

Abstract
This patient presented on the first day of life with pronounced lactic acidosis with an elevated lactate/pyruvate ratio. Urine organic acids showed Krebs cycle metabolites and mildly elevated methylmalonate and methylcitrate. The acylcarnitine profile showed elevated propionylcarnitine and succinylcarnitine. Amino acids showed elevated glutamic acid, glutamine, proline, and alanine. From age 2 months on, she had elevated transaminases and intermittent episodes of liver failure. Liver biopsy showed steatosis, and a decrease of mitochondrial DNA to 50% of control. She had bilateral sensorineural hearing loss. Over the course of the first two years of life, she developed a progressively severe myopathy with pronounced muscle weakness eventually leading to respiratory failure, Leigh disease, and recurrent hepatic failure. The hepatic symptoms and the metabolic parameters temporarily improved upon treatment with aspartate, but neither muscle symptoms nor brain lesions improved. Laboratory testing revealed a deficiency of succinyl-CoA ligase enzyme activity and protein in fibroblasts due to a novel homozygous mutation in the SUCLG1 gene: c.40A>T (p.M14L). Functional analysis suggests that this methionine is more likely to function as the translation initiator methionine, explaining the pathogenic nature of the mutation. Succinyl-CoA ligase deficiency due to a SUCLG1 mutation is a new cause for mitochondrial hepatoencephalomyopathy.
Introduction
Maintenance of the mitochondrial integrity requires replication of its own mitochondrial DNA (mtDNA). This requires both an intanct replication process and an available pool of its substrates such as nucleotide triphosphates. Defects in the enzymes associated with mtDNA replication and with maintenance of the nucleotide triphosphate pool are associated with mtDNA depletion syndromes, in which the amount of mtDNA decreases below a clinically necessary threshold (1). One such recently described gene is succinyl-CoA ligase, defects of which have been associated with mtDNA depletion in muscle.
Succinyl-CoA ligase, also called succinate synthase, is an enzyme in the Krebs cycle that converts succinyl-CoA to succinate and free coenzyme A, and converts ADP or guanosine diphosphate (GDP) to ATP or guanosine triphosphate (GTP) respectively (2,3). It is a mitochondrial matrix enzyme composed of two subunits. The substrate specificity for GDP or ADP is determined by the β-subunit, whereas the α-subunit is shared. The α-subunit is coded by the gene SUCLG1, whereas the β-subunit is encoded by SUCLA2 for the subunit with specificity for ADP (enzyme EC 6.2.1.5), and by SUCLG2 for the subunit with specificity for GDP (EC 6.2.1.4) (3,4). The substrate for the enzyme, succinyl-CoA, is made from α-ketoglutarate by α-ketoglutarate dehydrogenase and from methylmalonyl-CoA by methylmalonyl-CoA mutase (Figure 1). SUCLG1 is ubiquitously expressed, but is particularly high in heart, brain, kidney, and liver (3,4). The SUCLA2 protein is mostly present in brain, skeletal muscle, and heart, and the SUCLG2 protein in liver and kidney (3,4). The enzyme is activated by mitochondrial phosphate levels (5) and is associated with the nucleotide diphosphate kinase (6.2.1.4) (6).
Figure 1.
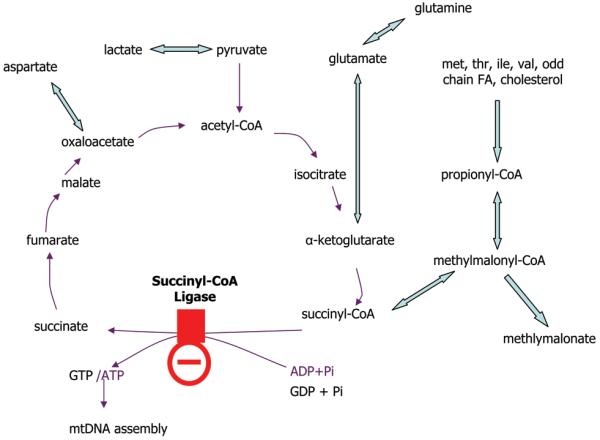
Biochemical pathway
Legend: This biochemical pathway shows the interrelationship of the Krebs cycle and the methylmalonate pathway as it relates to the metabolic block in this condition.
