

Abstract
The karlotoxins (KmTxs) are a family of compounds produced by the dinoflagellate Karlodinium veneficum that cause membrane permeabilization. The structure of KmTx 1, determined using extensive 2D NMR spectroscopy, is very similar to the amphidinols and related compounds, though KmTx 1 features unique structural modifications of the conserved core region. The structure of KmTx 1 differs from that reported for KmTx 2, the only other reported karlotoxin to date, in lacking chlorination at its terminal alkene and possessing a hydrophobic arm that is two carbons longer.
Keywords: karlotoxin, hemolytic toxin, dinoflagellate, amphidinol
Karlotoxins (KmTxs) are a group of potent amphipathic ichthyotoxins produced by the dinoflagellate Karlodinium veneficum. This organism, first described as Gymnodinium galatheanum collected from Walvis Bay, Namibia in 1950 during the second famed Danish “Galathea” expedition1 and as Gymnodinium veneficum collected from the English Channel in the same year,2 has been associated with fish kills world-wide ever since.3-6 The taxonomic identity of the organism has changed several times in recent years with synonyms now including Gymnodinium/Gyrodinium galatheanum, Gymnodinium micrum, Gymnodinium veneficum, and Karlodinium micrum.7,8 Several hemolytic, cytotoxic, and ichthyotoxic compounds were first described from then K. micrum following an investigation of a large mortality event at HyRock fish farm, Maryland, USA in 1996.9 Karlotoxins appear to function by non-specifically increasing the ionic permeability of biological membranes resulting in osmotic cell lysis.10 They kill fish through damage to sensitive gill epithelial tissues.11 The physiological effects of the karlotoxins suggest similarities with the amphidinols, a series of amphipathic linear polyketides isolated from various species of the dinoflagellate Amphidinium (Figure 1). The amphidinols have been shown to adopt a hairpin-like conformation when associated with micelles and to form pores in membranes that promote cellular lysis.12 The improved lytic activity of several amphidinols in the presence of even very low levels of unsaturated sterols in lipid bilayers has been proposed to suggest direct interactions between amphidinols and sterols.13 The allelopathic, cytotoxic, and anti-fungal properties of karlotoxins are likewise believed to be due to differential membrane specificity determined by membrane sterol composition, which also appears to be responsible for the apparent immunity of K. veneficum to the membrane-disrupting properties of its own toxins.14-18 KmTx 2 (2), found in isolates and fish kill waters from outside of the Chesapeake Bay watershed, Maryland, USA was recently reported to be a linear polyketide with a molecular weight of 1344.8 Da.19,20 We report here for the first time the planar structure of KmTx 1 (1), isolated from a K. veneficum culture originating from the inland bays of Delaware, USA. This compound is characterized by a close structural similarity to the amphidinols.
Figure 1.

Structures of the karlotoxins and amphidinol 3. KmTx 1 (1), KmTx 2 (2), and amphidinol 3 (3).
The strain of Karlodinium veneficum investigated in these studies (CCMP 2936) was originally isolated from the Delaware Inland Bays, USA, and provided as a gift from Kathy Coyne (U. Delaware, Lewes, DE). Cultures were grown using an f/2-Si based medium.21 Culture filtrate was subjected to solid phase extraction and desalting on octadecyl bonded phase silica and the organic material recovered by elution with methanol and 1:1 acetone/methanol. The extract (125 mg) was subjected to reversed phase flash chromatography (1.2 × 19 cm) using stepwise elution with 20 mL each of 30, 45, 60, and 80% aqueous methanol followed by 60 mL of methanol. Fractions derived from the 80% and 100% methanol elutions were combined (10.2 mg) and subjected to purification by HPLC using isocratic elution with 38% acetonitrile in water at 0.8 mL/min to give pure karlotoxin 1 (1) as a colorless amorphous solid (1.0 mg); [α]D22 +9° (c 0.1, MeOH); UV λmax (MeOH) 228 nm (ε 23,000); IR (KBr film) νmax 3354, 1066 cm−1.
