

Abstract
Objective
Pericytes are critical cellular components of the microvasculature that play a major role in vascular development and pathologies, yet their study has been hindered by lack of a standardized method for their isolation and growth. Here we report a method for culturing human pericytes from a readily available tissue source, placenta, and provide a thorough characterization of resultant cell populations.
Methods
We developed an optimized protocol for obtaining pericytes by outgrowth from microvessel fragments recovered after enzymatic digestion of human placental tissue. We characterized outgrowth populations by immunostaining, by gene expression analysis, and by functional evaluation of cells implanted in vivo.
Results
Our approach yields human pericytes that may be serially expanded in culture and that uniformly express the cellular markers NG2, CD90, CD146, α-SMA, and PDGFR-β , but lack markers of smooth muscle cells, endothelial cells, and leukocytes. When co-implanted with human endothelial cells into C.B-17 SCID/bg mice, human pericytes invest and stabilize developing human endothelial cell-lined microvessels.
Conclusions
We conclude that our method for culturing pericytes from human placenta results in the expansion of functional pericytes that may be used to study a variety of questions related to vascular biology.
Keywords: microvessel, endothelial cell, placenta, pericyte isolation
Introduction
Pericytes (PC) are contractile vascular cells that play an integral role in stabilizing and remodeling capillaries and other small-caliber vessels throughout life. PC loss or dysfunction is involved in numerous pathologies, such as hypertension, diabetic microangiopathy and tumor angiogenesis [6, 8, 5]. PC interact with endothelial cells (EC), the other major component of the microvasculature, both through paracrine growth factor- and cell-cell contact-dependent mechanisms [6, 10, 4]. While EC biology has progressed dramatically over the past three decades, largely due to the availability of a standardized system for the isolation and culture of human EC from umbilical veins [7, 15], much less is known about human PC biology. This is at least in part because most tissue sources for isolating PC are difficult to obtain and individual research groups have used a variety of complex methods for their isolation and culture.
PC have been routinely isolated from bovine retina, where PC coverage of the endothelium is particularly high [6]. However, many properties of vascular cells are species-dependent and increased access to human PC may significantly advance the field. While a similar approach may be applied to human retina, providing PC of particular interest for the study of diabetic retinopathy [5], this tissue is extremely scarce and not practical for routine isolation. Others have described culture of PC from human skeletal muscle, skin, and fetal tissues [1, 3, 14], but these sources are also limited. In general, regardless of the starting tissue material, PC isolation protocols rely on enzymatic tissue digestion followed by either positive immunoselection (with magnetic beads or by flow cytometry) or microvessel outgrowth. The former approach is complex, expensive and yields relatively low numbers of cells while the latter often results in mixed populations.
Human placenta is an abundantly available tissue source from which PC have been isolated [2, 11], yet the methods to do so have not been fully described and the resultant cell populations have not been fully characterized. Furthermore, isolates from such methods have been contaminated by other cell types and the yields were not reported. Nonetheless, this appeared to be a reasonable starting point for developing a standardized approach to PC isolation. Here we present a straightforward protocol for culturing human PC from placenta, demonstrating that the microvessel outgrowth populations acquired by our procedure are free of contaminating cell types, readily expand in culture, uniformly express characteristic PC markers in vitro, and behave functionally to stabilize EC-lined structures in vivo.