Patients with deficient activity of the succinyl-CoA ligase due to mutations in the SUCLA2 gene present with hypotonia and muscle weakness, Leigh disease, dystonia, sensorineural hearing loss, and sometimes polyneuropathy and renal Fanconi syndrome (7,8,9,10). Biochemical findings are mild lactic acidosis and mild elevations of methylmalonic acid and succinylcarnitine. The disorder is progressive with muscle weakness and mitochondrial DNA depletion in muscle.
Patients with mutations in SUCLG1 were described from the Faroe Islands (11). All patients had severe lactic acidosis, and elevated methylmalonate, methylcitrate, and pyruvate in blood and urine resulting in neonatal death. Another patient was described with failure to thrive, progressive muscle weakness, atrophy, and hypotonia, hearing loss, hypoglycemia (12). He died at age 2 years 9 months. His brain MRI showed lesions in the basal ganglia. He had moderate lactic acidosis, mild elevated methylmalonate, and mild increase in transaminases, but with preserved liver function.
In this paper, we present a patient with succinyl-CoA ligase deficiency due to a mutation in the SUCLG1 gene.
Case report
The patient was born by Cesarean section at 36 weeks gestation with normal Apgars and birth weight of 2.290 kg, length 46.5 cm, and head circumference 33.5 cm. She was the sixth child of unrelated parents of Hispanic ethnicity. Her siblings were unaffected. She presented at 17 hours of life with Kussmaul breathing and severe metabolic acidosis, pH 7.15, with an anion gap of 28 mEq/L and lactic acidosis of 15 mM. Lactic acid decreased with fasting and increased with carbohydrate feeding. During the first months, she had normal tone, reflexes, was fixing and following with her eyes, and did not have organomegaly. She was tachycardic 160 to 180/minute, a symptom that always persisted. The acidosis was treated with bicarbonate then oral citrate, and by carbohydrate restriction.
Laboratory investigations showed normal ammonia, liver function tests, uric acid, vitamin B12 level, and absent urine sulfocysteine. Urine organic acids showed increased levels of lactate, ketones, and to a lesser extent increased fumarate, malate, α-ketoglutarate, and small amounts of methylmalonate and methylcitrate. The acylcarnitine profile showed increased propionylcarnitine 8.6 and 20.7 μM (normal < 7 μM), and slightly increased C4-dicarboxylcarnitine 1.0 μM (normal < 0.6 μM). Serum amino acids showed elevated glutamine 1030 μM, glutamic acid 320 μM, proline 820 μM, and alanine 720 μM. The enzyme activities of propionyl-CoA carboxylase, 3-methylcrotonyl-CoA carboxylase, pyruvate carboxylase, biotinidase, pyruvate dehydrogenase, and lipoamide dehydrogenase (E3) were all normal. Serum lactate ranged between 9.9 and 19.0 mM with a lactate:pyruvate ratio ranging between 25 and 26. Cardiac evaluation showed a normal cardiac muscle, and liver and renal ultrasounds were normal. A screen for common mitochondrial DNA mutations was negative as was sequencing of the POLG1 gene. At 2 months of age, succinyl-CoA ligase deficiency was suspected.
At age 2 months, she had vomiting, raised transaminases (aspartate transaminase (AST) 164 IU/L, alanine transaminase (ALT) 122 IU/L), low albumin 2.8 g/dl, but not hepatomegaly. A gastric tube was placed with fundoplication, and a liver biopsy taken. Hearing loss was identified by failed newborn hearing screen. At age 5 months, she developed axial hypotonia and lost head control, but smiled appropriately. Her liver was not enlarged but lactate was still elevated at 17 mM. Feedings provided carbohydrates restricted to 37.5% of calories, lipid 55.9% of calories, and protein 7.2% (2 g/kg/day). At age 7 months, she developed poor swallowing and muscle weakness but still had antigravity movements. At age 11 months, liver symptoms developed with the liver edge increased to 6 cm, visible vertical abdominal veins, and persistently elevated transaminases (AST 159 IU/L) and lactate between 6 and 10 mM. Her muscle weakness was marked with profound hypotonia, and areflexia. She was tachycardic 160/min and tachypneic 60/min. Her growth had stagnated, and she developed fatty cheeks and a hypoplastic nose.