The positive ion ESIMS of 1 exhibited an [M+H]+ molecular ion (m/z 1339), an [M+Na]+ ion (m/z 1361), and a series of losses of water from the molecular ion (1321, 1303). In addition, the spectra exhibited a complex and characteristic series of apparently doubly charged ions and fragments, many resulting from consecutive losses of water. A molecular formula of C69H126O24 was determined from HRESIMS analysis (m/z 1339.8682 [M+H]+; Calcd. 1339.8717; Δ −2.6 ppm). The combined mass spectral data suggested a highly oxygenated molecule with multiple hydroxyl groups. A total of seven double bond equivalents are expected from the molecular formula.
Initial 500 MHz NMR spectra showed considerable overlap for several 1H and 13C resonances. A complete 2D NMR data set was subsequently recorded at 800 MHz to resolve resonances and derive the structure and NMR assignments for 1 (Table 1). A multiplicity-edited HSQC spectrum of 1 revealed sixty-two resolved and six overlapped protonated carbon cross peaks. These arise from the following types of carbons: seven sp2 methines, two sp2 methylenes, twenty-five oxygenated sp3 methines, one oxygenated sp3 methylene, three sp3 methines, twenty-one resolved sp3 methylenes, three methyls, and six overlapped sp3 methylenes near 29-31 ppm (Table 1). The 29-31 ppm carbon signals correlate to overlapped 1.1-1.2 ppm proton resonances. Integration of these overlapped 1H resonances in the 1D NMR spectrum showed the presence of twelve protons, i.e. six methylene groups. With firm evidence for sixty-eight protonated carbons, only a single carbon remains unaccounted for. This is a quaternary sp2 carbon resonating at 151.7 ppm, whose assignment to C-40 is confirmed by HMBC (Figure 2).
Table 1.
1H and 13C NMR data for KmTx-1 (1) a
| posn. | δ H | (mult, Hz) | δC (mult) |
|---|---|---|---|
| la | 3.59 | (dd, 4.6, 11.0) | 66.8 (t) |
| lb | 3.55 | (dd, 6.3, 11.1) | |
| 2 | 3.73 | m | 72.8 (d) |
| 3a | 2.32 | m | 37.4 (t) |
| 3b | 2.22 | m | |
| 4 | 5.75 | (dt, 15.5, 7.3) | 127.9 (d) |
| 5 | 5.59 | (dd, 15.5, 6.7) | 136.9 (d) |
| 6 | 4.05 | (q, 6.7) | 73.1 (d) |
| 7a | 1.55 | m | 38.0 (t) |
| 7b | 1.51 | m | |
| 8a | 1.60 | m | 22.8 (t) |
| 8b | 1.42 | m | |
| 9 | 1.45 | m | 38.1 (t) |
| 10 | 3.56 | m | 72.0 (d) |
| 11 | 1.41 | m | 38.1 (t) |
| 12a | 1.66 | m | 23.0 (t) |
| 12b | 1.42 | m | |
| 13 | 1.47 | m | 39.8 (t) |
| 14 | 3.68 | m | 70.1 (d) |
