

Abstract
The following article describes a concise synthesis of a collection of 4,5-dihydro-1H-benzo[e][1,4]diazepines fused to a hydantoin ring. Molecular complexity and biological relevance is high and structures are generated in a mere three steps, employing the Ugi reaction to assemble diversity reagents. The protocol represents a novel UDC (Ugi-deprotect-cyclize) strategy employed in the Ugi-5-component CO2 mediated condensation, followed by further cyclization under basic conditions, to afford the fused hydantoin. Mechanistic caveats, dependent on aldehydes of choice will be revealed and a facile oxidation of final products to imidazolidenetriones briefly discussed.
Operationally friendly protocols to produce libraries of novel small molecules of high molecular complexity are in huge demand for the interrogation of biological systems.1 As such, development of new MCRs (multi-component reactions) and functional group modification of MCR products have proven fruitful tools in the quest for new molecular probes and their expedited progression along the drug discovery value chain.2 Such products, of high-iterative-efficiency potential2 have found their way into numerous corporate compound collections and examples exist of hit to clinic campaigns were final drugs resided in the virtual diversity space of the original hit generation library.3 This communication describes the development of a concise three step synthesis of novel tricyclic 4,5-dihydro-1H-benzo[e][1,4]diazepines fused to an hydantoin ring and employs the rarely used 5-component Ugi reaction (U-5-CR), Scheme 1. Essentially, CO2 in MeOH produces carbonic acid and the reaction follows the widely accepted classical Ugi mechanism, even though the condensation product differs in that a urethane is now encapsulated within the final skeleton 1. Prior reports on applications of this reaction are scarce4, although our planned strategy builds on an early report from this laboratory which employed the amidic NH of U-5-CR as an internal nucleophile to afford fully functionalized libraries of hydantoins in a mere two steps.4a
Scheme 1.
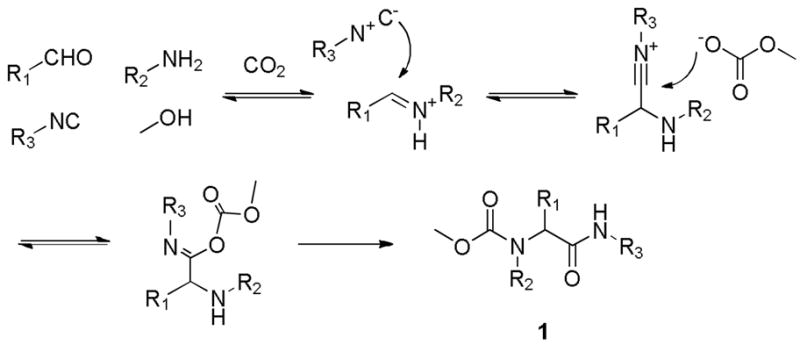
5-component CO2 modified Ugi reaction
A summary of the generic scaffolds 2 and 3 made accessible and introduced in this article is shown in Figure 1. Thus, construction of the desired Ugi precursor 6 is achieved via condensation of ortho-N-Boc benzylamines 4, phenylglyoxaldehydes 5, isonitriles and a saturated solution of CO2 in methanol, Scheme 2. Note that 4 was prepared in 3 steps from commercially available 2-aminobenzylamine as portrayed in Scheme 3 according to the referenced procedure.5 Purified Ugi product is subsequently treated with trifluoroacetic acid promoting amine deprotection and cyclization to the 4,5-dihydro-1H-benzo[e][1,4]diazepine, 7 typically in good yield (> 90%). Note that this transformation extends the repertoire of available chemotypes from UDC (Ugi/DeBoc/Cyclize) methodology and libraries of this benzodiazepine should now be readily accessible. Final ring construction was achieved by treatment of 7 with KOH, thus promoting cyclization and fusion of a hydantoin-like ring whilst simultaneously initiating a 1,3-H shift to give the tricyclic chemotype 8 in good yield. As such, the methodology represents an example of a post-condensation Ugi modification4a that employs two internal nucleophiles in distinct operations, generating a novel scaffold of high complexity in a succint 3 functional operations.
Figure 1.
