATCATCGTAGTAATAATCTAAC-3′ from cDNAs provided by Ben Cravatt (Scripps Research Institute, La Jolla, CA, USA). The K108A and E171Q mutation torsinA clones were provided by Phyllis Hanson (Washington University) and the hydrophobic region deletion torsinA clone was provided by Michal Zolkiewski (Kansas State University). For cloning, these mutants were also amplified using the above primers. The N-terminal 40 amino acid deletion were created using primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCCCGTCTGTACTGCCTCTTC-3′ and 5′-GGGGACTTTGTACAAGAAAGCTGGGTCTCAATCATCGTAATAATC-3′ on either WT or torsinA(ΔE) templates. The H216 mutation was introduced into cDNA clones of torsinA by site-directed mutagenesis using the following primers: TorA/attb-F: GGGGACAAGTTTGTACAAAAAAGCAGGCTAAATGAAGCTGGGCCGGGCC; TorA/attb-R2: GGGGACCACTTTGTACAAGAAAGCTGGGTATCATCGTAGTAATAATCTAACTTGG; TorA/216-F: GCAGAAAGGATCACACATGTGGCTTTGGATTTCTGG; TorA/216-R: CCAGAAATCCAAAGCCACATG-TGTGATCCTTTCTGC.
Expression plasmids were created with Gateway recombinational cloning (Invitrogen). PCR products were initially recombined with the pDONR221 vector to create pENTRY clones.
TorsinA variants were expressed under the promoter from the gut-specific type B carboxylesterase (ges-1) gene. Briefly, a Gateway recombination cassette was inserted downstream of the ges-1 promoter in plasmid pJM16 (56) to create the pDEST-JM16 vector. All torsinA pENTRY plasmids (above) were then recombined into the pDEST-JM16 vector and these recombination plasmid products were then subsequently used to create transgenic worms.
C. elegans strains and protocols
Nematodes were grown and maintained using standard procedures (57). Strains used were hsp-4::GFP (SJ4005), xbp-1(zc12) III; zcls4 V (SJ17) and ire-1(zc14) II; zcIs4 V (SJ30). For new strains generated in the course of this work, each plasmid was injected at a total concentration of 50 µg/ml. Either WT or torsinA(ΔE) alone (50 µg/ml) or together (25 µg/ml) each were co-injected into the hsp 4::GFP strain with rol-6 (50 µg/ml) (58), which was used as the selective marker for transformation. For each plasmid mixture, at least three stable lines were generated and analyzed. Representative lines were selectively integrated by using UV irradiation either using a Spectroline UV cross-linker with 254 nm bulbs and an energy setting of 20 mJ/cm2 or with 365 nm bulbs at 300 mJ/cm2. The integrated heterozygous torsinA strain was generated directly by integrative microinjection (58). The integrated transgenic lines were designated as follows: WT torsinA [strain UA97 (baIn16)], mutant torsinA(ΔE) [strain UA98 (baIn17)], heterozygous D216H/WT torsinA [strain UA124 (baIn21)], heterozygous D216H/torsinA(ΔE) [strain UA125 (baIn22)] and heterozygous WT/torsinA(ΔE) [strain UA99 (baIn18)]. The following torsinA strains were maintained as stable lines: 1–40 amino acid deletion WT torsinA [UA100 (baEx75)], 1–40 amino acid deletion torsin(ΔE) [strain UA101 (baEx76)], 1–40 amino acid deletion heterozygous torsinA [strain UA102 (baEx77)], K108A mutation [strain UA103 (baEx78)], E171Q mutation [strain UA104 (baEx79)], hydrophobic amino acid 24–40 deletion of WT torsinA [strain UA105 (baEx80)], hydrophobic amino acid 24–40 deletion of torsinA(ΔE) [strain UA106 (baEx81)], hydrophobic amino acid 24–40 deletion of heterozygous torsinA [strain UA107 (baEx82)] and mutant torsinA (D216H) [strain UA123 (baEx97)]. All torsinA expression cassettes encode the D126 version of the protein, except where noted.