| 15a | 1.76 | m | 41.0 (t) |
| 15b | 1.29 | m | |
| 16 | 2.09 | m | 33.3 (d) |
| 17 | 3.19 | (dd, 4.2, 6.1) | 79.6 (d) |
| 18 | 3.68 | m | 72.8 (d) |
| 19a | 1.70 | m | 32.0 (t) |
| 19b | 1.62 | m | |
| 20a | 1.72 | m | 31.4 (t) |
| 20b | 1.45 | m | |
| 21 | 2.02 | m | 34.8 (d) |
| 22 | 3.86 | (dd, 2.3, 9.0) | 73.8 (d) |
| 23 | 3.47 | (dd, 0.9, 9.0) | 73.6 (d) |
| 24 | 4.02 | m | 71.4 (d) |
| 25a | 1.78 | m | 35.4 (t) |
| 25b | 1.58 | m | |
| 26a | 1.53 | m | 24.2 (t) |
| 26b | 1.46 | m | |
| 27a | 1.72 | m | 32.7 (t) |
| 27b | 1.27 | m | |
| 28 | 1.78 | m | 36.4 (d) |
| 29 | 3.12 | (dd, 3.3, 7.2) | 79.9 (d) |
| 30 | 4.03 | m | 68.1 (d) |
| 31a | 1.93 | m | 34.6 (t) |
| 31b | 1.80 | m | |
| 32 | 4.34 | (dt, 10.9,3.1) | 74.6 (d) |
| 33 | 3.88 | (dd, 3.1, 3.1) | 73.0 (d) |
| 34 | 3.91 | (dd, 3.3, 7.8) | 73.0 (d) |
| 35 | 4.11 | (dd, 7.8, 7.8) | 71.2 (d) |
| 36 | 3.57 | m | 77.9 (d) |
| 37 | 4.19 | m | 71.7 (d) |
| 38a | 2.16 | m | 31.8 (t) |
| 38b | 1.82 | m | |
| 39a | 2.63 | (ddd, 5.0, 10.8, 15.8) | 27.4 (t) |
| 39b | 2.26 | (ddd, 6.0, 10.0, 15.8) | |
| 40 | 151.7 (s) | ||
| 41 | 4.39 | (d, 8.7) | 76.7 (d) |
| 42 | 3.51 | (dd, 1.1, 8.8) | 74.9 (d) |
| 43 | 4.21 | (dt, 12.2, 1.8) | 70.7 (d) |
| 44a | 2.32 | m | 31.7 (t) |
| 44b | 1.63 | m | |
| 45 | 4.17 | m | 67.2 (d) |
| 46 | 4.31 | (t, 2.3) | 68.5 (d) |
| 47 | 3.99 | (dd, 1.2, 10.0) | 80.6 (d) |
| 48 | 4.19 | m | 72.5 (d) |
| 49 | 4.61 | (dd, 2.9, 7.4) | 74.1 (d) |
| 50 | 5.74 | (dd, 8.1, 15.6) | 128.6 (d) |
| 51 | 5.81 | (dt, 15.6, 6.8) | 135.1 (d) |
| 52 | 1.94 | m | 33.4 (t) |
| 53 | 1.26 | m | 30.1 (t) |
| 54 | 1.12-1.20 | m | 29.0-31.4 (t) |
| 55 | 1.12-1.20 | m | 29.0-31.4 (t) |
| 56 | 1.12-1.20 | m | 29.0-31.4 (t) |
| 57 | 1.12-1.20 | m | 29.0-31.4 (t) |
| 58 | 1.12-1.20 | m | 29.0-31.4 (t) |
| 59 | 1.12-1.20 | m | 29.0-31.4 (t) |
| 60 | 1.27 | m | 30.0 (t) |
| 61 | 1.98 | m | 33.4 (t) |
| 62 | 5.63 | (dt, 15.0, 7.0) | 136.2 (d) |
| 63 | 6.00 | (dd, 10.5, 15.0) | 132.1 (d) |
| 64 | 6.27 | (ddd, 10.3, 10.5, 17.0) | 138.5 (d) |
| 65a | 5.03 | (dd, 1.3, 17.0) | 114.7 (t) |
| 65b | 4.88 | (dd, 1.3, 10.3) | |
| 66 | 0.98 | (d, 7.0) | 17.8 (q) |
| 67 | 0.97 | (d, 7.0) | 13.5 (q) |
| 68 | 0.92 | (d, 6.7) | 16.8 (q) |
| 69a | 5.10 | s | 112.9 (t) |
| 69b | 5.01 | s |
Recorded in 2:1 CD3OD/pyridine-d5 at 800 MHz for 1H. Referenced to residual CD3OD peak at δH 3.30 ppm and δC 49.0 ppm.
