

Abstract
Flavin mononucleotide adenylyltransferase (FMNAT) catalyzes the formation of the essential flavocoenzyme FAD and plays an important role in flavocoenzyme homeostasis regulation. By sequence comparison, bacterial and eukaryotic FMNAT enzymes belong to two different protein superfamilies and apparently utilize different set of active site residues to accomplish the same chemistry. Here we report the first structural characterization of a eukaryotic FMNAT from a pathogenic yeast Candida glabrata (CgFMNAT). Four crystal structures of CgFMNAT in different complexed forms were determined at 1.20–1.95 Å resolutions, capturing the enzyme active site states prior to and after catalysis. These structures reveal a novel flavin-binding mode and a unique enzyme-bound FAD conformation. Comparison of the bacterial and eukaryotic FMNATs provides a structural basis for understanding the convergent evolution of the same FMNAT activity from different protein ancestors. Structure-based investigation of the kinetic properties of FMNAT should offer insights into the regulatory mechanisms of FAD homeostasis by FMNAT in eukaryotic organisms.
Keywords: flavocoenzymes, FAD biosynthesis, adenylyltransferase, Rossmann-like fold, convergent evolution
INTRODUCTION
Flavin nucleotides FMN and FAD are essential cofactors involved in many redox reactions in the cell.1 The chemical and functional versatility of these cofactors, in association with various flavoproteins, allows them to be involved in a large variety of different types of reactions and to participate in many cellular processes ranging from energy production, light emission, DNA repair, chromatin remodeling, and protein folding to detoxification, neural development, and apoptosis.2; 3; 4 Riboflavin, also known as vitamin B2, is the universal precursor for the synthesis of FMN and FAD, the primary form of flavins in cells.5; 6 Due to the involvement of flavocofactors in wide-ranging metabolic processes, riboflavin deficiency leads to a multitude of physiological aberrations, such as abnormal fetal development, inadequate ion absorption, cardiovascular disease and corneal defects.7
In prokaryotes, yeast and plants, riboflavin is either synthesized de novo, or obtained from the environment and transported into the cells. 6,8 Higher eukaryotes, such as humans, lack the de novo riboflavin synthesis machinery and the only means of obtaining riboflavin is from the diet.5; 6 The riboflavin transporter for eukaryotes has been identified in Saccharomyces cerevisiae and is encoded by gene mch5.9 Converting riboflavin to FAD involves two universally conserved enzymes, riboflavin kinase (RFK) and flavin mononucleotide adenylyltransferase (FMNAT) (Fig. 1). Riboflavin kinase (ATP:riboflavin 5′-phosphotransferase, EC 2.7.1.26) phosphorylates riboflavin to generate flavin mononucleotide (FMN), while FMNAT (ATP:FMN adenylyltransferase, EC 2.7.7.2) adenylates FMN to form FAD. In bacteria, RFK and FMNAT are encoded in the same gene, ribF or ribC,10; 11 and the protein product is referred to as FAD synthetase (FADS) with the FMNAT domain located at the N-terminus and the RFK domain at the C-terminus. In eukaryotes, RFK and FMNAT are encoded by separate genes.12; 13 In higher eukaryotes, the gene encoding FMNAT also contains a second domain with sequence similarity to proteins involved in molybdenum-cofactor (MoCo) biosynthesis, such as MogA and MoeA.14; 15 The potential function of this MoCo-binding protein-like domain is unknown.
Figure 1.

Reactions catalyzed by riboflavin kinase (RFK) and FMN adenylyltransferase (FMNAT).
Bacterial and eukaryotic RFKs are similar in sequence and structure, and they belong to a unique protein family containing only riboflavin kinases.16; 17 In contrast, the evolutionary link between bacterial and eukaryotic FMNATs is less clear as they show little sequence similarity and are classified in different protein superfamilies in SCOP18 or different clans in pfam19 databases. The bacterial FMNAT domain of the bifunctional RFK/FMNAT belongs to the (H/T)xGH motif containing nucleotidylyl transferase superfamily, while eukaryotic FMNAT is currently classified as a member of the 3′-phosphoadenosine 5′-phosphosulfate (PAPS) reductase-like family belonging to the “adenine nucleotide α hydrolase-like” superfamily, which has conserved motifs different from those of nucleotidylyl transferases. Despite substantial differences in sequence and structure, the mammalian and bacterial FMNAT enzymes have similar kinetic properties.20; 21 Both enzymes catalyze the formation of FAD through an ordered bi-bi mechanism and have the same substrate binding and product release order where ATP binds first to the enzyme followed by FMN, and product inorganic pyrophosphate (PPi) is released first followed by the release of FAD. Pronounced product feedback inhibition was observed for rat liver FMNAT, and it was suggested that such a property would enable FMNAT to play a role in regulating cellular FAD homeostasis as the Ki values of FAD against FMN (0.75 μM) and Mg2+ATP (1.3 μM) are close to the concentration of free FAD (0.4 μM).21 Maintenance of FAD homeostasis is important, as several cellular processes, such as oxidative protein folding and homocysteine metabolism, are sensitive to FAD levels.22; 23
Both RFK and FMNAT are needed for generating the indispensable flavocofactors FMN and FAD.6 The essentiality of the two enzymes has been established experimentally in bacterial24 and yeast species,12; 13; 22 and has been inferred to all other organisms. The significant differences between eukaryotic and bacterial FMNAT makes it a particularly attractive target for developing selective anti-infectious drugs.24 Structural analysis of both eukaryotic and bacterial FMNATs will reveal the different configurations of the substrate binding and catalytic sites, which may benefit a structure-based inhibitor development effort. Such analysis will also address two fundamental questions: how eukaryotic and bacterial FMNAT accomplish the same chemistry with different active site architectures and what mechanistic controls are embedded in the enzyme to influence FAD homeostasis. Here we report the first structural characterization of a eukaryotic FMNAT from Candida glabrata, an opportunistic yeast pathogen causing candidemia and invasive candidiasis.25; 26 We have determined the crystal structures of Candida glabrata FMNAT (CgFMNAT) in the apo-form and in three different complexed forms (with ATP, with substrate FMN and the ATP analog AMPCPP, and with products FAD and PPi, respectively). These structures reveal a novel flavin-binding mode and the detailed catalytic site configuration that are likely shared among all eukaryotic FMNATs. Combined with the results from steady-state kinetic analysis, a mechanism for eukaryotic FMNAT catalyzed adenylyl transfer reaction is proposed.