At age 13.5 months she was admitted with liver failure, presenting with hypoglycemia (7 mg/dl), respiratory distress and apnea requiring intubation and ventilation. She had very elevated transaminases (AST 1065, ALT 573), albumin 3.6 g/dl, bilirubin 0.5 mg/dl, and abnormal coagulation. The creatine kinase was 1400 IU/L, and serum triglycerides ranged between 400 and 900 mg/dl. After stabilization, she was started on aspartate 3 mmol/kg/day, increased two days later to 6 mmol/kg/day as a source of anaplerotic support (13), with subsequent decrease in lactate from 14.6 to 8.5 mM (average of each 29 measurements before and after start of treatment, p< 10−7), decrease in liver size from 6 cm to 4 cm, and decrease in triglyceride levels by nearly 8 fold (Table 1). She had moderate hearing loss evidenced by absent auditory brain stem response, and absent distortion product otoacoustic emissions. She had a normal eye exam. She developed Leigh disease on brain MRI. Echocardiography showed mild ventricular hypertrophy. She had mild apneic episodes. She had demineralization on wrist X-ray but not rachitic changes. As part of her supportive care, she received a subclavian port. A new diet included 25% of calories as long-chain fat, 21% of calories as medium-chain fat, 47% of calories as carbohydrates, and protein at 1.7 g/kg/day. She did not gain weight despite a total caloric intake of 93 kcal/kg/day.
TABLE 1.
| Aspartate treatment | 0 mmol/kg/day | 3 mmol/kg/day | 6 mmol/kg/day |
|---|---|---|---|
| Methylmalonate μM | 4.6 | 5.5 | 4.4 |
| Propionylcarnitine μM | 29.7 | 40.1 | 44.7 |
| Alanine μM | 549 | 509 | 568 |
| Glutamate μM | 147 | 288 | 249 |
| Glutamine μM | 678 | 1177 | 1061 |
| Proline μM | 578 | 553 | 718 |
| Lactate mM | 8.2 | 2.6 | 3.9 |
| AST IU/L | 314 | 265 | 209 |
| ALT IU/L | 185 | 176 | 123 |
| Triglycerides mg/dL | 639 | 223 | 87.5 |
| Liver size cm BRCM | 6 | 4 cm | |
| Carbohydrate intake | 37% of calories | 47% of calories |
She was hospitalized repeatedly for vomiting and respiratory distress. At age 1 year 5 months she had severe hypotonia, absent reflexes, atrophic muscle, and rare dystonic posturing with no antigravity strength. Her liver was 5 cm below the costal margin. Four months later, she experienced an acute deterioration, rapidly rising lactate, triglycerides, and transaminases, and sepsis episodes. She died two weeks later from respiratory weakness after failing multiple extubation trials. A postmortem evaluation was not allowed.
Methods
A liver needle biopsy was obtained at 3 months of age at the time of the Nissen fundoplication. Formalin-fixed, paraffin embedded sections were stained with hematoxylin and eosin, with additional sections stained with Masson’s trichrome and periodic acid Shiff (PAS) with and without diastase. A small portion was fixed in 2.5% glutaraldehyde and prepared for routine electron microscopy. Brain magnetic resonance imaging was obtained with a 1.5 T magnet at ages 2 months and 14 months. Fibroblasts were grown from a skin biopsy and cultured in DMEM medium with 4.5g/L glucose and L- glutamine (BioWhittaker) supplemented with 10% fetal bovine serum (FBS, BioWhittaker), 100 U/ml penicillin, 100 μg/ml streptomycin and 25 mM Hepes buffer (DMEM media only) in a humidified atmosphere of 5% CO2 and at 37°C.
The enzyme activity of succinyl-CoA ligase was measured in fibroblast homogenates prepared by sonication in phosphate buffered saline. Homogenate (1 mg protein/mL final concentration) was incubated for 10 minutes at 37 °C in 50 mM TRIS buffer, pH 8.0 with 1 mM MgCl2, 1 mM ATP or GTP, 1 mM coenzyme A, and 10 mM succinate. The reaction was stopped with 30 μL 2 M perchloric acid. The supernatant was brought to pH 3 with 30 μL 2 M KOH/1M citrate. After centrifugation, 100 μL samples were injected for HPLC analysis on a C18 reverse phase column (Supelcosil LC-18-DB), flow 0.75 ml/min, buffer A 100 mM potassium phosphate buffer pH 4.0, buffer B 20% acetonitrile in 100 mM potassium phosphate buffer pH 4.0, with a linear gradient from 20 % to 50% B over 30 minutes with absorbance detection at 260 nm.