Figure 2.

The two major substructures as determined by TOCSY and COSY NMR for 1 with selected HMBC (solid arrow) and HSQC-TOCSY (dashed arrow) correlations shown.
Comparison of these features with the molecular formula reveals that the molecule contains a minimum of five double bonds, leaving two units of unsaturation to account for. Furthermore, the number of oxygenated carbons (twenty-six) is two more than the number of oxygens (twenty-four), suggesting that the two remaining units of unsaturation must be accounted for by ether rings. Moreover, the analysis above shows the presence of 104 protons attached to carbon, leaving twenty-two protons attached to oxygen atoms, which is again consistent with the molecular formula and the presence of two ether rings.
Structural fragments were assembled through the combined use of COSY, TOCSY, and two-dimensional HSQC-TOCSY spectra. Substructure A-1 (Figure 2) was deduced directly from the COSY and TOCSY data and found to feature the primary alcohol group, two secondary alcohols, and one of the olefinic bonds. The olefinic proton coupling (3J=15.5 Hz) indicated an E configuration (Table 1). Analysis beyond H2-7 in substructure A-1 was hampered by overlap of the NMR signals of H2-8 with those of H2-9. However, HSQC-TOCSY and HMBC correlations from the resolved C-8 to H2-7 and from C-10 to H2-8 and H2-9 in concert with the homonuclear data allowed extension of the spin system to H2-11 in substructure A-2 (Figure 2). Likewise, HSQC-TOCSY correlations from C-12 to H2-11 and H2-13 enabled connection of the spin system through to H-14 (Figure 2). Further support for the presence of the two consecutive 1,5-pentanediol fragments in 1 is provided by comparison of the 1H and 13C chemical shifts observed for C-6 through C-14 with the assignments for similar fragments in structurally related compounds such as amphidinols 3 (3),22 5, 6,23 9, and 10,24 lingshuiol A,25 and luteophanol D.26
Substructure A-3 spans C-13 to C-39 (Figure 2). TOCSY and COSY correlations to a methine signal at δH 2.09 ppm (H-16) indicated adjacency to a methyl group (C-66), a methylene (C-15) and a vic-diol group (C-17 and C-18). The latter group is part of another 1,5-pentanediol fragment that contains a methyl substituent at C-21. The moiety C-27 through C-30 is structurally similar to that at C-15 through C-18, and indeed there are distinct similarities between the chemical shift values for H-15 through H-17 and the corresponding values at H-27 through H-29 (Table 1). For example, both structural moieties contain oxymethine groups that exhibit relatively high-field δH values (3.19 and 3.12 ppm, respectively for H-18 and H-28) and relatively low-field δC values (79.6 and 79.9 ppm for C-18 and C-28, respectively). C-30 is connected by a methylene group to a string of six consecutive oxymethines (C-32 through C-37). TOCSY correlations were crucial in establishing the linkages of H-33 and H-34 as their direct COSY correlation was not resolved from the spectrum diagonal. The C-37 oxymethine is linked via two methylene groups to an olefinic carbon (C-40) bearing an exo-methylene substituent that terminates substructure A-3. Connectivity of substructures A-3 and A-4 through quaternary carbon C-40 was established by HMBC correlations between C-40 and protons H-39b and H-42 (Figure 2). Additionally, close scrutiny of the TOCSY revealed weak correlation peaks between the olefinic methylene protons (H-69a and H-69b) and H2-38, H2-39, H-41, and H-42.