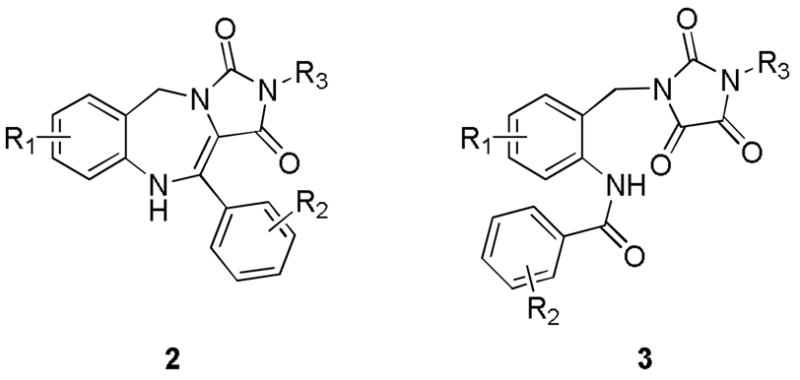
Generic scaffolds of 4,5-dihydro-1H-benzo[e][1,4]diazepines fused to a hydantoin ring 2 and imidazolidenetriones 3
Scheme 2.

Preparation of fused benzodiazepine-hydantoins
Scheme 3.

Synthesis of Boc-2-aminobenzylamine 4
With a satisfactory protocol to the generic structure 8 in place6, a small collection of these molecules were prepared to demonstrate the generality of the reaction sequence, Figure 2. Diversification was based on the commercial availability of different isonitriles and substituted phenylglyoxaldehydes. Reported percent yields represent conversions of the two combined steps from the Ugi product 6 to scaffold 8. In essence, scaffold 7 did not require purification, thus simplifying the production protocol. Unequivocal evidence for the structure of this chemotype was provided by X-ray crystallography for 9.7
Figure 2.

Example Analogs (x% = Ugi yield, x% = yield of 8 from 6)
Interestingly, the tri-cyclic scaffolds 8 underwent a chemical oxidative transformation to the pharmacologically relevant imidazolidinetriones8 3 (Scheme 4) on standing in CDCl3. One particular example 9 showed 75% conversion to its imidazolidinetrione congener after 10 days in CDCl3. Oxidative carbon-carbon double bond cleavage of similar hydantoin derivatives has been previously reported9 and compound 9 was successfully proven to undergo such oxidation upon treatment with KMnO4.10 Encouragingly for future screening efforts the fused hydantoin compounds are stable in DMSO and other solvents, with no oxidation detected over prolonged periods in solution. As supported by a previous study11, this finding exemplifies the phenomenom of air oxidation in chloroform, suggested to be far more facile than in other regularly used solvents. Oxidative rate acceleration of 9 by light suggests a singlet oxygen mechanism may be involved in this process.
Scheme 4.

Aerobic chemical transformations of 8 on standing in CDCl3
Note an exception was found with compound 16, an analog derived from 2-methoxyphenylglyoxaldehyde. Interestingly, a second product was also observed during exposure to CDCl3 (16a:16b = 1:4.5). Tentatively, the following mechanism, Scheme 5, is proposed that involves aromatic substitution with water, tautomerization/rearomatization and oxidation to the imine, 16b. Evidence for this structure was provided by detailed NMR studies.
Scheme 5.

Proposed mechanism involving (i) aromatic substitution, (ii) tautomerization-rearomatization and (iii) oxidation to the imine
In summary, a concise 3-step synthesis of a collection of fused 4,5-dihydro-1H-benzo[e][1,4]diazepines-hydantoins has been successfully developed that utilizes the scarcely employed 5-component CO2 modified Ugi reaction as the diversity generating event followed by two subsequent cyclization transformations. The first transformation occurs under acidic conditions to construct the benzodiazepine ring and is followed by a second cyclization under basic conditions to afford the fused hydantoin. Because of the uniqueness of these scaffolds, the desirable drug-like properties of the molecules generated, and the ease of synthesis, this methodology represents a viable strategy for future enrichment of small molecule compound libraries.
Figure 3.