For immunoblot analysis, fibroblast homogenates were subjected to 12% SDS-PAGE and transferred onto nitrocellulose by semidry blotting. Membranes were then incubated with a SUCLG1 polyclonal antibody (dilution 1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA) and, for verification of equal loading, a β-actin monoclonal antibody (dilution 1:10,000; Sigma-Aldrich, St. Louis, MO). Antigen-antibody complexes were visualized with IRDye 800CW goat anti-mouse secondary antibody (for monoclonal antibodies) or goat anti-rabbit secondary antibody (for polyclonal antibodies) using the Odyssey Infrared Imaging System (LI-COR Biosciences, Nebraska, USA).
Sequence analysis of the SUCLA2 and SUCLG1 genes was performed by sequencing all exons plus flanking intronic sequences of both genes amplified by PCR from genomic DNA isolated from fibroblasts (primer sequences available on request). All forward and reverse primers were tagged with a −21M13 (5-′TGTAAAACGACGGCCAGT-3′) sequence or M13rev (5′-CAGGAAACAGCTATGACC-3′) sequence, respectively. PCR fragments were sequenced in two directions using ‘-21M13’ and ‘M13rev’ primers by means of BigDye Terminator v1.1 Cycle Sequencing Kits (Applied Biosystems, Foster City, CA, USA) and analyzed on an Applied Biosystems 377A automated DNA sequencer, following the manufacturer’s protocol (Applied Biosystems, Foster City, CA, USA). The SUCLA2 and SUCLG1 sequences were compared to published reference sequences (SUCLA2, GenBank accession number NM_003850 and SUCLG1, GenBank accession number NM_003849) with nucleotide numbering starting at the first adenine of the indicated translation initiation ATG codons.
To determine the effect of the c.40A>T mutation in the SUCLG1 gene on post-transcriptional level, we prepared cDNA from total RNA isolated from cultured fibroblasts and determined the relative expression levels of SUCLG1 mRNA to cyclophylin mRNA by quantitative RT-PCR analysis using the LightCycler® system (Roche, Mannheim, Germany). Analysis for mtDNA content in liver was done by quantitative PCR using probes for mtDNA and nuclear DNA (14). Deletions in mtDNA were analyzed by Southern blot and by long range PCR as described (14).
Results
The initial brain MRI at 2 months of age was normal (Figure 2A). At 14 months (Figure 2B), brain MRI showed bilateral abnormal increased T2 signal and volume loss in the caudate, globus pallidus and putamen nuclei. There was enlargement of the frontal and temporal horns, the third and fourth ventricle indicating volume loss in frontal and termporal lobes. New cortical and subcortical volume loss was present predominantly anterior. Myelination is appropriate for age, and there is no signal abnormality in the thalami or brain stem. Diffusion weighted images show restricted diffusion in bilateral frontal and periatrial white matter with low signal on apparent diffusion coefficient (ADC) map images, consistent with cytotoxic edema. There is no restricted diffusion within the basal ganglia or brainstem. On magnetic resonance spectroscopy she had elevated lactate and reduced N-acetylaspartate signals.
Figure 2.
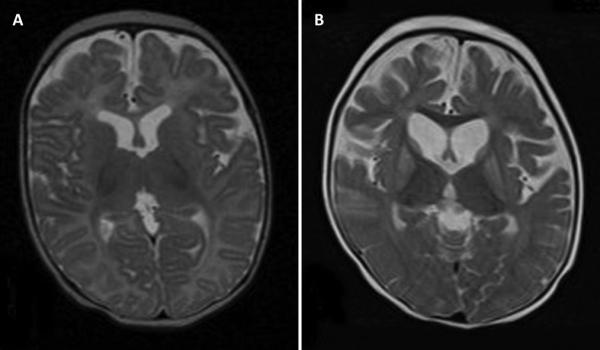
Brain imaging
Legend: A. Normal brain MRI at age two months. Axial T2 image at the level of the foramen of Monroe shows normal signal intensity and volume of bilateral basal ganglia, normal ventricular size and normal cerebral volume. B. Abnormal brain MRI at age 14 months. Axial T2 image at the level of the foramen of Monroe shows symmetric abnormal increased signal and volume loss in bilateral basal ganglia. There is cortical and subcortical volume loss with enlargement of the lateral and third ventricles and enlarged cerebral sulci. Myelination is normal.
The liver biopsy showed both micro and macrosteatosis, with the latter most marked in the periportal areas (Figure 3). No inflammation, cholestasis, giant cell formation or fibrosis was seen. Electron microscopy confirmed the presence of steatosis without significant mitochondrial abnormalities or lysosomal storage products.