Substructure A-4 (Figure 2) begins with three consecutive oxymethines (C-41 through C-43) bridged by a methylene to a system with five consecutive oxymethines (C-45 through C-49). C-49 is linked to an E-double bond (C-50 and C-51), as determined from 3J values (15.6 Hz; Table 1). The spin system can be further traced to two additional methylenes (C-52 and C-53) until the signals enter a highly congested region of the spectrum at δH 1.12-1.20 ppm, which integrates for twelve protons. Substructure B (Figure 2) contains a terminal conjugated diene system with an E double bond, as indicated by the 3J value (15.0 Hz; Table 1). TOCSY and COSY data showed that this moiety is linked to two methylene groups that correlate with the same group of unresolved signals in the region δH 1.12-1.20 ppm. Since no other signals exhibit COSY correlations with these signals, they were identified as a linear chain of six methylene groups connecting substructures A-4 and B which, as will be discussed below, was consistent with mass spectral fragmentation data.
The combined analytical data had indicated the presence of two ether rings and their locations were determined by comparison of the NMR data of 1 with similar compounds such as the amphidinols and by detailed analysis of the ROESY and NOESY data. The occurrence of an ether ring containing C-43 through 47 was supported by the strong agreement in 1H and 13C chemical shifts for the corresponding region in other amphidinol-like compounds.27-30,24,26,31 The observed NOESY/ROESY data and 3J couplings are consistent with the presence of a tetrahydropyran ring in a chair conformation (substructure C; Figure 3). The multiplet structure of H-43 (δH 4.21, dt, 12.2 Hz, 1.8 Hz) suggested the presence of one strong and two weak coupling interactions, one of the latter occurring with H-42. Comparison of the antiphase active couplings of the DQF-COSY cross peaks H-43/H-44a and H-43/H-44b (Supporting Data, Figure S1) revealed a strong coupling (12 Hz) for the former and a weaker coupling (< 6 Hz) for the latter, suggesting an axial location for H-43.32,33 Comparison of the cross peak between H-45 and H-44a to that of H-43 and H-44a indicated that the 3J value for the former is of a similar magnitude to the latter, suggesting an axial location for H-45 as well (Figure S1). The 3J value between H-45 and H-46 is near 2.2 Hz (Table 1), suggesting an equatorial location for the latter. An axial location for the alkyl side chain at C-47 is suggested by the presence of a NOESY correlation between H-43 and H-49, and provides additional evidence for an ether linkage between C-43 and C-47 (Figure 3). Direct cross peaks between any of the three axial groups (H-43, H-45, and H-48) were not observed due to overlap with the spectrum diagonal. These combined data established that the location and relative stereochemical arrangement of this ring is identical to those of other amphidinol-like compounds.22,29,26,31
Figure 3.
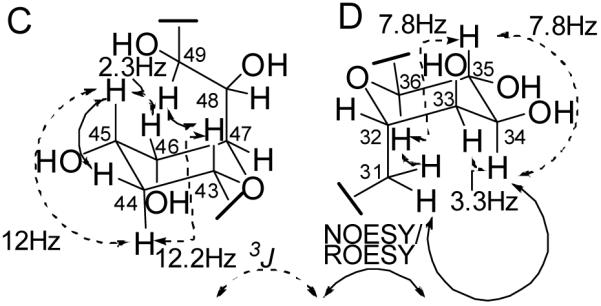
NMR evidence for the relative stereochemical configuration of the two tetrahydropyran rings in 1.
A likely location for the second ether ring was suggested by ROESY correlations between H-36 and both H2-31 protons, indicating that both H-36 and C-31 are located axially on a chair-conformation tetrahydropyran ring (substructure D; Figure 3). ROESY and coupling constant (3JHH) data for positions 31-36 strongly support the local stereochemical arrangement shown for substructure D in Figure 3. Axial locations for H-34, H-35, and H-36 are suggested by the presence of a triplet-like multiplet with 3J=7.8 Hz for H-35,34 while a 3.3 Hz coupling between H-34 and H-33 indicates an equatorial location for the latter (Table 1). Note that this ring configuration, aside from the additional alcohol group at C-35 which is absent from most other amphidinol-like compounds, is identical to that determined for amphidinol 3 (3),22 luteophanols B, C, and D,29,26 and lingshuiol.30 The only other amphidinol-like compounds that contain a hydroxy substituent at C-35 are the karatungiols.31 However, in the karatungiols the orientation of the hydroxyl group at the equivalent position in the pyran ring is axial rather than equatorial. Significantly, the agreement in 3J couplings and the 1H and 13C chemical shifts for this ring between 1 and the karatungiols is poor, particularly when compared to the agreement observed for the other ring described above.