Fluorescent analysis of hsp-4::GFP worms
ER stress response was examined in late L4 stage animals that were transferred manually to nematode growth medium (NGM) plates spread with concentrations of tunicamycin or DTT indicated within the results and figure legends. Tunicamycin (MP Biomedicals) was made at a stock concentration of 1 mg/ml in dimethylsulfoxide (DMSO), and DTT (BioRad) was made at a stock concentration of 1 m in water. After exposure to either treatment for the durations indicated within the figure legends, worms were mounted on a 2% agarose pad and analyzed with a charge-coupled device camera (Photometrics CoolSnapHQ) on a Nikon (E800) microscope at 40X magnification. GFP intensity was measured in pixels following image capture using consistent conditions and assigned arbitrary units from a 100 × 100 µm region of the anterior-most intestine, directly behind the pharynx of each animal using MetaMorph software (Molecular Devices). For each strain and condition, at least 30 animals were quantitated in three independent replicates.
Stress survival assay
Thirty L4 stage worms of each strain [hsp-4::GFP alone or with WT torsinA, mutant torsinA(ΔE) or xbp-1 mutation] were placed on 60 mm NGM plates spread with 3 mm DTT or water solvent control. After 16 h, the number of living worms was counted. The determination of survival was made as described in Bischof et al. (28). The assay was repeated three times.
Worm extracts and western blot analysis
Each worm strain was grown up on ten 150 mm plates, washed five times using M9 buffer, washed once with protein extraction buffer [150 mm KCl, 1 mm EDTA, 0.25% sodium dodecylsulfate (SDS), 1% NP-40, 50 mm Tris–HCl (pH 7.4) and Roche complete protease inhibitor cocktail], then collected in 15 ml conical tubes. The worm pellets were frozen in liquid N2 overnight and then sonicated on ice. The supernatants were collected after centrifugation and protein concentrations were determined using BCA kit (Sigma). The supernatants were mixed with protein-loading buffer [95% protein sample buffer (BioRad), 5% β-ME], boiled for 5 min and then centrifuged at 16 100 g for 5 min before loading on a gel. The western blot was performed as previously described (20), with the following changes: (i) 200–300 mg total worm protein for each strain was loaded in each well, (ii) a polyclonal peptide rabbit anti-torsinA antibody (Santa Cruz Biotechnology) was diluted 1:200, (iii) the horse anti-rabbit IgG (Amersham Biosciences) was diluted in 1:5000 and used as the secondary antibody to probe the rabbit anti-torsinA. The control actin antibody was the monoclonal C4 (MB Biomedicals). In the case of HSP-4 protein detection, an antibody to human BiP [GRP78] (Santa Cruz Biotechnology) that recognizes a shared epitope with worm HSP-4 (amino acids 19–50 near the N-terminus of GRP78) was used at a 1:1000 dilution. Western blots were blocked using SuperBlock blocking buffer (Pierce). To compare amounts of protein among integrated transgenic lines on western blots, X-ray film was scanned using a FujiFilm LAS-4000 digital imaging system and analyzed with Multi Gauge v.3.0 software. Protein intensities were determined (i.e. torsinA values were divided by the actin values) and values were then normalized to 1 (i.e. using the lowest torsinA sample within a single blot).
RNAi and RT–PCR
RNAi feeding was performed as described (59) except NGM media was used and 20 gravid animals per plate were allowed to lay eggs for 12 h before removing them. Approximately 4 days later, the late L4 stage was analyzed for hsp-4::GFP expression. The bacterial clone producing dsRNA for xbp-1 was obtained from the MRC RNAi library (Cambridge, UK). The control bacteria contained the empty RNAi expression vector pL4440. For monitoring spliced xbp-1 transcript expression, 50 late L4 stage worms were transferred to tunicamycin (7 µg/ml) or DMSO controls for 5 h followed by RNA isolation and RT–PCR.