RESULTS
Quality of the structures
The crystal structure of apo-CgFMNAT was solved by the single-wavelength anomalous dispersion (SAD) phasing method using selenomethione as the source of anomalous dispersion and was refined against a native dataset to the resolution of 1.20 Å (Table 1). There is one CgFMNAT molecule in the asymmetric unit. Crystal-packing analysis suggests that the functional unit of CgFMNAT is a monomer, which is consistent with the gel filtration result that showed CgFMNAT is monomeric in solution (see Fig. S1 available online). The refined model contains residues -3–304, except for residues 85–101, for which we found no associated electron density, and which are presumably disordered. Residues -3–0 (Gly-Ala-Met-Val) were introduced during cloning (see Supplementary Methods). The CgFMNAT-ATP complex crystal is isomorphous to the apo-CgFMNAT, and its model was refined to 1.87 Å resolution. This model contains residues -3–83, 103–304, and an ATP molecule with well defined density (Fig. 2a). Crystals of the substrate and product ternary complexes are isomorphous to each other and belong to the space group C2 with six monomers in the asymmetric unit. They were refined to resolutions of 1.95 Å and 1.35 Å, respectively. For the substrate ternary complex, the densities for AMPCPP and Mg2+ ion are well defined and double conformations of the AMPCPP phosphate tail are observed (Fig. 2b). The density for the phosphoribityl tail of FMN, on the other hand, is discontinuous, indicating significant conformational flexibility (Fig. 2b). For the product ternary complex, the densities for both products FAD and pyrophosphate (PPi) are well defined (Fig. 2c). The flexible loop region (residues 84–100) disordered in the apo- and ATP-complexed CgFMNAT structures is ordered in two of the six crystallographically independent CgFMNAT monomers in the ternary complexes, presumably due to crystal-packing interactions. For all models, the main chain dihedral angles (φ, ψ) for each residue are in the allowed Ramachandran area. The exception is Gly224 in the ATP complex structure, which is associated with well defined electron density. Superposition of the Cα backbones of apo-protein, ATP complex and substrate ternary complex (monomer C) to product ternary complex (monomer B) gives a root mean square deviation (rmsd) of 0.42 Å, 0.41 Å, and 0.27 Å, respectively, indicating no substantial conformational changes among these structures (Fig. 2d).
Table 1.
Data Collection and Refinement Statistics
| Protein | Selenomethionyl apo-CgFMNAT | apo-CgFMNAT | ATP complex | Ternary complex with substrates | Ternary complex with products |
|---|---|---|---|---|---|
| Space Group/Monomer per Asymmetric Unit | P3221/1 | P3221/1 | P3221/1 | C2/6 | C2/6 |
| Data | SAD | Native | Native | Native | Native |
| Wavelength (Å) | 0.97927 | 0.97931 | 1.54178 | 1.54178 | 0.97874 |
| Resolution (Å) | 50.0−2.18 | 50.0−1.20 | 50.0−1.87 | 50.0−1.95 | 50.0−1.35 |
| Unit cell dimensions | |||||
| a, b, c (Å) | 80.31, 80.31, 78.27 | 80.09, 80.09, 78.09 | 79.79, 79.79, 77.94 | 207.83, 81.75, 136.70 | 206.58, 81.48, 136.60 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 129.67, 90.0 | 90.0, 129.79, 90.0 |
| Total no. of reflections | 200196 | 1036966 | 124769 | 312183 | 2800906 |
| No. of unique reflections | 15582 | 90728 | 24161 | 123738 | 380943 |
| % Completeness | 99.9 (100.0)a | 99.9 (99.7) | 99.9 (99.6) | 96.4 (97.1) | 100 (99.8) |
| Rsymb | 0.091 (0.335) | 0.070 (0.442) | 0.078 (0.549) | 0.088 (0.447) | 0.077 (0.551) |
| I/σ | 38.9 (8.5) | 48.2 (2.3) | 22.0 (2.5) | 11.5 (2.1) | 35.9 (2.4) |
| Mosaicity (°) | 0.6 | 0.4 | 0.7 | 0.5 | 0.4 |
| Model Refinement | Anisotropice | Isotropic | Isotropic | Anisotropic | |
| Resolution range (Å) | 50.0−1.20 | 50.0−1.87 | 35.1−1.95 | 32.2−1.35 | |
| Rworkc (%) | 16.2 | 17.4 | 17.5 | 15.3 | |
| Rfreed (%) | 17.8 | 22.2 | 23.6 | 18.7 | |
| No. of protein atoms | 2502 | 2397 | 14700 | 14968 | |
| No. of ligand atoms | 13 | 44 | 397 | 417 | |
| No. of water molecules | 390 | 303 | 1790 | 1870 | |
| Average B-factor (Å2) | |||||
| Protein atoms | 17.1 | 19.3 | 18.2 | 17.1 | |
| Ligand atoms | 21.3 | 23.5 | 17.5 | 15.8 | |
| Water molecules | 29.3 | 30.8 | 28.5 | 30.4 | |
| Rmsd bond length (Å) | 0.015 | 0.015 | 0.014 | 0.014 | |
| Rmsd bond angle (°) | 1.621 | 1.536 | 1.458 | 1.523 | |
| Ramachandran Plot | |||||
| % favored region | 98.6 | 98.6 | 98.9 | 99.1 | |
| % allowed region | 1.4 | 1.05 | 1.1 | 0.9 | |
| % outliers | 0.0 | 0.35 | 0.0 | 0.0 | |
Values in parenthesis are for highest-resolution shell.
Rsym = Σhkl [(Σj (|Ij − <I>|)/ΣjIj ]. <I> is the average for all j measurements of reflection hkl.
Rwork = Σhkl |Fo − kFc|/Σhkl |Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
R-factor calculated from randomly selected 1.5% (apo-CgFMNAT) or 5% (complexes) reflections that are excluded from refinement for cross-validation.
Figure 2. Electron densities of the bound ligands and comparison of CgFMNAT structures.

Simulated-annealing Fo-Fc omit maps of (a) ATP in the binary complex; (b) AMPCPP and FMN in the substrate ternary complex, and (c) FAD and PPi in the product ternary complex. The maps are contoured at 3.0σ for (a) and (c), and 2.5σ for (b). (d) Superposition of apo-form (magenta), ATP complex (tan), substrate ternary complex monomer C (dark yellow), and product ternary complex monomer B (dark blue).
Overall Structure
CgFMNAT is composed of two domains (Fig. 3a). The N-terminal domain has an α/β fold with a central, twisted six-stranded β-sheet sandwiched by α-helices. The topology of this domain is a modified Rossmann-fold where the 5th β-strand of the central β-sheet is anti-parallel to the rest of the strands (Fig. 3a). Comparison of CgFMNAT with known protein structures using the Dali server27 indicates that the core of CgFMNAT is most similar to the members of the PAPS reductase-like protein family. These include bacterial adenosine 5′-phosphosulfate (APS) reductase28 (PDB Id 2goy; Z-score of 15.4 and rmsd of 2.5 Å for 174 superimposed Cα atoms), PAPS reductase29 (1sur, Z-score of 14.0 and rmsd of 2.4 Å for 159 superimposed Cα atoms), and ATP sulfurylase30 (1zun, Z-score of 13.1 and rmsd of 2.9 Å for 160 superimposed Cα atoms). Similar to CgFMNAT, these protein also have an N-terminal domain of a modified Rossmann-fold topology (Fig. 3b). The C-terminal domain of CgFMNAT is composed largely of loops (64%) interlaced by α6–α9 helices and two short 310-helices (Fig. 3a). This domain is much longer than that of APS and PAPS reductases and appears to be uniquely elaborated and expanded in yeast FMNAT (Supplementary Fig. S2). As will be discussed later, residues from this domain are also involved in interactions with ATP.