Figure 3.
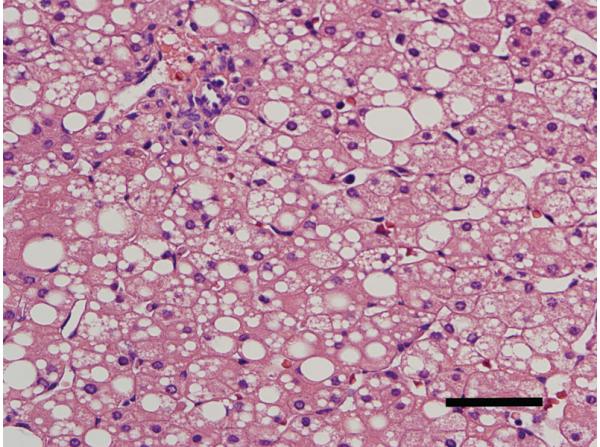
Pathology of the liver biopsy
Legend: A liver needle biopsy taken at age 3 months showed marked macro- and microvesicular steatosis, more marked around the portal areas, without inflammation, cholestasis or fibrosis. Hematoxylin and eosin staining, original magnification 400X, scale bar is 100 μm.
Because the clinical presentation and clinical chemistry suggested succinyl-CoA ligase deficiency as the underlying cause, we performed mutation analysis of the SUCLA2 gene of the patient, which did not reveal any potential pathogenic mutations. Subsequent mutation analysis of the SUCLG1 gene showed that the proband was homozygous for a c.40A>T mutation (Figure 4A). Homozygosity was confirmed by parental gene analysis. Based on the human SUCLG1 reference sequence, the c.40A>T mutation results in a substitution of the methionine at position 14 for a leucine. However, quantitative RT-PCR analysis revealed markedly decreased levels of the mutated SUCLG1 mRNA in the patient’s fibroblasts (Figure 4C), indicating that the c.40A>T mutation has an effect on SUCLG1 mRNA stability. The finding that incubation of the patient’s fibroblasts with emitine, a compound known to block mRNA decay, leads to restoration of the SUCLG1 mRNA levels confirms that the c.40A>T mutation makes the SUCLG1 mRNA substrate for mRNA decay (Figure 4C).
Figure 4.

Molecular analyses
Legend: A. Sequencing of the SUCLG1 gene. The patient shows homozygosity for the c.40A>T mutation, whereas each parent shows heterozygosity for this mutation. B. Analysis of the enzyme activity in fibroblasts. C. SUCLG1 mRNA in fibroblasts of patient and control with and without incubation with emetine. D. Western blot of SUCLG1 protein shows decreased amount of normal sized peptide compared to controls. (Pat = patient, Ctr = control)
As would be expected from a defect of SUCLG1, the enzyme analysis for succinyl-CoA ligase showed markedly deficient activities of both the ATP- and the GTP-dependent enzyme activities in the patient’s fibroblasts (0.2 nmol.min−1.mg−1 protein, controls 2.4-4.8 for ATP, and 0.1 controls 4.7-9.6 for GTP) (Figure 4B). In agreement with the decreased levels of SUCLG1 mRNA as well as the marked deficiency of SUCLG1 enzyme activity, we found that the SUCLG1 protein was severely reduced in the patients’ fibroblasts compared to controls (Figure 4D).
Because the succinyl-CoA ligase has been claimed to form a complex with the nucleoside diphosphate kinase and in previous patients depletion of mtDNA was documented, we quantified the mtDNA content in the liver biopsy. At 3 months of age the liver showed levels of mtDNA decreased by 50% compared to controls (ratio of mtDNA/nuclear DNA: patient 2.1, controls 4.23±0.8). No deletions were identified by Southern blotting or by the more sensitive long range PCR assay.
Discussion
Four patients with succinyl-CoA ligase deficiency due to mutations in SUCLG1 gene have been reported. Three patients were homozygous for a two base pair deletion and had no residual protein or activity (11). They presented with intrauterine growth retardation, hypotonia, hypothermia, abnormal EEG, and severe metabolic acidosis with lactic acidosis in the first day of life, leading to death within four days. A fourth patient had hepatomegaly. Another patient was homozygous for the missense mutation p.G72A (12). Western blot analysis in fibroblasts demonstrated decreased protein, but with some preserved residual protein. This patient had a normal birth weight and was described as normal for the first three months; then, developed failure to thrive, hypotonia and motor delay, progressive muscle atrophy and absent tendon reflexes, and severe hearing loss. There was no spasticity, no consistent dystonia, and no clinical liver disease. Lactic acidosis was mild 3 to 5 mM, and methylmalonic acid was mild 4 to 13 μM. On brain MRI there was progressive abnormal signal in putamen, globus pallidus, and caudate with brain atrophy. The child died at age 2 year 8 months of lactic acidosis following a gastroenteritis episode. This child’s phenotype was milder and similar to that of patients with a defect in the SUCLA2 gene.