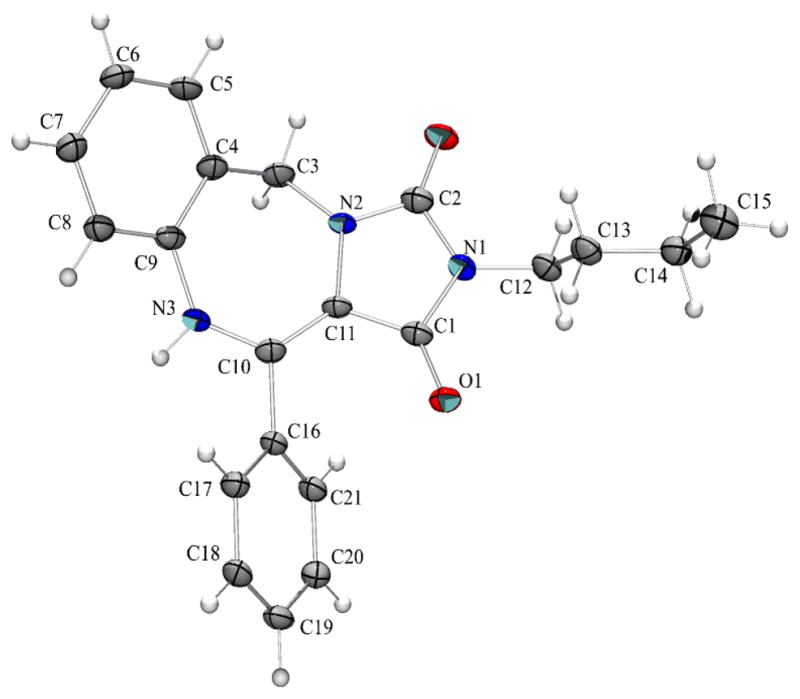
X-ray crystal structure of 9
Acknowledgments
We would like to thank the Office of the Director, NIH and the National Institute of Mental Health for funding (1RC2MH090878-01). Abbott Laboratories are also thanked for a New Faculty Award to CH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Steven Gunawan, The Southwest Comprehensive Center for Drug Discovery and Development, College of Pharmacy, Department of Pharmacology/Toxicology, The University of Arizona, Tucson, AZ 85721.
Gary S. Nichol, Department of Chemistry and Biochemistry, The University of Arizona, Tucson, AZ 85721
Shashi Chappeta, The Southwest Comprehensive Center for Drug Discovery and Development, College of Pharmacy, Department of Pharmacology/Toxicology, The University of Arizona, Tucson, AZ 85721.
Justin Dietrich, The Southwest Comprehensive Center for Drug Discovery and Development, College of Pharmacy, Department of Pharmacology/Toxicology, The University of Arizona, Tucson, AZ 85721.
Christopher Hulme, The Southwest Comprehensive Center for Drug Discovery and Development, College of Pharmacy, Department of Pharmacology/Toxicology, The University of Arizona, Tucson, AZ 85721.
References
- 1.(a) Hulme C. Multicomponent Reactions. 2005:311–341. [Google Scholar]; (b) Bienayme H, Hulme C, Oddon G, Schmitt P. Chem Eur J. 2000;6(10):3321. doi: 10.1002/1521-3765(20000915)6:18<3321::aid-chem3321>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dolle RE, La Bourdonnec B, Goodman AJ, Morales GA, Thomas CJ, Zhang W. J Comb Chem. 2009;11:739–790. doi: 10.1021/cc9000828. [DOI] [PubMed] [Google Scholar]; (b) Dőmling A. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; (c) Hulme C, Gore V. Curr Med Chem. 2003;10:51–80. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]; (d) Hulme C, Lee YS. Mol Div. 2008;12:1–15. doi: 10.1007/s11030-008-9072-1. [DOI] [PubMed] [Google Scholar]; (e) Hulme C, Nixey T. Curr Opin Drug Discovery Dev. 2003;6:921–929. [PubMed] [Google Scholar]
- 3.(a) Hulme C, Peng J, Tang SY, Burns CJ, Morize I, Labaudiniere R. J Org Chem. 1998;63:8021–8023. [Google Scholar]; (b) Nixey T, Hulme C. Tetrahedron Lett. 2002;43:6833–6835. [Google Scholar]; (c) Habashita H, Kokubo M, Hamano SI, Hamanaka N, Toda M, Shibayama S, Tada H, Sagawa K, Fukushima D, Maeda K, Mitsuya H. J Med Chem. 2006;49:4140–4152. doi: 10.1021/jm060051s. [DOI] [PubMed] [Google Scholar]; (d) Nishizawa R, Nishiyama T, Hisaichi K, Matsunaga N, Minamoto C, Habashita H, Takaoka Y, Toda M, Shibayama S, Tada H, Sagawa K, Fukushima D, Maeda K, Mitsuya H. Bioorg Med Chem Lett. 2007;17:727–731. doi: 10.1016/j.bmcl.2006.10.084. [DOI] [PubMed] [Google Scholar]
- 4.(a) Hulme C, Ma L, Romano J, Morton G, Tang S, Cherrier M, Choi S, Salvino J, Labaudiniere R. Tetrahedron Lett. 2000;41:1889–1893. [Google Scholar]; (b) Keating TA, Armstrong RW. J Org Chem. 1998;63:867–871. doi: 10.1021/jo971463z. [DOI] [PubMed] [Google Scholar]