For semi-quantitative RT–PCR of the above RNAi-treated animals, about 30 µl of worms were washed into a microcentrifuge tube and lysed in Trizol (Invitrogen) by freezing in liquid nitrogen and thawing at 42°C repeatedly four times. Phases were separated using 1-bromo-3-chloropropane (Sigma) and the top phase was removed to a separate microcentrifuge tube and precipitated in isopropanol at −20°C for 1 h. Glycoblue (Ambion) was added before isopropanol to a concentration of 50 µg/ml to allow for easy viewing of the RNA pellet. After precipitation, RNA was washed in 75% ethanol and resuspended in RNase-free water. RNA was converted to cDNA using Superscript III RT (Invitrogen) and the PCR was performed using Phusion Polymerase (New England Biolabs) on a PTC-200 thermocycler. RT–PCR of gene upregulation was performed as above with the following changes: 50 worms were picked into the cap of an RNase-free microcentrifuge tube containing 10 µl of M9. These were pulse spun to bring the worms to the bottom of the tube and lysed with Trizol and extracted as described above. The precipitation was at −20°C for 3 h. The RNA was resuspended in 10 µl of RNase-free water and put directly into a DNase-I reaction (Invitrogen) to eliminate any contaminating DNA, followed by RT–PCR. The following primers were used for RNAi and/or gene upregulation PCR. For xbp-1, the forward primer was 5′-TTTGAATCAGCAGTGGGAAC-3′, and the reverse primer was 5′-CTTGGGCTCTTGAGATGTTC-3′. These primers were designed to specifically and only detect the alternative stress-spliced version of xbp-1 (25,27). For the ama-1 (RNA polymerase II large subunit) loading control, the forward primer was 5′-CGAGTCCAACGTACTCTCC-3′, and the reverse primer was 5′-GATGTTGGAGAGTACTGAGC-3′.
Mammalian cell culture and analysis
Primary MEFs were generated from individual E13–14 embryos of crosses between heterozygous +/torsinA-null mice (kindly provided by Dr Yuqing Li, UAB). Cultures were maintained as described (10), genotyped (60), treated with Plasmocin™ (InvivoGen) to avoid mycoplasm contamination, and used within the first four passages. The torsinA+/+ and torsinA−/− MEFs were plated at 105 cells per well and incubated at 37°C overnight. The next day, both types of MEFs were treated with 0, 0.125, 0.25, 0.5, 1 and 2 µm of DTT for 4 h or 0, 1, 3 or 5 µg/ml tunicamycin for 5 h. After 20 h, the cells were washed in PBS and resuspended in 200 µl RIPA lysis buffer (10). The protein concentration was measured by Bradford's method (Sigma). Proteins were resolved by electrophoresis in 10% SDS–polyacrylamide stained for protein and immunoblotted as described (61). The antibody used to detect GRP78 (BiP) (Santa Cruz) was diluted 1:300, for control GAPDH (Chemicon) was diluted 1:500 and for actin control (Sigma monoclonal AC-74) was diluted 1:3000. Band densities were determined by scanning the blots using a Perfection 2480/2580 scanner (Epson America Inc., Long Beach, CA, USA), quantifying band intensity using Quantity One 4.6.1 software (BioRad) and normalizing levels of BiP to GAPDH (Fig. 5A) or BiP to actin (Fig. 5C) for each lane in three experiments.
Statistical analysis
For the nematode experiments, comparisons were done on GFP pixel intensity between strains and between drug and solvent controls unless otherwise noted. For each assay, the data presented are the normalized fold change mean ± SD of three independent trials for every strain or condition unless otherwise noted. Each trial consists of the GFP intensity from 30 animals averaged. Normalization compares GFP intensity for all the samples (hsp-4::GFP alone or with torsinA versions, untreated or treated) divided by the average of untreated hsp-4::GFP alone. Statistics were performed using ANOVA (www.physics.csbsju.edu/stats/anova.html) and post-test Bonferroni correction (http://www.graphpad.com/quickcalcs/posttest1.cfm). Statistical analysis on MEF western blots was performed using Prisma software (version 3.0). Results are expressed as mean ± SEM. Comparison between groups was evaluated by the two-tailed Student's t-test.
AUTHOR CONTRIBUTIONS
P.C., A.J.B. and F.C.N. developed concepts/approaches, performed experiments, contributed reagents/data analysis and edited manuscript. C.J.P. and J.H. developed concepts/approaches, performed experiments and contributed reagents/data analysis. J.C.R. and S.A.F. performed experiments and contributed reagents/data analysis. L.A.B. contributed reagents/data analysis, wrote and edited manuscript. X.O.B. developed concepts/approaches, analyzed data and edited the manuscript. K.A.C. and G.A.C. developed concepts/approaches, analyzed data, wrote and edited the manuscript.
SUPPLEMENTARY MATERIAL
FUNDING
All C. elegans mutant strains came from the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources. This work was supported by grants from the Bachmann-Strauss Dystonia and Parkinson Foundation (X.O.B., K.A.C. and G.A.C.), NINDS NS037409 (X.O.B.), the Jack Fasciana Fund for Support of Dystonia Research (X.O.B.) and a sponsored research agreement from QRxPharma, Ltd. (K.A.C. and G.A.C.). Thanks to Dr Mark Ricketts and Modern Movers, Inc. for generous scholarship support and donation of equipment funds, respectively. S.A.F. was supported by a scholarship from the Parkinson Association of Alabama. J.C.R. was an Alabama Dystonia Scholar supported by members and friends of the Alabama Dystonia Support Group of Birmingham.