Our patient’s phenotype is intermediate between both phenotypes. Similar to the first three patients, there was dysmaturity and severe lactic acidosis from birth, but in our patient this was manageable with judicious treatment. Similar to the milder affected patient with the missense mutation, our patient developed severe and ultimately fatal muscle disease with weakness, hypotonia, muscle atrophy, and respiratory failure. She developed MRI signs of Leigh disease, although she exhibited very limited signs of dystonia. She also developed hearing loss, but vision was spared. This is similar to previous patients. However, our patient also developed progressive liver disease with intermittent liver failure, which only temporarily responded to anaplerotic therapy with aspartate. From 2 months on, the child had increased transaminases, intermittently low albumin, hepatomegaly, occasional hypoglycemia, and disturbed coagulation. The biopsy was consistent with steatotis, but was taken very early before most signs of liver disease had developed. Chronic liver disease has not yet been reported in this condition. Previously reported patients had liver involvement limited to hepatomegaly in one severely affected patient, and intermittent mild elevation of transaminases and PT in a mildly affected patient (11,12). Similar phenotype has been described in other causes of liver based mtDNA depeletion syndromes such as caused by mutations in DGUOK and MPV17. The clinical fully developed phenotype of the fifth SUCLG1 deficient patient included a hepatic, a myopathic, and an encephalopathic component.
Previous reports on biochemical abnormalities in SUCLG1 deficiency have noted elevated lactate, pyruvate, methylmalonate and methylcitrate with mild increases in Krebs cycle metabolites fumarate, malate, citrate, and alpha-ketoglutarate (11,12). This patient’s biochemical phenotype was more striking, and included strongly elevated glutamate and alpha-ketoglutarate, in addition to the small but persistent elevations of methylmalonate and propionylcarnitine. Secondary reflecting these metabolites were the elevations in glutamine, proline, and alanine. These findings are strongly suggestive for succinyl-CoA ligase deficiency when present in a patient with lactic acidosis. The lactate to pyruvate ratio was only mildly increased. The metabolite profile reflects an interruption of the Krebs cycle rather than respiratory chain dysfunction. Particularly the elevation in glutamic acid is very striking (tenfold) and should lead one to review this condition.
Using a new enzyme assay, the diagnosis could be confirmed enzymatically with substrate linked to either GTP or ATP. Since the alpha-subunit encoded by SUCLG1 gene is in common for both subunits, the enzyme activity was deficient with both substrates GTP and ATP. In brain, muscle, and testis, the primary expression of beta-subunits of succinyl-CoA ligase is mostly the ADP-linked, SUCLA2-encoded beta-subunit (2,3,4). Liver has primarily GDP-linked activity with the SUCLG2 encoded beta-subunit, and heart and kidney have fairly equal amounts of ADP and GDP-linked activities (2,3,4). This tissue distribution explains why patients with a mutation in the SUCLA2 gene and hence only deficiency in the ADP-linked activity have symptoms in muscle and brain, but not in liver. Patients with a mutation in the SUCLG1 gene also have deficient GDP-linked activity and can present with liver dysfunction, which would not be expected for patients with a mutation in the SUCLA2 gene.