- 5.For the preparation of 4: 2-aminobenzyl-Cbz-amine (4a): To a solution of 2-aminobenzylamine (5.70 g, 46.7 mmol) in anhydrous dichloromethane (150 ml) was added DIPEA (16.30 ml, 93.0 mmol). Next, benzyl chloroformate (6.66 ml, 46.7 mmol) in anhydrous dichloromethane (45 ml) was added drop-wise via syringe. The reaction proceeded for 2 h. Then, reaction mixture was poured into a separatory funnel and washed with brine solution (3 x 80 ml). Organic layer was dried over MgSO4 and concentrated in vacuo to give yellowish white solid which was subsequently purified by Teledyne Isco CombiFlash Rf (Hexane/EtOAc 5–50%) to afford 4a (10.82g, 42.2 mmol, 90%) of yellowish white solid product. 1H NMR (300 MHz, CDCl3) δ 7.47 – 7.30 (m, 5H), 7.13 (td, J = 7.7, 1.6 Hz, 1H), 7.06 (dd, J = 7.4, 1.3 Hz, 1H), 6.65 – 6.75 (m, 2H), 5.15 (s, 2H), 5.08 (s, 1H), 4.33 (d, J = 6.2 Hz, 2H), 4.05 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 157.34, 145.75, 136.74, 130.68, 129.63, 128.94, 128.59, 128.47, 122.53, 118.48, 116.33, 67.44, 42.92. [M+H]+ = 257. 2-Boc-aminobenzyl-Cbz-amine (4b): To a solution of 4a (10.74 g, 41.9 mmol) and DIPEA (14.64 ml, 84 mmol) in anhydrous dichloromethane (100 ml) was added Boc2O (10.97 g, 50.3 mmol). Reaction was then refluxed for 3 days. Reaction was then concentrated in vacuo and toluene was added to pull out t-BuOH giving 19.76 g light yellowish white solid which was then purified by Teledyne Isco CombiFlash Rf (Hexane/EtOAc 5–50%) to afford 4b (14.30 g, 40.1 mmol, 96%) of yellowish white solid product. 1H NMR (300 MHz, CDCl3) δ 7.86 (s, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.34 – 7.40 (d, J = 2.6 Hz, 5H), 7.31 (dd, J = 7.8, 1.7 Hz, 1H), 7.21 (dd, J = 7.5, 1.3 Hz, 1H), 7.06 (t, J = 7.4 Hz, 1H), 5.33 (s, 1H), 5.15 (s, 2H), 4.33 (d, J = 6.4 Hz, 2H), 1.55 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 157.58, 154.23, 137.14, 136.58, 130.47, 129.20, 128.95, 128.65, 128.56, 124.40, 123.22, 80.66, 67.60, 42.35, 28.77. [M+Na]+ = 379. 2-Boc-aminobenzylamine (4): To a solution of 4b (14.2 g, 39.8 mmol) in methanol (50 ml) was added 0.5 g Pd/C (10%). H2 (g) was flown into glass reactor at 40 psi. The reaction was then allowed to run at room temperature over night. Pd/C was removed using celite then solution was collected using vacuum filtration to yield 4 (8.80 g, 39.6 mmol, 99%) of yellow sticky product. 1H NMR (300 MHz, CDCl3) δ 9.48 (s, 1H), 7.99 (d, J = 8.1 Hz, 1H), 7.27 (td, J = 7.7, 1.6 Hz, 1H), 7.11 (dd, J = 7.4, 1.4 Hz, 1H), 6.96 (td, J = 7.4, 1.1 Hz, 1H), 3.97 (s, 2H), 1.74 (s, 2H), 1.54 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 153.86, 139.46, 129.37, 129.00, 128.66, 122.73, 120.72, 80.17, 45.91, 28.83. [M+H]+ = 223.