Figure 3. Overall structure of CgFMNAT.

(a) Ribbon diagram of CgFMNAT structure shown in complex with the products FAD, PPi, and Mg2+. Secondary structure elements are labeled. Structure motifs involved in substrate binding and catalysis are labeled and highlighted in different colors with PP-loop in dark blue, ADE motif dark green, ARG1 motif magenta, γ-Phosphate motif red, and Flavin Motif brown. The magnesium ion is shown as a green sphere.
(b) Comparison of CgFMNAT with closely related APS reductase. Helix α1 and the last ~70 residues of CgFMNAT are removed for clarity. Bound ligand FAD in CgFMNAT, and APS in APS reductase are shown as sticks colored by atom type with carbon atoms colored yellow and orange, respectively. Equivalent structural motifs are colored similarly. The characteristic PP-loop motifs are colored red. Regions that deviate from the typical Rossmann-fold topology are shown in magenta.
Mg2+ATP Binding Site
The structures of the CgFMNAT-ATP binary complex and the substrate ternary complex reveal details about the interactions between the enzyme and the ATP substrate. Five structural motifs are identified to be involved in ATP and Mg2+ binding (Fig. 3a). The PP-loop motif, extending from β1 to the N-terminus of α3, has the sequence of 60SYNGGKDC67 and is generally conserved in the superfamily. The ADE motif, named for ADEnine binding, corresponds to the LDTG motif in APS reductase28 and consists of a short stretch of four residues, 107FIDH110, following strand β2. The first arginine-containing ARG1 motif (named Arg-loop in ref. 28) is located in the loop connecting β4 to β5 and consists of residues 163GIRHTD168. The γ-Phosphate motif encompasses part of α7 and the following hairpin loop from the C-terminal domain situated above the nucleotide-binding site. Near the C-terminus of the protein, another arginine-rich motif, ARG2, of sequence 296ERAGR300 is also involved in nucleotide binding.
The ATP nucleotide binds in a crevice formed between β1 and β4, with its βγ-phosphate tail positioned in an anion-binding pocket near the N-terminus of α3 (Fig. 4a). A hydrogen-bond network is formed between ATP and residues from the five motifs (Fig. 4a). The adenine N1 nitrogen and N6 amino groups are hydrogen bonded to the main chain amide and carbonyl of Ile108 (part of the ADE motif), respectively. The N3 group of adenine forms a hydrogen bond to the side chain of Ser60 of the PP-loop motif, while the O2′ hydroxyl of ATP ribose interacts with the main chain carbonyl of Ser60. The O2′ hydroxyl forms another hydrogen bond with the main chain amide of Gly163 of the ARG1 motif. The PP-loop motif residues interacts extensively with the phosphates of ATP through both main chain and side chain moieties. The side chain of Asn62 interacts with the α-phosphate; the main chain amide of Cys67 interacts with the β-phosphate; and the side chain of Lys65 provides two hydrogen bonds to the γphosphate. Additional hydrogen-bonds to the γ-phosphate are formed with the Tyr216 hydroxyl and the Leu223 main chain amide. Both residues are from the γ-Phosphate motif (Fig. 4a).
Figure 4. Substrate and product binding in CgFMNAT.


(a) Details of the Mg2+ATP-binding site. Mg2+AMPCPP in the catalytically relevant conformer I is shown. Protein residues interacting with bound subtrates are shown as sticks. The Mg2+ ion is shown as a green sphere and water ligands are shown as red spheres. Hydrogen bonds are shown as dash lines and metal ligands are indicated with solid lines.
(b) Details of FMN binding site.
(c) Stereo view of FAD and PPi product binding site.
In the presence of magnesium ions, as in the case of the substrate ternary complex structure, the AMPCPP phosphate tail adopts two distinct conformations (conformer I and II, Fig. 2b & Fig. 5) in different monomers of the six CgFMNAT substrate ternary complexes in the asymmetric unit, both of which differ from that observed in the Mg2+-free ATP binary complex structure (Fig. 5). The largest difference between these conformations is in the position of the β-phosphate, which moves 5.1 Å between conformer I and II, while the α- and γ-phosphates move 1.6 Å and 2.2 Å, respectively. In both conformers, the bound Mg2+ ion remains in the same position and maintains a six-ligand octahedral configuration (Fig. 5). In the first conformer, the Mg2+ is liganded to the β- and γ-phosphate oxygens, the Asp66 side chain carboxyl and three conserved water molecules, w2, w3, and w4 (Fig. 4a). W2 and w3 are further coordinated to the carboxylate group of Asp168 of the ARG1 motif. In the second conformer, all three AMPCPP phosphoryl moieties are coordinated to the Mg2+ ion, with the β-phosphate oxygen substituting the w4 water ligand. The β-phosphate in this conformation also interacts with Arg279 of the ARG2 motif through a bifurcated hydrogen bond (not shown). In the Mg2+-free ATP binary complex structure, the position of the β-phosphate largely overlaps with the Mg2+ binding site (Fig. 5).
Figure 5. Stereo view of the superposition of bound ATP, Mg2+, FMN and FAD.

The dual conformations of AMPCPP (light green) in the substrate ternary complex are marked as I and II. The ATP molecule in the Mg2+-free binary complex structure is shown as blue thin lines. The substrate FMN is colored pink, while the product FAD is colored by atom types with carbon atoms in yellow. The Mg2+ ion (green sphere) and corresponding water ligands (red spheres) are also shown.
Compared to other PAPS reductase-like proteins, three of the five ATP binding motifs in CgFMNAT, PP-loop, ADE and ARG1 motifs, are conserved (Fig. S2). Although the γ-Phosphate motif is also present in members of the PAPS reductase-like family such as APS reductase and bacterial ATP sulfurylase, its role in substrate binding and catalysis in these enzymes is not clear. Unique to CgFMNAT and APS reductase is the presence of the ARG2 motif (Supplementary Fig. S2). In APS reductase, Arg242 and Arg245 near the protein C-terminus, corresponding to Arg297 and Arg300 of CgFMNAT, provide important interactions with the phosphosulfate group of APS. Our preliminary kinetic analysis of the R297A mutant shows that the apparent Km,ATP and Km,FMN of the mutant increased ~5 and ~3 times, respectively, compared to the wild-type enzyme (data not shown), indicating that this Arg residue is indeed involved in substrate binding, presumably through interactions with the phosphoryl groups of the substrates.