Further support for structure 1 was provided by electrospray tandem MS/MS of the [M+Na]+ ion (m/z 1361; Figure 4). In particular, fragmentation at C-29/C-30 (m/z 675) supported the bulk composition of the polyol arm as assigned. Additionally, the length of the lipophilic arm was confirmed by the complementary fragment ions resulting from scission of C-41/C-42 (m/z 937 and 447, respectively). The charged polyol fragment from the latter fragmentation was much more intense, likely due to the presence of a higher number of sodium coordinating sites.
Figure 4.

Fragment ions observed by tandem MS/MS of [M+Na]+ (m/z 1361). All m/z values correspond to ion fragments containing sodium ions.
The structure for KmTx 1 (1) differs from that of KmTx 2 (2) in that it features a terminal monosubstituted alkene rather than a chloroalkene and that its lipophilic arm contains an additional two methylene groups. In addition, the previously reported molecular formula and mass spectral fragmentation data for hydroxy-KmTx 1-3, a compound of unknown structure, suggests that it is either identical in structure to or isomeric with 1.19 With a few exceptions, the core substructure for positions 28 through 49 is nearly identical for most amphidinol-like compounds. The modifications present in the core structure for KmTx 1 are unique, suggesting that the karlotoxins are a distinct family within amphidinol-like compounds. For example, there is no double bond at C-28-C-29, additional hydroxyl groups at C-29 and C-35, and no hydroxyl group at C-31.
While much of the structural variety found in the amphidinols arises from differences in the polyol arm, there are some similarities between 1 and other amphidinol-like compounds in this region. The portion of the polyol arm of 1 between C-1 and C-14 is identical to desulfoamphidinol 1.35 Interestingly, the karlotoxins are the only amphidinol-like compounds to contain a terminal diene on the lipophilic arm with no corresponding internal triene, though the absence of a conjugated triene in the lipophilic arm of lingshuiol did not lead to a loss of hemolytic potency.36 Increasing the length of the lipophilic arm has been reported to increase hemolytic activity among the amphidinols and related compounds.24,13,12 In this respect, 1 is similar to amphidinols 1, 3 (3), 5, 9, and 13, which all contain the longest known lipophilic arms.
The amphidinols display a variety of biological properties including hemolytic, antifungal and cytotoxic effects, and inline with these observations previous studies with 19 and with 215,11 have demonstrated hemolytic, antifungal, and ichthyotoxic activities for these compounds. Measurements of lysis of human erythrocytes in the presence of varying concentrations of purified 1 showed an EC50 of 63 nM. This high level of potency is comparable to that of 3, the most hemolytic amphidinol, which has been reported in different studies with human erythrocytes to have EC50 values of 9 nM,23 150 nM,12 250 nM,37 and 400 nM.38 The discovery of KmTx 1 adds to the growing list of amphipathic toxins produced by dinoflagellates, suggesting that the production of these compounds is an effective strategy adopted by these organisms to avoid predation and, in the case of K. veneficum, for prey capture.17
Supplementary Material
Acknowledgements
We express gratitude to C. Tomas and A.O. Tatters (UNCW-CMS) for performing hemolytic assays on purified KmTx 1. Funding supporting this research was provided by the NIH (5P41GM076300-01, JLCW; R01 A1057588, AAR), the North Carolina Dept. of Health and Human Services (01505-04, JLCW), NOAA-ECOHAB (MML-106390A, JLCW), NCI (P30-CA-14236, AAR), NOAA Coastal Ocean Program (NA04NOS4780276, University of Maryland Biotechnology Institute), the CDC (U50/CCU 323376, ARP), and the Maryland Department of Health and Mental Hygiene (ARP). This is contribution #08-192 from the Center of Marine Biotechnology and contribution #289 from the ECOHAB program. NMR instrumentation in the Duke NMR Center was funded by the NSF, the NIH, the NC Biotechnology Center and Duke University. We thank Mr. F. Sun (University of Illinois at Urbana-Champaign) for collecting high-resolution MS data. The Q-Tof Ultima mass spectrometer (UIUC) was purchased in part with a grant from the NSF, Division of Biological Infrastructure (DBI-0100085).