ACKNOWLEDGEMENTS
We would like to acknowledge the outstanding collegiality of all members of the Caldwell Laboratory and the participants in joint UA-UAB Dystonia Research Group meetings for their insights. Special thanks to David Ron for the hsp-4::GFP strain, as well as Phyllis Hanson, Laurie Ozelius, Michal Zolkiewski and Ben Cravatt for torsinA-encoding cDNA clones. We thank Yuqing Li for providing the colony of heterozygous torsinA knock-out mice used to generate MEF cultures.
Conflict of Interest statement. G.A.C. and K.A.C. are scientific advisors to QRxPharma, Ltd., from whom they receive compensation and manage a sponsored research agreement for The University of Alabama.
REFERENCES
- 1.Dobson C.M. Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. doi:10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Ellis R.J., Minton A.P. Protein aggregation in crowded environments. Biol. Chem. 2006;387:485–497. doi: 10.1515/BC.2006.064. doi:10.1515/BC.2006.064. [DOI] [PubMed] [Google Scholar]
- 3.Rao R.V., Bredesen D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell. Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. doi:10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozelius L.J., Hewett J.W., Page C.E., Bressman S.B., Kramer P.L., Shalish C., de Leon D., Brin M.F., Raymond D., Corey D.P., et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. doi:10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 5.Neuwald A.F., Aravind L., Spouge J.L., Koonin E.V. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 6.Hanson P.I., Whiteheart S.W. AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell. Biol. 2005;6:519–529. doi: 10.1038/nrm1684. doi:10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 7.Ozelius L.J., Page C.E., Klein C., Hewett J.W., Mineta M., Leung J., Shalish C., Bressman S.B., de Leon D., Brin M.F., et al. The TOR1A (DYT1) gene family and its role in early onset torsion dystonia. Genomics. 1999;62:377–384. doi: 10.1006/geno.1999.6039. doi:10.1006/geno.1999.6039. [DOI] [PubMed] [Google Scholar]
- 8.Kustedjo K., Bracey M.H., Cravatt B.F. TorsinA and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J. Biol. Chem. 2000;275:27933–27939. doi: 10.1074/jbc.M910025199. [DOI] [PubMed] [Google Scholar]
- 9.Hewett J.W., Tannous B., Niland B.P., Nery F.C., Zeng J., Li Y., Breakefield X.O. Mutant torsinA interferes with protein processing through the secretory pathway in DYT1 dystonia cells. Proc. Natl Acad. Sci. USA. 2007;104:7271–7276. doi: 10.1073/pnas.0701185104. doi:10.1073/pnas.0701185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hewett J.W., Nery F.C., Niland B., Ge P., Tan P., Hadwiger P., Tannous B.A., Sah D.W., Breakefield X.O. siRNA knock-down of mutant torsinA restores processing through secretory pathway in DYT1 dystonia cells. Hum. Mol. Genet. 2008;17:1436–1445. doi: 10.1093/hmg/ddn032. doi:10.1093/hmg/ddn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Callan A.C., Bunning S., Jones O.T., High S., Swanton E. Biosynthesis of the dystonia-associated AAA+ ATPase torsinA at the endoplasmic reticulum. Biochem. J. 2007;401:607–612. doi: 10.1042/BJ20061313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Augood S.J., Penney J.B., Jr., Friberg I.K., Breakefield X.O., Young A.B., Ozelius L.J., Standaert D.G. Expression of the early-onset torsion dystonia gene (DYT1) in human brain. Ann. Neurol. 1998;43:669–673. doi: 10.1002/ana.410430518. doi:10.1002/ana.410430518. [DOI] [PubMed] [Google Scholar]
- 13.Shashidharan P., Kramer B.C., Walker R.H., Olanow C.W., Brin M.F. Immunohistochemical localization and distribution of torsinA in normal human and rat brain. Brain Res. 2000;853:197–206. doi: 10.1016/s0006-8993(99)02232-5. doi:10.1016/S0006-8993(99)02232-5. [DOI] [PubMed] [Google Scholar]