FMN/FAD Binding Site
Unexpectedly, the flavin-binding site, as revealed in the substrate and product ternary complexes, is located on the same side of the central β-sheet as the adenosine moiety of ATP, where a deep trough is formed between the face of the β-sheet, helix α5, and the loop connecting the anti-parallel β5 to β6 (Fig. 3 & 4). This pocket forms a unique binding site for the flavin isoalloxazine ring that is different from those observed in any other FMN- or FAD-binding protein.31 Residues from a broad range of structural elements are involved in the interaction with the isoalloxazine ring (Fig. 3a, & 4c). These include Met143 and Phe147 from helix α5, Ile160 to Ile162 from strand β4; Asp181, Trp184 and Phe187 from the loop connecting β5 to β6, and Arg189 from strand β6. These residues are highly conserved among eukaryotic FMNATs (Supplementary Fig. S3) and are collectively referred to as the Flavin motif. The isoalloxazine ring is sandwiched between the indole ring of Trp184 and the planar guanidinium group of Arg189. Deeply buried in the flavin-binding pocket is the hydrophobic dimethylbenzene moiety of the isoalloxazine ring, forming van der Waals contacts with hydrophobic side chains of Met143, Phe147, Ile160, Ile162, and Phe187 (Fig. 4b & 4c). The hydrophilic lumazine side of the ring forms two specific hydrogen bonds with the enzyme, between its C4 carbonyl and the main chain amide of Asp181, and between its N3 amide and the side chain of Asp 181, respectively. In both substrate and product ternary complex structures, the isoalloxazine group is also in van der Waals contact with the adenosine moiety of either AMPCPP or FAD, suggesting that the adenosine group is part of the isoalloxazine-binding pocket.
In contrast to the extensive interactions with the isoalloxazine ring, there are few contacts between the enzyme and the FMN phosphoribityl tail (Fig. 3b). As a result, the conformation of this part of the substrate is not well defined as indicated by the high B-factors, partial occupancy, and discontinuous density (Fig. 2b), In five of the six monomers in the asymmetric unit, FMN phosphoribityl tail is seen pointing toward the solvent, positioned over the N-terminus of helix α5 and away from the bound AMPCPP, probably due to electrostatic repulsion (Fig. 3b and 5). This observed conformation of FMN is apparently not in the catalytically ready state, as the phosphate is too far from the bound AMPCPP. Clearly the flexibility of FMN phosphoribityl tail would allow it to adopt multiple conformations and to move close to ATP so that the adenylyltransferase reaction could occur. Comparing the substrate and product ternary complexes, the positions of the isoalloxazine ring and the adenosine moieties remain essentially unchanged (Fig. 5). The phosphoribityl group of the product FAD becomes well ordered when covalently linked to the adenylyl group of ATP. The position of the PPi product is also well defined in the crystal structure and is identical to that of the βγ-phosphates of AMPCPP in conformer I. The Mg2+ ion in the product ternary complex is coordinated to the PPi, the adenylyl phosphate of FAD, Asp66 and two water molecules in a configuration with features of both AMPCPP conformers in the substrate ternary complex (Fig. 5). Arg297 from the C-terminal ARG2 motif is found to interact with the diphosphate moiety of FAD (Fig. 3c), supporting its potential involvement in binding the phosphate groups of both ATP and FMN substrates, and positioning them for the adenylyl transfer reaction.
Steady-state kinetic properties of CgFMNAT
The steady-state kinetic parameters of CgFMNAT were determined using a continuous coupled assay. The Lineweaver-Burk reciprocal plots of 1/v vs. 1/[FMN] and 1/v vs. 1/[ATP] (Fig. 6a and b) are consistent with an ordered bi-bi system,32 with the substrate binding order being ATP first, followed by FMN. The same mechanism and substrate binding order were also proposed for rat liver and bacterial FMNATs in early studies.20; 21 The initial rates of the reaction were globally fitted to the general equation for an ordered steady-state bireactant model (inserts of Fig. 6a and b). The steady-state kinetic parameters obtained for CgFMNAT are: Km for ATP 10.7 ± 2.3 μM, Km for FMN 0.76 ± 0.15 μM, and kcat 0.087 s−1. The Km, FMN value for CgFMNAT is similar to that obtained for human FMNAT isoform II in a recent study (apparent Km, FMN 0.36 ± 0.06 μM), while the kcat appears to be more than ten times higher than that of the human enzyme (0.0036 ± 0.0001 s−1).33 These values are somewhat different from those obtained for the endogenous rat-liver FMNAT, where the apparent Km for ATP and FMN are 71 μM and 9.1 μM, respectively, and Vmax is 345 nmol FAD/min/mg protein, corresponding to a kcat of 0.15 s−1.21
Figure 6. Adenylyl transfer mechanism for CgFMNAT.



(a) Initial rates represented by Lineweaver-Burk plot of 1/v versus 1/[ATP] at fixed FMN concentrations. Insert shows the same data represented by a hypernolic plot (insert) as a function of ATP. (b) Initial rates represented by Lineweaver-Burk plot of 1/v versus 1/[FMN] at fixed ATP concentrations. Insert shows the same data represented by a hypernolic plot (insert) as a function of FMN. The rates in (a) and (b) were globally fitted to the velocity equation for an ordered bi-bi (1) reactant system. (c) Proposed catalytic mechanism of CgFMNAT.