Footnotes
Supplementary data
Experimental procedures, full NMR assignments, and annotated 1H NMR, NOESY, ROESY, TOCSY, and HSQC spectra are available online with the paper at ScienceDirect.
References and notes
- 1.Braarud T. Galathea Report. 1957;1:137–138. [Google Scholar]
- 2.Ballantine D. J. Mar. Biol. Assoc. U.K. 1956;35:467–474. [Google Scholar]
- 3.Pieterse F, Van Der Post DC. Adm. So. West Afr. Mar. Res. Lab. Invest. Rep. 1967;14:8–27. [Google Scholar]
- 4.Nielsen MV. Mar. Ecol. Progr. Ser. 1993;95:273–277. [Google Scholar]
- 5.Kempton JW, Lewitus AJ, Deeds JR, Law JM, Place AR. Harmful Algae. 2002;1:233–241. [Google Scholar]
- 6.Goshorn D, Deeds J, Tango P, Poukish C, Place A, McGinty M, Butler W, Luckett C, Magnien R. “Occurrence of Karlodinium micrum and its association with fish kills in Maryland estuaries”; Harmful Algae 2002; Proceedings of the Xth International Conference on Harmful Algae; St. Petersburg, FL. 2004. [Google Scholar]
- 7.Daugbjerg N, Hansen G, Larsen J, Moestrup Ø. Phycologia. 2000;39:302–317. [Google Scholar]
- 8.Bergholtz T, Daugbjerg N, Moestrup Ø , Fernández-Tejedor M. J. Phycol. 2006;42:170–193. [Google Scholar]
- 9.Deeds JR, Terlizzi DE, Adolf JE, Stoecker DK, Place AR. Harmful Algae. 2002;1:169–189. [Google Scholar]
- 10.Deeds JR. Dissertation. University of Maryland; 2003. [Google Scholar]
- 11.Deeds JR, Reimschuessel R, Place AR. J. Aq. Anim. Health. 2006;18:136–148. [Google Scholar]
- 12.Houdai T, Matsuoka S, Morsy N, Matsumori N, Satake M, Murata M. Tetrahedron. 2005;61:2795–2802. [Google Scholar]
- 13.Morsy N, Houdai T, Konoki K, Matsumori N, Oishi T, Murata M. Bioorg. Med. Chem. 2008;16:3084–3090. doi: 10.1016/j.bmc.2007.12.029. [DOI] [PubMed] [Google Scholar]
- 14.Adolf JE, Bachvaroff TR, Krupatkina DN, Nonogaki H, Brown PJP, Lewitus AJ, Harvey HR, Place AR. Afr. J. Mar. Sci. 2006;28:415–419. [Google Scholar]
- 15.Deeds JR, Place AR. Afr. J. Mar. Sci. 2006;28:421–425. [Google Scholar]
- 16.Place AR, Harvey HR, Bai X, Coats DW. Afr. J. Mar. Sci. 2006;28:347–351. [Google Scholar]
- 17.Adolf JE, Krupatkina D, Bachvaroff T, Place AR. Harmful Algae. 2007;6:400–412. [Google Scholar]
- 18.Bai X, Adolf JE, Bachvaroff T, Place AR, Coats DW. Harmful Algae. 2007;6:670–678. [Google Scholar]
- 19.Bachvaroff TR, Adolf JE, Squier AH, Harvey HR, Place AR. Harmful Algae. 2008;7:473–484. [Google Scholar]
- 20.Place AR, Saito K, Deeds JR, Robledo JAF, Vasta GR. In: Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection. Botana LM, editor. Taylor and Francis; Boca Raton, FL, USA: 2008. pp. 717–752. [Google Scholar]