- 14.Rostasy K., Augood S.J., Hewett J.W., Leung J.C., Sasaki H., Ozelius L.J., Ramesh V., Standaert D.G., Breakefield X.O., Hedreen J.C. TorsinA protein and neuropathology in early onset generalized dystonia with GAG deletion. Neurobiol. Dis. 2003;12:11–24. doi: 10.1016/s0969-9961(02)00010-4. doi:10.1016/S0969-9961(02)00010-4. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Alegre P., Paulson H.L. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J. Neurosci. 2004;24:593–601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodchild R.E., Dauer W.T. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc. Natl Acad. Sci. USA. 2004;101:847–852. doi: 10.1073/pnas.0304375101. doi:10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naismith T.V., Heuser J.E., Breakefield X.O., Hanson P.I. TorsinA in the nuclear envelope. Proc. Natl Acad. Sci. USA. 2004;101:7612–7617. doi: 10.1073/pnas.0308760101. doi:10.1073/pnas.0308760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLean P.J., Kawamata H., Shariff S., Hewett J., Sharma N., Ueda K., Breakefield X.O., Hyman B.T. TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J. Neurochem. 2002;83:846–854. doi: 10.1046/j.1471-4159.2002.01190.x. doi:10.1046/j.1471-4159.2002.01190.x. [DOI] [PubMed] [Google Scholar]
- 19.Caldwell G.A., Cao S., Sexton E.G., Gelwix C.C., Bevel J.P., Caldwell K.A. Suppression of polyglutamine-induced protein aggregation in Caenorhabditis elegans by torsin proteins. Hum. Mol. Genet. 2003;12:307–319. doi: 10.1093/hmg/ddg027. doi:10.1093/hmg/ddg027. [DOI] [PubMed] [Google Scholar]
- 20.Hamamichi S., Rivas R.N., Knight A.L., Cao S., Caldwell K.A., Caldwell G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson's disease model. Proc. Natl Acad. Sci. USA. 2008;105:728–733. doi: 10.1073/pnas.0711018105. doi:10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breakefield X.O., Blood A.J., Li Y., Hallett M., Hanson P.I., Standaert D.G. The pathophysiological basis of dystonias. Nat. Rev. Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. doi:10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- 22.Travers K.J., Patil C.K., Wodicka L., Lockhart D.J., Weissman J.S., Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. doi:10.1016/S0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 23.Shen X., Ellis R.E., Sakaki K., Kaufman R.J. Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet. 2005;1:e37. doi: 10.1371/journal.pgen.0010037. doi:10.1371/journal.pgen.0010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin J.H., Walter P., Yen T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. doi:10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calfon M., Zeng H., Urano F., Till J.H., Hubbard S.R., Harding H.P., Clark S.G., Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. doi:10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 26.Cao S., Gelwix C.C., Caldwell K.A., Caldwell G.A. Torsin-mediated neuroprotection from cellular stresses to dopaminergic neurons of C. elegans. J. Neurosci. 2005;25:3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005. doi:10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen X., Ellis R.E., Lee K., Liu C.-Y., Yang K., Solomon A., Kurnit D.M., Mori K., Kaufman R.J. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107:893–903. doi: 10.1016/s0092-8674(01)00612-2. doi:10.1016/S0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- 28.Bischof L.J., Kao C.-Y., Los F.C.O., Gonzalez M.R., Shen Z., Briggs S.P., van der Goot F.G., Aroian R.V. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008;4:e1000176. doi: 10.1371/journal.ppat.1000176. doi:10.1371/journal.ppat1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burdette A.J., Churchill P.F., Caldwell G.A., Caldwell K.A. The early-onset torsion dystonia-associated protein, torsinA, displays molecular chaperone activity in vitro. Cell Stress Chaperones. 2010 doi: 10.1007/s12192-010-0173-2. Epub ahead of print (19 Feb 2010), doi:10.1007/s12192-010-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z., Zolkiewska A., Zolkiewski M. Characterization of human torsinA and its dystonia-associated mutant form. Biochem. J. 2003;374:117–122. doi: 10.1042/BJ20030258. doi:10.1042/BJ20030258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torres G.E., Sweeney A.L., Beaulieu J.M., Shashidharan P., Caron M.G. Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc. Natl Acad. Sci. USA. 2004;101:15650–15655. doi: 10.1073/pnas.0308088101. doi:10.1073/pnas.0308088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Risch N.J., Bressman S.B., Senthil G., Ozelius L.J. Intragenic cis and trans modification of genetic susceptibility in DYT1 torsion dystonia. Am. J. Hum. Genet. 2007;80:1188–1193. doi: 10.1086/518427. doi:10.1086/518427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kock N., Naismith T.V., Boston H.E., Ozelius L.J., Corey D.P., Breakefield X.O., Hanson P.I. Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum. Mol. Genet. 2006;15:1355–1364. doi: 10.1093/hmg/ddl055. doi:10.1093/hmg/ddl055. [DOI] [PubMed] [Google Scholar]
- 34.Kojima E., Takeuchi A., Haneda M., Yagi A., Hasegawa T., Yamaki K., Takeda K., Akira S., Shimokata K., Isobe K. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J. 2003;11:1573–1575. doi: 10.1096/fj.02-1184fje. [DOI] [PubMed] [Google Scholar]
- 35.Hewett J., Ziefer P., Bergeron D., Naismith T., Boston H., Slater D., Wilbur J., Schuback D., Kamm C., Smith N., et al. TorsinA in PC12 cells: localization in the endoplasmic reticulum and response to stress. J. Neurosci. Res. 2003;72:158–168. doi: 10.1002/jnr.10567. doi:10.1002/jnr.10567. [DOI] [PubMed] [Google Scholar]
- 36.Kuner R., Teismann P., Trutzel A., Naim J., Richter A., Schmidt N., Bach A., Ferger B., Schneider A. TorsinA, the gene linked to early-onset dystonia, is upregulated by the dopaminergic toxin MPTP in mice. Neurosci. Lett. 2004;355:126–130. doi: 10.1016/j.neulet.2003.10.069. doi:10.1016/j.neulet.2003.10.069. [DOI] [PubMed] [Google Scholar]
- 37.Shashidharan P., Paris N., Sandu D., Karthikeyanm L., McNaught K.S., Walker R.H., Olanow C.W. Overexpression of torsinA in PC12 cells protects against toxicity. J. Neurochem. 2004;88:1019–1025. doi: 10.1046/j.1471-4159.2003.02233.x. [DOI] [PubMed] [Google Scholar]
- 38.Bragg D.C., Camp S.M., Kaufman C.A., Wilbur J.D., Boston H., Schuback D.E., Hanson P.I., Sena-Esteves M., Breakefield X.O. Perinuclear biogenesis of mutant torsin-A inclusions in cultured cells infected with tetracycline-regulated herpes simplex virus type 1 amplicon vectors. Neuroscience. 2004;125:651–661. doi: 10.1016/j.neuroscience.2004.01.053. doi:10.1016/j.neuroscience.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 39.Vander Heyden A.B., Naismith T.V., Snapp E.L., Hodzic D., Hanson P.I. LULL1 retargets torsinA to the nuclear envelope revealing an activity that is impaired by the DYT1 dystonia mutation. Mol. Biol. Cell. 2009;20:2661–2672. doi: 10.1091/mbc.E09-01-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giles L.M., Chen J., Li L., Chin S. Dystonia-associated mutations cause premature degradation of torsinA protein and cell-type specific mislocalization to the nuclear envelope. Hum. Mol. Genet. 2008;17:2712–2722. doi: 10.1093/hmg/ddn173. doi:10.1093/hmg/ddn173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gordon K.L., Gonzalez-Alegre P. Consequences of the DYT1 mutation on torsinA oligomerization and degradation. Neuroscience. 2008;157:588–595. doi: 10.1016/j.neuroscience.2008.09.028. doi:10.1016/j.neuroscience.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grundmann K., Reischmann B., Vanhoutte G., Hubner J., Teismann P., Hauser T.K., Bonin M., Wilbertz J., Horn S., Nguyen H.P., et al. Overexpression of human wild-type torsinA and human DeltaGAG torsinA in a transgenic mouse model causes phenotypic abnormalities. Neurobiol. Dis. 2007;27:190–206. doi: 10.1016/j.nbd.2007.04.015. doi:10.1016/j.nbd.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Esapa C.T., Waite A., Locke M., Benson M.A., Kraus M., McIlhinney R.A., Sillitoe R.V., Beesley P.W., Blake D.J. SGCE missense mutations that cause myoclonus-dystonia syndrome impair episolon-sarcoglycan trafficking to the plasma membrane: modulation by ubiquitination and torsinA. Hum. Mol. Genet. 2007;16:327–342. doi: 10.1093/hmg/ddl472. doi:10.1093/hmg/ddl472. [DOI] [PubMed] [Google Scholar]