Proposed mechanism for CgFMNAT
The four high-resolution structures of CgFMNAT (apo form, complexes with ATP, with substrate FMN+AMPCPP, and with products FAD+PPi) along with the kinetic data allow us to envision the events in the enzyme active site during catalysis (Fig. 6c and Supplementary Movie). In this process, ATP binds preferably first to the enzyme, because its binding pocket would be partially blocked by FMN, which binds at a site closer to the surface (Fig. 7). Additionally, binding ATP first may help to properly position the FMN substrate as the C8M methyl group of the isoalloxazine ring is packed against the adenosine moiety of the bound ATP. These observations are consistent with the substrate-binding order deduced from the kinetic data. The binding of ATP induces small adjustment (0.3–0.5 Å) of several surrounding residues, including Asn62, Lys65, Asp66, Ile108, and Asp168, presumably to optimize their interactions with Mg2+ and ATP. In the presence of Mg2+, the phosphate tail of ATP can adopt either of the two discrete conformations I or II, in which the Mg2+ ion position remains the same (Fig. 5). Upon subsequent binding of FMN, side chains of several residues around the isoalloxazine ring, e.g., Met143, Phe147, Asp181 and Trp184, also make small adjustments to optimally interact with the substrate. Due to the lack of interaction between the enzyme and the phosphoribityl tail of FMN, this part of the FMN substrate is highly flexible and able to adopt multiple conformations. For the adenylation reaction to occur, the FMN phosphate would move close to the α-phosphate of ATP for the ensuing nucleophilic attack. The presence of Mg2+ ion and interaction with Arg297 (and potentially Arg300) of the ARG2 motif may help to overcome the electrostatic repulsion between the phosphate groups of the two substrates, and position FMN phosphate for the attack on the α-phosphate of ATP (Fig. 6c). The cleavage of the αβ-phosphodiester bond is facilitated by the coordination of the Mg2+ ion, which is required for the reaction. The Mg2+ATP in the Conformer I position appears to be the catalytically competent conformation which allows the FMN phosphate group to approach the α-phosphate from the direction opposite the β-phosphate for the direct in-line nucleophilic attack. Minimal structural rearrangements are observed after product formation. The leaving diphosphate group is practically in the same position as the βγ-phosphates of the Conformer I of the nucleotide and interacts with the same set of protein residues and Mg2+ ion. The transferred α-phosphate moves about 2.5 Å away from its original position and is now directly liganded to the Mg2+ ion (Fig. 5 and see Supplementary Movie). Earlier kinetic studies of rat liver FMNAT revealed that the enzyme is markedly inhibited by the product FAD with a Ki against FMN of 0.75 μM and a Ki against Mg2+ATP of 1.3 μM.21 It has been suggested that the biosynthesis of FAD is most likely regulated by product FAD at the last FMNAT step of the pathway.21 Although not yet determined explicitly, our observation that CgFMNAT copurifies with intrinsically bound FAD, along with the extensive interactions between the enzyme and FAD seen in the crystal structure, suggests that FAD binds to CgFMNAT with high affinity and likely exerts a feedback inhibitory effect as well.
Figure 7. Electrostatic surface potential representation of CgFMNAT substrate binding pocket.
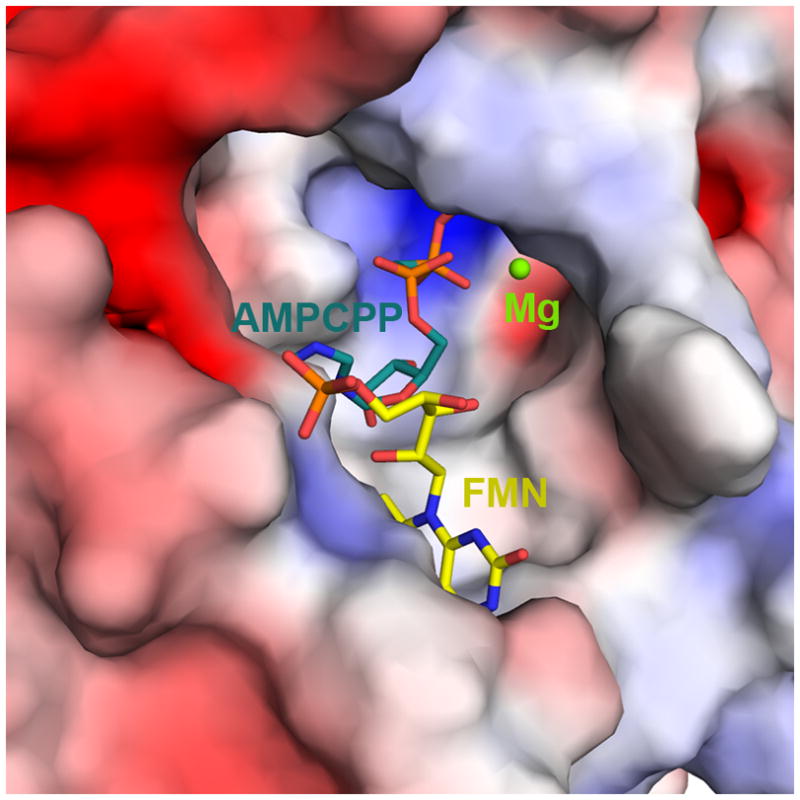
The electrostatic potential is color ramped from −5 kT/e (red) to +5 kT/e (blue). FMN and AMPCPP are represented by sticks and Mg2+ ion is shown as a green sphere.
DISCUSSION
Our high-resolution structures of yeast FMNAT present the first characterization of a eukaryotic version of this essential enzyme. Although the overall structure of CgFMNAT shares significant similarity with PAPS reductase-like family of proteins, as reflected in several shared nucleotide binding motifs. The mode of flavin binding in CgFMNAT has not been observed before. In a 2001 survey by Dym and Eisenberg,31 all FAD-binding proteins with known 3D structures were categorized into four different groups, represented by glutathione reductase, ferredoxin reductase, p-cresol methylhydroxylase and pyruvate oxidase, respectively. CgFMNAT is clearly distinct from any of these FAD-binding proteins in both the conserved sequence motifs and flavin-binding mode. In other Rossmann-fold FAD-binding proteins (e.g., glutathione reductase and pyruvate oxidase groups) the isoalloxazine ring of FAD invariably binds on the side of the central β-sheet opposite from the adenine moiety and across the top of the sheet between β1 and β4. It often interacts with residues from another domain. In contrast, in CgFMNAT, the isoalloxazine ring binds to the same side of the central β-sheet as the adenine, and the binding involves exclusively residues from the Rossmann-like N-terminal domain. The deviation from the typical Rossmann-fold topology, characterized by an anti-parallel strand β5 at the edge of the β-sheet, opens up the side of the β-sheet to form the flavin-binding pocket. In the CgFMNAT product ternary complex structure, FAD adopts a bent conformation with adenosine moiety and isoalloxazine ring pack against each other. While the FAD cofactor in most flavoproteins adopts an extended conformation, bent FAD has been observed in flavodoxin reductase and DNA photolyase.31 Yet, the conformations of these bent FADs are very different from that in CgFMNAT (Fig. 8), emphasizing further the unique flavin-binding mode of eukaryotic FMNAT.
Figure 8. Comparison of protein bound FAD conformations.