The pathogenesis of this disorder is likely complex, and can be different in different organs. The succinyl-CoA ligase affects the Krebs cycle, but through its interaction with nucleoside diphosphate kinase might also affect the salvage pathways for nucleotides in the mitochondria (7). Previous studies have documented decreased mtDNA in muscle in patients with mutations in SUCLA2 (7,8,9) and in SUCLG1 (11, 12). This has been related to the interaction of the succinyl-CoA ligase with nucleoside diphosphate kinase gene and imbalances in nucleotide triphosphates has been proposed, but not yet proven, as the cause for mtDNA depletion and subsequent dysfunction of respiratory chain enzyme activities, similar to other mtDNA depletion disorders (15). In the liver of a SUCLG1 patient mtDNA was reduced to 65% of normal (12) and in our patient to 50% of normal. Such reduction is not sufficient to cause liver dysfunction, but this was obtained very early in the course of the disease. Succinyl-CoA ligase is a part of the Krebs cycle, and is instrumental in the generation of succinyl-CoA for use in ketone utilization and heme formation (2,6). The metabolite pattern, with accumulation of metabolites prior to succinyl-CoA in the Krebs cycle, suggests that there was an excess of succinyl-CoA in the liver, and ketones were not excessive as would expected for insufficient function of the succinyl-CoA:3-ketoacid coenzyme A transferase (Figure 1). Thus, the deficit highlights the impact on the forward catabolic, rather than the reverse anabolic reaction. The methylmalonyl-CoA pathway also provides anaplerosis to the Krebs cycle. When the amount of such metabolites decreased, liver dysfunction worsened, and substitution with aspartate improved, but did not correct, the liver function.
We found that the patient was homozygous for a c.40A>T mutation, which according to the reference human SUCLG1 sequence is predicted to result in the substitution of the methionine at position 14 for a leucine. The methionine at position 14 is located within the predicted mitochondrial targeting sequence of SUCLG1, which comprises the first 41 amino acids of the encoded protein. Bioinformatic analysis using the MitoProt II 1.0a4 software (16) indicates that the M14L substitution does not significantly alter the probability of the amino terminus to function as a mitochondrial targeting sequence (0.3309 for M14 and 0.3366 for L14). However, when we compared the amino terminus of the predicted human SUCLG1 coding region with the amino terminus of the predicted SUCLG1 coding regions of chimpanzee, rat, and mouse (Figure 5), it appeared that the affected methionine is more likely to serve as the translation initiator methionine. When M14 is the translation initiation methionine, the probability of the shorter amino terminus to function as a mitochondrial targeting sequence is increased to 0.6946. Thus the c.40A>T mutation in fact would prevent proper translation initiation at this site. Consequently, translation initiation may occur at the next downstream AUG (i.e. 67 nucleotides downstream of this AUG), which then results in premature translation termination and which would make the mRNA substrate for the so-called nonsense-mediated mRNA decay (or NMD) pathway (17,18), a cellular quality-control mechanism which results in destabilization of the mRNA (18,19). Indeed, this is supported by the observation of very low SUCLG1 mRNA levels in the patient’s fibroblasts. Moreover, when the NMD pathway in the patient’s fibroblasts is inhibited with emitine, we observed a marked increase in mRNA levels.
Figure 5.

Comparison of the amino terminus of SUCLG1 proteins
Legend: Alignment of the predicted amino-terminal coding sequences of the SUCLG1 proteins from human, chimpanzee, rat and mouse.
* denotes M14, which is mutated in the patient. ↓ indicates the predicted mitochondrial targeting sequence cleavage site.
In conclusion, patients with succinyl-CoA ligase deficiency present with a progressive hepatoencephalomypathy. The pathogenesis of the disease is complex and involves anaplerosis and disruption of the Krebs cycle as well as mtDNA depletion.
Acknowledgments
The authors thank Dr. James Zauche for referral and care of the patient.
This study was supported by a grant from the National Institutes of Health U54 DK078377 (Cholestatic Liver Disease Consortium) and U01 DK062453 (Cholestatic Liver Disease Research and Education Network).