- 44.Zimprich A., Grabowski M., Asmus F., Naumann M., Berg D., Bertram M., Scheidtmann K., Kern P., Winkelmann J., Müller-Myhsok B., et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat. Genet. 2001;29:66–69. doi: 10.1038/ng709. doi:10.1038/ng709. [DOI] [PubMed] [Google Scholar]
- 45.Morimoto R.I., Cuervo A.M. Protein homeostasis and aging: taking care of proteins from the cradle to the grave. J. Gerontol. A. Biol. Sci. Med. Sci. 2009;64A:167–170. doi: 10.1093/gerona/gln071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rane N.S., Kang S.W., Chakrabarti O., Feigenbaum L., Hegde R.S. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev. Cell. 2008;15:359–370. doi: 10.1016/j.devcel.2008.06.015. doi:10.1016/j.devcel.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muraro N.I., Moffat K.G. Down-regulation of torp4a, encoding the Drosophila homologue of torsinA, results in increased neuronal degeneration. J. Neurobiol. 2006;12:1338–1353. doi: 10.1002/neu.20313. [DOI] [PubMed] [Google Scholar]
- 48.Kang S.W., Rane N.S., Kim S.J., Garrison J.L., Taunton J., Hegde R.S. Substrate-specific translocation attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127:999–1013. doi: 10.1016/j.cell.2006.10.032. doi:10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oberlin S.R., Konakova M., Pulst S., Chesselet M.F. Development and anatomic localization of torsinA. Adv. Neurol. 2004;94:61–65. [PubMed] [Google Scholar]
- 50.Xiao J., Gong S., Zhao Y., LeDoux M.S. Developmental expression of rat torsinA transcript and protein. Brain Res. Dev. Brain Res. 2004;152:47–60. doi: 10.1016/j.devbrainres.2004.05.012. doi:10.1016/j.devbrainres.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 51.Vasudevan A., Breakefield X.O., Bhide B. Developmental patterns of torsinA and torsinB expression. Brain Res. 2006;1073–1074:139–145. doi: 10.1016/j.brainres.2005.12.087. doi:10.1016/j.brainres.2005.12.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balch W.E., Morimoto R.I., Dillin A., Kelly J.W. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. doi:10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 53.Morimoto R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. doi:10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saxena S., Cabuy E., Caroni P. A role for motorneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. doi:10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 55.Cao S., Hewett J.S., Fumiaki Y., Lu J., Clark Buckley A., Burdette A.J., Chen P., Nery F.C., Li Y., Breakefield X.O., et al. Chemical enhancement of torsinA function in animal models of torsion dystonia. Dis. Model. Mech. 2010;3:386–396. doi: 10.1242/dmm.003715. doi:10.1242/dmm.003715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aamodt E.J., Chung M.A., McGhee J.D. Spatial control of gut-specific gene expression during Caenorhabditis elegans development. Science. 1991;252:579–582. doi: 10.1126/science.2020855. doi:10.1126/science.2020855. [DOI] [PubMed] [Google Scholar]
- 57.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mello C.C., Kramer J.M., Stinchcomb D., Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Locke C.J., Williams S.N., Schwarz E.M., Caldwell G.A., Caldwell K.A. Genetic interactions among cortical malformation genes that influence susceptibility to convulsions in C. elegans. Brain Res. 2006;1120:23–34. doi: 10.1016/j.brainres.2006.08.067. doi:10.1016/j.brainres.2006.08.067. [DOI] [PubMed] [Google Scholar]
- 60.Dang M.T., Yokoi F., McNaught K.S., Jangelley T.A., Jackson T., Li J., Li Y. Generation and characterization of Dyt1 deltaGAG knock-in mouse as a model for early-onset dystonia. Exp. Neurol. 2005;196:452–463. doi: 10.1016/j.expneurol.2005.08.025. doi:10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 61.Hewett J.W., Zeng J., Niland B.P., Bragg D.C., Breakefield X.O. Dystonia-causing mutant torsinA inhibits cell adhesion and neurite extension through interference with cytoskeletal dynamics. Neurobiol. Dis. 2006;22:98–111. doi: 10.1016/j.nbd.2005.10.012. doi:10.1016/j.nbd.2005.10.012. [DOI] [PubMed] [Google Scholar]