Representative protein bound FAD molecules were superimposed over the isoalloxazine rings. FAD bound to CgFMNAT (yellow) is compared to FAD from (a) flavodoxin reductase (blue) (PDB code 1fdr);48 (b) DNA photolyase (magenta) (PDB code 1dnp);49 and (c) glutathione reductase (green) (PDB code 3grs).50
A comparison of the structures of CgFMNAT, a prototypical eukaryotic FMNAT, and bacterial FMNAT as exemplified by TmFADS34 revealed remarkable differences in substrate binding modes and in the catalytic site configurations. TmFADS belongs to the large nucleotidylyl transferase superfamily with the signature (H/T)xGH motif located between the end of the first β-strand and the first helix of the Rossmann-fold core. It has a different conformation from that of the corresponding PP-loop region of CgFMNAT (Fig. 9). The second conserved motif of the nucleotidylyl transferase superfamily, ISSTxxR, is located at the N-terminal end of an α-helix in a C-terminal subdomain and interacts with the β- and γ-phosphates of the ATP nucleotide. No equivalent structural motif corresponding to ISSTxxR motif exists in CgFMNAT, though the γ-Phosphate motif appears to perform a similar role. Most strikingly, the bound adenine nucleotides in the two proteins are orientated in opposite directions with regard to the plane of the central β-sheet (Fig. 9), delineating two completely different nucleotide-binding modes in these two protein superfamilies. Currently, no flavin-bound bacterial FMNAT structure is available. Based on the structural similarity of TmFADS to other members of nucleotidylyl transferases, such as nicotinamide mononucleotide adenylyltransferases (NMNAT) for which extensive structural information are available,35; 36; 37 the FMN substrate likely binds to a site corresponding to the NMN-binding site in NMNAT on the opposite side of the central β-sheet from ATP so that the product FAD adopts a largely extended conformation (Fig. 9, right). Again, this arrangement is very different from the flavin-binding mode observed in CgFMNAT (Fig. 9, left). Thus, eukaryotic and bacterial FMNAT present a remarkable case of ancient Rossmann-fold proteins that, after first diverging into two distinct protein families with different nucleotide-binding modes, have developed the same enzymatic activity via different active site configurations.
Figure 9. Comparison of eukaryotic and bacterial FMNAT structures.

Ribbon diagrams of the Rossmann-like fold core of the CgFMNAT-FAD complex (left) and the FMNAT domain of TmFADS (right) are shown in roughly the same orientation. An FAD molecule is modeled in the TmFMNAT active site based on the TmFADS-AMP complex structure (PDB identifier 1t6y)34 and homologous NMNAT-NAD complex structures. Corresponding structural elements are colored identically in the two structures.
In summary, we present here high-resolution structures of a eukaryotic FMN adenylyltransferase in different complexed states, which revealed details about the active site configuration and a unique FAD-binding mode. The extensive interactions observed between the enzyme and the product FAD, and the lack of conformational changes after catalysis may explain why the enzyme is likely inhibited by the product. These structures lay a foundation for future investigation of the functional roles of active-site residues and the mechanism by which FMNAT influences FAD homeostasis in cells.
MATERIALS AND METHODS
Protein Expression and Purification
The predicted gene encoding CgFMNAT (gi|50291750) was amplified from Candida glabrata genomic DNA (strain NCYC, ATCC36909D) by PCR and cloned into the NcoI and SalI restriction sites of the pHIS parallel vector.38 The plasmid was transformed into E. coli BL21(DE3) (Novagen), and the His6-CgFMNAT protein was expressed at 20 °C. His6-CgFMNAT was first loaded on a nickel-sepharose affinity column (GE Healthcare) equilibrated in Buffer A (20 mM HEPES, pH 8.0, 300 mM NaCl, 20 mM imidazole, 5% (v/v) glycerol and 1 mM DTT) and eluted with a gradient of 20–500 mM imidazole in Buffer A. The His6-tag was cleaved by tobacco etch virus (TEV) protease during overnight dialysis at 4 °C and was removed from CgFMNAT by passing through nickel-sepharose column a second time. As a second purification step, protein was loaded onto a Resource Q anion exchange column (GE Healthcare) equilibrated with Buffer B (20 mM HEPES, pH 7.5 and 5% (v/v) glycerol) and eluted with a 0–350 mM NaCl gradient, which yielded two pools. The first pool had a bright yellow color and was later shown to contain the CgFMNAT-FAD complex. The second pool had a light yellow color, indicating the presence of flavin with partial occupancy. To remove the flavin and obtain homogeneous apo-CgFMNAT, the second pool was incubated with 1.5 M (NH4)2SO4 and purified by phenyl-sepharose hydrophobic-interaction chromatography (GE Healthcare). The final purification step for both CgFMNAT-FAD and apo-CgFMNAT consisted of a size exclusion chromatography column (Superdex 75 16/60, GE Healthcare) and the protein was eluted in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM DTT. The selenomethionyl apo-CgFMNAT was expressed in E. coli BL21(DE3) grown in minimal media supplemented with selenomethionine and other nutrients, and purified using the same procedure as for apo-CgFMNAT.
Crystallization
All crystals were grown using hanging-drop vapor diffusion methods. For apo-CgFMNAT and ATP complex crystallization, the reagents and greased 24-well plates were chilled on ice before setting up crystallization drops. The apo-CgFMNAT crystals were grown by mixing 1.5 μl of protein (24 mg/ml in the gel filtration column buffer) with 1.5 μl of reservoir solution composed of 0.1 M Na acetate, pH 4.4–5.4 and 6–12% (w/v) PEG 4000, and equilibrating against the reservoir at 16°C. Prism-shaped apo-CgFMNAT crystals grew to a maximum size of 0.55 × 0.35 × 0.35 mm3 within several days. Selenomethionyl apo-CgFMNAT crystals were grown under similar conditions as the native protein. The CgFMNAT-ATP complex crystals were grown in the presence of 10 mM ATP under similar conditions as those for apo-CgFMNAT. The co-crystals of CgFMNAT substrate ternary complex were grown at 20 °C in the presence of 0.2 mM FMN and 5 mM AMPCPP under similar conditions as those for apo-CgFMNAT, except that consecutive micro-seeding procedure was performed in order to obtain single crystals. The CgFMNAT product ternary complex was obtained by adding 2 mM Na pyrophosphate to the CgFMNAT-FAD pool (final protein concentration ~20 mg/ml in gel filtration buffer). The complex crystals were grown by mixing 1.5 μl CgFMNAT-FAD-PPi complex with 1.5 μl reservoir solution composed of 0.1 M Na acetate, pH 4.4–5.4, 0.2 M MgSO4 and 20–26% (w/v) PEG MME 2000, and equilibrating against the reservoir at 20°C. All crystals were cryoprotected in a solution containing all the reservoir components and increments of glucose (10%, 20% and 30%), flash-frozen in liquid propane and stored in liquid nitrogen. For cryoprotecting the substrate ternary complex crystals, 5 mM MgSO4 was also added along with glucose.