- 21.Guillard RRL. In: Culture of marine invertebrate animals. Smith WL, Chanley MH, editors. Plenum Press; New York, NY: 1975. pp. 26–60. [Google Scholar]
- 22.Murata M, Matsuoka S, Matsumori N, Paul GK, Tachibana K. J. Am. Chem. Soc. 1999;121:870–871. [Google Scholar]
- 23.Paul GK, Matsumori N, Konoki K, Murata M, Tachibana K. J. Mar. Biotechnol. 1997;5:124–128. [Google Scholar]
- 24.Echigoya R, Rhodes L, Oshima Y, Satake M. Harmful Algae. 2005;4:383–389. [Google Scholar]
- 25.Huang X-C, Zhao D, Guo Y-W, Wu H-M, Trivellone E, Cimino G. Tetrahedron Lett. 2004;45:5501–5504. [Google Scholar]
- 26.Kubota T, Takahashi A, Tsuda M, Kobayashi J. Mar. Drugs. 2005;3:113–118. [Google Scholar]
- 27.Paul GK, Matsumori N, Murata M, Tachibana K. Tetrahedron Lett. 1995;36:6279–6282. [Google Scholar]
- 28.Doi Y, Ishibashi M, Nakamichi H, Kosaka T, Ishikawa T, Kobayashi J. J. Org. Chem. 1997;62:3820–3823. [Google Scholar]
- 29.Kubota T, Tsuda M, Doi Y, Takahashi A, Nakamichi H, Ishibashi M, Fukushi E, Kawabata J, Kobayashi J. Tetrahedron. 1998;54:14455–14464. [Google Scholar]
- 30.Huang X-C, Zhao D, Guo Y-W, Wu H-M, Lin L-P, Wang Z-H, Ding J, Lin Y-S. Bioorg. Med. Chem. Lett. 2004;14:3117–3120. doi: 10.1016/j.bmcl.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 31.Washida K, Koyama T, Yamada K, Kita M, Uemura D. Tetrahedron Lett. 2006;47:2521–2525. [Google Scholar]
- 32.Freeman R, McIntyre L. Isr. J. Chem. 1992;32:231–244. [Google Scholar]
- 33.Delaglio F, Wu Z, Bax A. J. Magn. Reson. 2001;149:276–281. doi: 10.1006/jmre.2001.2297. [DOI] [PubMed] [Google Scholar]
- 34.Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. J. Org. Chem. 1999;64:866–876. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- 35.Satake M, Murata M, Yasumoto T, Fujita T, Naoki H. J. Am. Chem. Soc. 1991;113:9859–9861. [Google Scholar]
- 36.Qi X-M, Yu B, Huang X-C, Guo Y-W, Zhai Q, Jin R. Toxicon. 2007;50:278–282. doi: 10.1016/j.toxicon.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Houdai T, Matsuoka S, Matsumori N, Murata M. Biochim. Biophys. Acta, Biomembr. 2004;1667:91–100. doi: 10.1016/j.bbamem.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 38.Morsy N, Houdai T, Matsuoka S, Matsumori N, Adachi S, Oishi T, Murata M, Iwashita T, Fujita T. Bioorg. Med. Chem. 2006;14:6548–6554. doi: 10.1016/j.bmc.2006.06.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.