Abbreviations
- AST
aspartate transaminase
- ALT
alanine transaminase
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- mtDNA
mitochondrial DNA
- NMD
nonsense-mediated mRNA decay
Footnotes
J.L.K.H. and M.S.S.contributed equally to this study.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alberio S, Mineri R, Tiranti V, Zeviani M. Depletion of mtDNA: syndromes and genes. Mitochondrion. 2007;7:6–12. doi: 10.1016/j.mito.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JD, Muhonen WW, Lambeth DO. Characterization of the ATP- and GTP-specific succinyl-CoA synthetases in pigeon. J Biol Chem. 1998;273:27573–27579. doi: 10.1074/jbc.273.42.27573. [DOI] [PubMed] [Google Scholar]
- 3.Johnson JD, Mehus JG, Tews K, Milavetz BL, Lambeth DO. Genetic evidence for the expression of ATP- and GTP-specific succinyl-CoA synthetases in multicellular eucaryotes. J Biol Chem. 1998;273:27580–27586. doi: 10.1074/jbc.273.42.27580. [DOI] [PubMed] [Google Scholar]
- 4.Lambeth DO, Tews KN, Adkins S, Frohlich D, Milavetz BL. Expression of two succinyl-CoA synthetases with different nucleotide specificities in mammalian tissues. J Biol Chem. 2004;279:36621–36624. doi: 10.1074/jbc.M406884200. [DOI] [PubMed] [Google Scholar]
- 5.Philips D, Aponte AM, French SA, Chess DJ, Balaban RS. Succinyl-CoA synthetase is a phosphate target for the activation of mitochondrial metabolism. Biochemistry. 2009;48:7140–7149. doi: 10.1021/bi900725c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kowluru A, Tannous M, Chen H-Q. Localization and characterization of the mitochondrial isoform of the nucleoside diphosphate kinase in the pancreatic β cell: evidence for its complexation with mitochondrial succinyl-CoA synthetase. Arch Biochem Biophys. 2002;398:160–169. doi: 10.1006/abbi.2001.2710. [DOI] [PubMed] [Google Scholar]
- 7.Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, Eshhar S, Saada A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081–1086. doi: 10.1086/430843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostergaard E, Hansen FJ, Sorensen N, Duno M, Vissing J, Larsen PL, Faeroe O, Thorgrimsson S, Wibreand F, Christensen E, Schwartz M. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–861. doi: 10.1093/brain/awl383. [DOI] [PubMed] [Google Scholar]
- 9.Carrozzo R, Dionisi-Vici C, Steuerwald U, Lucioli S, Deodato F, Giandomenico SD, Bertini E, Franke B, Kluijtmans LA, Meschini MC, Rizzo C, Piemonte F, Rodenburg R, Santer R, Santorelli FM, Van Rooij A, Vermunt-de Koning D, Morava E, Wevers RA. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130:862–874. doi: 10.1093/brain/awl389. [DOI] [PubMed] [Google Scholar]
- 10.Morava E, Steuerwald U, Carrozzo R, Kluijtmans LA, Joensen F, Santger R, Dionisi-Vici C, Wevers RA. Dystonia and deafness due to SUCLA2 defect: clinical course and biochemical markers in 16 children. Mitochondrion. 2009;9:438–442. doi: 10.1016/j.mito.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Ostergaard E, Christensen E, Kristensen E, Mogensen B, Duno M, Shoubridge EA, Wibrand F. Deficiency of the α-subunit of succinate-coenzyme A ligase causes infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81:383–387. doi: 10.1086/519222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostergaard E, Schwartz M, Batbayli M, Christensen E, Hjalmarson O, Kollberg G, Holme E. A novel missense mutation in SUCLG1 associated with mitochondrial DNA depletion, encephalomyopathic form, with methylmalonic aciduria. Eur J Pediatr. 2010;106:201–205. doi: 10.1007/s00431-009-1007-z. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad A, Kahler SG, Kishnani PS, Artigas-Lopez M, Pappu AS, Steiner R, Millington DS, Van Hove JL. Treatment of pyruvate carboxylase deficiency with high doses of citrate and aspartate. Am J Med Genet. 1999;87:331–338. doi: 10.1002/(sici)1096-8628(19991203)87:4<331::aid-ajmg10>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 14.Naini A, Shanske S. Detection of mutations in mtDNA. Methods Cell Biol. 2007;80:437–463. doi: 10.1016/S0091-679X(06)80022-1. [DOI] [PubMed] [Google Scholar]
- 15.Saada A. Deoxyribonucleotides and disorders of mitochondrial DNA integrity. DNA Cell Biol. 2004;23:797–806. doi: 10.1089/dna.2004.23.797. [DOI] [PubMed] [Google Scholar]
- 16.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 17.Welch EM, Jacobson A. An internal open reading frame triggers nonsense-mediated decay of the yeast SPT10 mRNA. EMBO J. 1999;18:6134–6145. doi: 10.1093/emboj/18.21.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amrani N, Dong S, He F, Ganesan R, Ghosh S, Kervestin S, Li C, Mangus DA, Spatrick P, Jacobson A. Aberrant termination triggers nonsense-mediated mRNA decay. Biochem Soc Trans. 2006;34:39–42. doi: 10.1042/BST20060039. [DOI] [PubMed] [Google Scholar]
- 19.Amrani N, Sachs MS, Jacobson A. Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol. 2006;7:415–425. doi: 10.1038/nrm1942. [DOI] [PubMed] [Google Scholar]