Data collection, X-ray structure determination and refinement
The 2.18 Å single-wavelength anomalous diffraction (SAD) data from a selenomethionyl apo-CgFMNAT crystal was collected at the absorption edge (K-edge) of selenium at beam-line 19-BM; the 1.20 Å native apo-CgFMNAT and 1.35 Å CgFMNAT product ternary complex data sets were collected at beam-line 19-ID at the Advance Photon Source (APS), Argonne National Laboratory. The 1.87 Å dataset for the ATP complex and the 1.95 Å dataset for the substrate ternary complex were collected in-house with X-ray from a rotating anode generator (Rigaku FRE SuperBright), recorded on an RAXIS IV++ (Rigaku) image plate detector. All data were processed with either the HKL2000 or the HKL3000 package.39; 40 Data collection statistics are presented in Table 1. The initial phases for apo-CgFMNAT were obtained by the SAD phasing method (see Supplementary Methods for details), while the phases of all complexes were determined by the molecular replacement method using program MolRep41 and the refined apo-CgFMNAT as the search model. The refinements were performed using REFMAC42 or PHENIX,43 and manual model building was performed with Coot.44 Models were assessed by MolProbity.45
Steady-State Kinetics
Steady-state kinetic parameters were determined using a continuous coupled assay (EnzChek® pyrophosphate assay kit, Molecular Probes®) with modifications. The reaction mixture (0.5 ml) contained 20 mM HEPES, pH 7.5, 2 mM MgCl2, 0.5 units purine nucleoside phosphorylase, 0.015 units inorganic pyrophosphatase, 0.2 mM MESG (2-amino-6-mercapto-7-methylpurine riboside), 21 nM (10.5 pmol or 378 ng) apo-CgFMNAT, 2.5 to 20 μM FMN, and 1 to 250 μM ATP. All enzyme reactions were carried out in duplicates and performed at 25 °C. The reaction was initiated by the addition of ATP, and the progress was monitored at 360 nm for 5 minutes. Steady-state kinetic parameters were determined by fitting the initial rates to the general equation describing a bireactant ordered bi-bi system (1).32
| (1) |
KmATP and KmFMN are the Michaelis-Menten constants, and KiATP is the dissociation constant for an ATP-enzyme complex. A nonlinear least-squares method as implemented in the Sigma Plot Enzyme Kinetics module (Systat Software) was used to fit the data.
Accession codes
The coordinates and structures factors were deposited in the Protein Data Bank with accession codes 3FWK for apo-CgFMNAT, 3G59 for CgFMNAT·ATP complex, 3G5A for CgFMNAT·FMN·AMPCPP·Mg2+ complex and 3G6K for CgFMNAT·FAD·PPi·Mg2+ complex.
Supplementary Material
Acknowledgments
We thank Zbyszek Otwinowski for help with synchrotron data collection and initial SAD phasing. This work is supported by a grant from Welch Foundation (I-5105). C.H. is supported by National Institutes of Health Training Grant T32 GM008297. Results shown in this report are derived from work performed at Argonne National Laboratory (ANL), Structural Biology Center at the Advanced Photon Source. ANL is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Abbreviations
- FMNAT
flavin mononucleotide adenylyltransferase
- RFK
riboflavin kinase
- FADS
flavin adenine dinucleotide synthetase
- NMNAT
nicotinamide mononucleotide adenylyltransferase
- AMPCPP
α,β-methyleneadenosine 5′-triphosphate
- PAPS
3′-phospho-adenosine 5′-phosphosulfate
- APS
adenosine-5′-phosphosulfate
- SAD
single anomalous dispersion
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Massey V. The chemical and biological versatility of riboflavin. Biochem Soc Trans. 2000;28:283–96. [PubMed] [Google Scholar]
- 2.Müller F, editor. Chemistry and Biochemistry of Flavoenzymes. I. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 3.Joosten V, van Berkel WJ. Flavoenzymes. Curr Opin Chem Biol. 2007;11:195–202. doi: 10.1016/j.cbpa.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 4.van Berkel WJH. Wiley Encyclopedia of Chemical Biology. John Wiley & Sons, Inc; 2008. Aug 15, Chemistry of Flavoenzymes. [Google Scholar]
- 5.Merrill AH, Jr, Lambeth JD, Edmondson DE, McCormick DB. Formation and mode of action of flavoproteins. Annu Rev Nutr. 1981;1:281–317. doi: 10.1146/annurev.nu.01.070181.001433. [DOI] [PubMed] [Google Scholar]
- 6.Fischer M, Bacher A. Biosynthesis of flavocoenzymes. Nat Prod Rep. 2005;22:324–50. doi: 10.1039/b210142b. [DOI] [PubMed] [Google Scholar]
- 7.Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77:1352–60. doi: 10.1093/ajcn/77.6.1352. [DOI] [PubMed] [Google Scholar]
- 8.Vogl C, Grill S, Schilling O, Stulke J, Mack M, Stolz J. Characterization of riboflavin (vitamin B2) transport proteins from Bacillus subtilis and Corynebacterium glutamicum. J Bacteriol. 2007;189:7367–75. doi: 10.1128/JB.00590-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reihl P, Stolz J. The monocarboxylate transporter homolog Mch5p catalyzes riboflavin (vitamin B2) uptake in Saccharomyces cerevisiae. J Biol Chem. 2005;280:39809–17. doi: 10.1074/jbc.M505002200. [DOI] [PubMed] [Google Scholar]
- 10.Manstein DJ, Pai EF. Purification and characterization of FAD synthetase from Brevibacterium ammoniagenes. J Biol Chem. 1986;261:16169–16173. [PubMed] [Google Scholar]
- 11.Coquard D, Huecas M, Ott M, van Dijl JM, van Loon AP, Hohmann HP. Molecular cloning and characterisation of the ribC gene from Bacillus subtilis: a point mutation in ribC results in riboflavin overproduction. Mol Gen Genet. 1997;254:81–4. doi: 10.1007/s004380050393. [DOI] [PubMed] [Google Scholar]
- 12.Santos MA, Jimenez A, Revuelta JL. Molecular characterization of FMN1, the structural gene for the monofunctional flavokinase of Saccharomyces cerevisiae. J Biol Chem. 2000;275:28618–28624. doi: 10.1074/jbc.M004621200. [DOI] [PubMed] [Google Scholar]
- 13.Wu M, Repetto B, Glerum DM, Tzagoloff A. Cloning and characterization of FAD1, the structural gene for flavin adenine dinucleotide synthetase of Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:264–71. doi: 10.1128/mcb.15.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu MT, Wuebbens MM, Rajagopalan KV, Schindelin H. Crystal structure of the gephyrin-related molybdenum cofactor biosynthesis protein MogA from Escherichia coli. J Biol Chem. 2000;275:1814–22. doi: 10.1074/jbc.275.3.1814. [DOI] [PubMed] [Google Scholar]
- 15.Schrag JD, Huang W, Sivaraman J, Smith C, Plamondon J, Larocque R, Matte A, Cygler M. The crystal structure of Escherichia coli MoeA, a protein from the molybdopterin synthesis pathway. J Mol Biol. 2001;310:419–31. doi: 10.1006/jmbi.2001.4771. [DOI] [PubMed] [Google Scholar]