- 6.For the standard preparation of generic structures 8: CO2 gas was bubbled through a stirring solution of MeOH for 25 minutes to generate methyl carbonic acid. In a separate 25 ml flask, phenyl glyoxal 5 (226 mg, 1.687 mmol) was added to boc-2-aminobenyzlamine 4 (250 mg 1.125 mmol). Methyl carbonic acid (10 ml) and N-butyl isonitrile (0.237 ml, 2.252 mmol) were then added to the latter flask. The reaction was stirred at room temperature under an atmosphere of CO2 for 16 h. The solvent was evaporated in vacuo and crude product purified with a Biotage Isolera4TM system (hexane/EtOAc 10–30%) to afford the Ugi product (218 mg, 0.438 mmol, 39%) as a yellow oil. Ugi product (135 mg, 0.270 mmol) was treated with a 5 ml 10% TFA solution in 1,2-dichloroethane which was irradiated in a Biotage InitiatorTM at 80°C for 20 min. The resulting orange solution was washed with 1M NaHCO3 (4 x 2.5 ml) and the organic layer dried (Na2SO4), filtered and evaporated in vacuo. MeOH (1.50 ml), THF (0.75 ml), H2O (0.50 ml) were added to the crude product (102 mg, 0.269 mmol) followed by a 1 g/1 ml solution of KOH in H2O (0.03 ml). The solution was irradiated at 100°C for 20 min and resultant orange solution partitioned between EtOAc (5 ml) and 1M NaHCO3 (5 ml). The organic layer was dried (Na2SO4), filtered and evaporated in vacuo. Final crude product was purified with a Biotage Isolera4TM (hexane/EtOAc 30%) to afford the final product 9 (74 mg, 0.214 mmol, 79%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 7.59 – 7.44 (m, 5H), 7.35 (d, J = 7.5 Hz, 1H), 7.32 – 7.25 (td, J = 8.1, 1.3 Hz, 1H), 7.06 (t, J = 7.2 Hz, 1H),6.89 (d, J = 7.8 Hz, 1H), 5.95 (s, 1H), 4.98 (s, 2H), 3.49 (t, J = 7.4 Hz, 2H), 1.56 (m, 2H), 1.39 – 1.17 (m, 2H), 0.88 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 161.85, 153.72, 142.48, 135.43, 134.61, 130.66, 130.55, 129.71, 129.59, 129.11, 126.44, 123.92, 120.73, 109.36, 45.74, 39.01, 30.71, 20.47, 14.06. MS FT-ICR calculated for C21H22N3O2 [M+H]+: 348.1707, found: 348.1707.
- 7.Nichol GS, Gunawan S, Dietrich J, Hulme C. Acta Cryst. 2010;E66:o625. doi: 10.1107/S1600536810005477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Kerwin R. Ex Opin Investig Drugs. 2006;15:1487–1495. doi: 10.1517/13543784.15.12.1487. [DOI] [PubMed] [Google Scholar]; (b) Ishii A, Yamakawa M, Toyomaki Y. 4 985 453. US Patent. 1991
- 9.Kuninobu Y, Kikuchi K, Takai K. Chem Lett. 2008;37:740–741. [Google Scholar]
- 10.A thirty mmol batch of the reagent was prepared by combining KMnO4 (3 mmol, 0.474 g, finely ground) and alumina (Al2O3, neutral, 575 mg) in a mortar and pestle and grinding together until a free, homogeneous, purple powder is obtained (5–10 minutes). 9 (52 mg, 0.15 mmol) was dissolved in acetone and the KMnO4/Al2O3 powder (66.50 mg supported reagent, 0.20 mmol KMnO4) was added. After 15 min of vigorous stirring, the reaction mixture was filtered and the acetone filtrate condensed. The residue was taken up into Hexane/EtOAc 30%, washed with 0.1 M HCl, dried over MgSO4, and the solvent was evaporated in vacuo to give the crude material which was then purified by Biotage Isolera Four (Hexane/EtOAc 10–30%) to afford the oxidized product (21 mg, 0.055 mmol, 27%) of white solid.
- 11.Sideris TD, Ioannou PV. Phosphorus, Sulfur, and Silicon. 2006;181:751–762. [Google Scholar]