- 16.Karthikeyan S, Zhou Q, Mseeh F, Grishin NV, Osterman AL, Zhang H. Crystal structure of human riboflavin kinase reveals a beta barrel fold and a novel active site arch. Structure. 2003;11:265–73. doi: 10.1016/s0969-2126(03)00024-8. [DOI] [PubMed] [Google Scholar]
- 17.Cheek S, Zhang H, Grishin NV. Sequence and structure classification of kinases. J Mol Biol. 2002;320:855–881. doi: 10.1016/s0022-2836(02)00538-7. [DOI] [PubMed] [Google Scholar]
- 18.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 19.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–8. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efimov I, Kuusk V, Zhang X, McIntire WS. Proposed steady-state kinetic mechanism for Corynebacterium ammoniagenes FAD synthetase produced by Escherichia coli. Biochemistry. 1998;37:9716–23. doi: 10.1021/bi972817j. [DOI] [PubMed] [Google Scholar]
- 21.Yamada Y, Merrill AH, Jr, McCormick DB. Probable reaction mechanisms of flavokinase and FAD synthetase from rat liver. Arch Biochem Biophys. 1990;278:125–30. doi: 10.1016/0003-9861(90)90240-y. [DOI] [PubMed] [Google Scholar]
- 22.Tu BP, Weissman JS. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell. 2002;10:983–94. doi: 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- 23.Hustad S, Ueland PM, Vollset SE, Zhang Y, Bjorke-Monsen AL, Schneede J. Riboflavin as a determinant of plasma total homocysteine: effect modification by the methylenetetrahydrofolate reductase C677T polymorphism. Clin Chem. 2000;46:1065–71. [PubMed] [Google Scholar]
- 24.Gerdes SY, Scholle MD, D’Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, Campbell JW, Anantha S, Shatalin KY, Chowdhury SA, Fonstein MY, Osterman AL. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J Bacteriol. 2002;184:4555–4572. doi: 10.1128/JB.184.16.4555-4572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrosky-Zeichner L, Rex JH, Pappas PG, Hamill RJ, Larsen RA, Horowitz HW, Powderly WG, Hyslop N, Kauffman CA, Cleary J, Mangino JE, Lee J. Antifungal susceptibility survey of 2,000 bloodstream Candida isolates in the United States. Antimicrob Agents Chemother. 2003;47:3149–54. doi: 10.1128/AAC.47.10.3149-3154.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krcmery V, Barnes AJ. Non-albicans Candida spp. causing fungaemia: pathogenicity and antifungal resistance. J Hosp Infect. 2002;50:243–60. doi: 10.1053/jhin.2001.1151. [DOI] [PubMed] [Google Scholar]
- 27.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 28.Chartron J, Carroll KS, Shiau C, Gao H, Leary JA, Bertozzi CR, Stout CD. Substrate recognition, protein dynamics, and iron-sulfur cluster in Pseudomonas aeruginosa adenosine 5′-phosphosulfate reductase. J Mol Biol. 2006;364:152–69. doi: 10.1016/j.jmb.2006.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savage H, Montoya G, Svensson C, Schwenn JD, Sinning I. Crystal structure of phosphoadenylyl sulphate (PAPS) reductase: a new family of adenine nucleotide alpha hydrolases. Structure. 1997;5:895–906. doi: 10.1016/s0969-2126(97)00244-x. [DOI] [PubMed] [Google Scholar]
- 30.Mougous JD, Lee DH, Hubbard SC, Schelle MW, Vocadlo DJ, Berger JM, Bertozzi CR. Molecular basis for G protein control of the prokaryotic ATP sulfurylase. Mol Cell. 2006;21:109–22. doi: 10.1016/j.molcel.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 31.Dym O, Eisenberg D. Sequence-structure analysis of FAD-containing proteins. Protein Sci. 2001;10:1712–28. doi: 10.1110/ps.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley-Interscience; New York: 1993. [Google Scholar]
- 33.Galluccio M, Brizio C, Torchetti EM, Ferranti P, Gianazza E, Indiveri C, Barile M. Over-expression in Escherichia coli, purification and characterization of isoform 2 of human FAD synthetase. Protein Expr Purif. 2007;52:175–81. doi: 10.1016/j.pep.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Wang W, Kim R, Yokota H, Kim SH. Crystal structure of flavin binding to FAD synthetase of Thermotoga maritima. Proteins. 2005;58:246–8. doi: 10.1002/prot.20207. [DOI] [PubMed] [Google Scholar]
- 35.Saridakis V, Christendat D, Kimber MS, Dharamsi A, Edwards AM, Pai EF. Insights into ligand binding and catalysis of a central step in NAD+ synthesis: structures of Methanobacterium thermoautotrophicum NMN adenylyltransferase complexes. J Biol Chem. 2001;276:7225–32. doi: 10.1074/jbc.M008810200. [DOI] [PubMed] [Google Scholar]
- 36.Zhang H, Zhou T, Kurnasov O, Cheek S, Grishin NV, Osterman A. Crystal structures of E. coli nicotinate mononucleotide adenylyltransferase and its complex with deamido-NAD. Structure (Camb) 2002;10:69–79. doi: 10.1016/s0969-2126(01)00693-1. [DOI] [PubMed] [Google Scholar]
- 37.Zhou T, Kurnasov O, Tomchick DR, Binns DD, Grishin NV, Marquez VE, Osterman AL, Zhang H. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J Biol Chem. 2002;277:13148–54. doi: 10.1074/jbc.M111469200. [DOI] [PubMed] [Google Scholar]
- 38.Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr Purif. 1999;15:34–9. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–66. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 41.Teplyakov AVaA. MOLREP: an Automated Program for Molecular Replacement. Journal of Applied Crystallography. 1997;30:4. [Google Scholar]
- 42.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 43.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 46.Merritt EA. Expanding the model: anisotropic displacement parameters in protein structure refinement. Acta Crystallogr D Biol Crystallogr. 1999;55:1109–17. doi: 10.1107/s0907444999003789. [DOI] [PubMed] [Google Scholar]
- 47.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 2003;59:1131–7. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 48.Ingelman M, Bianchi V, Eklund H. The three-dimensional structure of flavodoxin reductase from Escherichia coli at 1.7 A resolution. J Mol Biol. 1997;268:147–57. doi: 10.1006/jmbi.1997.0957. [DOI] [PubMed] [Google Scholar]
- 49.Park HW, Kim ST, Sancar A, Deisenhofer J. Crystal structure of DNA photolyase from Escherichia coli. Science. 1995;268:1866–72. doi: 10.1126/science.7604260. [DOI] [PubMed] [Google Scholar]
- 50.Karplus PA, Schulz GE. Refined structure of glutathione reductase at 1.54 A resolution. J Mol Biol. 1987;195:701–29. doi: 10.1016/0022-2836(87)90191-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.