Materials and Methods
Culture of human pericytes
All human cells were obtained from discarded, anonymized tissue under protocols approved by the Yale Human Investigation Committee. Human placentas were obtained after vaginal delivery or caesarean section of healthy full-term infants and stored at 4°C for up to 48 hours before cell isolation. An approximately 10cm × 10cm × 3cm central section of human placental tissue, near the insertion of the umbilical cord, was manually dissected and freed from the amniotic sac under aseptic conditions. The tissue was washed three times in PBS and visible thrombi removed by aspiration. Washed tissue was minced continuously for 8–10 minutes with surgical scissors, placed into a sterile flask with an equal volume of Hanks Balanced Salt Solution (Gibco) containing 3mg/ml of collagenase type D (Sigma), and digested for 2 hours at 37°C on an orbital shaker at vigorous speed (180RPM). The digested suspension was first passed through a 100μm mesh filter (Falcon) to remove large vessel segments and fibrous tissue. The flow-through, containing microvessel fragments and single cells, was collected and passed a second time through a 40μm mesh filter (Falcon). The 40μm filter was then inverted over a 100cm tissue culture plate and reverse-washed with complete medium (Medium 199, Gibco, 20%FBS, 2mM L-glutamine, 100U/ml penicillin and 100ug/ml streptomycin) to collect microvessel fragments. Plates containing microvessels were left undisturbed for 7 days in a 37° C 95%O2/5%CO2 incubator in 8ml of medium to allow microvessel attachment and PC outgrowth.
On culture day 7 medium was carefully aspirated and replaced with 10ml of fresh complete medium, without dislodging adherent microvessel fragments. By day 10 extensive microvessel outgrowth colonies could be visualized by light microscopy, at which point cultures were washed twice with PBS and given fresh complete medium. Medium was changed every 48 hours thereafter until cells reached confluence, usually between culture days 14 and 21. Primary outgrowth cells were collected by aspirating medium, washing twice with PBS, and treating with 0.1% trypsin (Invitrogen) until detachment. Following trypsinization, cells were collected, centrifuged at 200 × g for 10 minutes, resuspended in complete medium, and passed a final time through a 40 μm mesh filter to remove residual microvessel fragments. Cells were thereafter propagated by passing them 1:2 and used between passages 1 and 8.
Isolation and culture of human endothelial and smooth muscle cells
Human umbilical vein EC were isolated and cultured as previously described[12] and used between passages 2 and 5. SMCs were isolated by explant outgrowth from human umbilical arteries. Artery segments were carefully dissected away from umbilical cords, cut into 1cm pieces, and placed into tissue culture flasks. SMC were propagated in Medium 199 containing 20%FBS, 2mM L-glutamine, 100U/ml penicillin and 100ug/ml streptomycin.
Immunophenotyping of cultured pericytes
Placental microvessel fragments were collected as described above and plated on glass slides for immunocytochemical analysis of primary outgrowth cells or serially propagated in complete medium on uncoated tissue culture plastic for flow cytometric characterization. For immunoflourescence microscopy, cells were fixed and stained with mouse anti-human primary antibodies (NG2 and Thy-1, Chemicon, Calponin and CD31 Abcam), then washed and stained with a fluorescently labeled goat anti-mouse secondary antibody (1:500, Invitrogen). Nuclei were counterstained with DAPI and appropriate isotype controls used in all experiments. Immunophenotyping of cell surface molecules was performed by labeling cells with directly conjugated antibodies (NG2, R&D, Thy-1, BioLegend, CD146, BD Biosciences, PDGFR-β , Abcam, CD31, CD34, CD14, CD45, Becton Coulter) and analysis by flow cytometry. For assessing intracellular protein expression, cells were fixed and permeabilized with Cytofix/Cytoperm Buffer (BD Biosciences), incubated with the primary antibody of interest (α-SMA, Novocastra, Calponin, SM22α and SMMHC, Abcam), stained with a species-appropriate fluorescently labeled secondary antibody (goat anti-mouse Alexa 488, 1:500, and goat anti-mouse Alexa 594, 1:500, Invitrogen), and collected by flow cytometry. All flow cytometry samples were acquired on an LSRII (BD Biosciences) and analyzed using FlowJo software (Tree Star). HUVEC and human umbilical artery SMC were analyzed in parallel for comparison.
Gene expression microarray
Primary placental outgrowth cells from three isolations were collected and plated onto 100mm tissue culture plates. RNA was isolated from subculture 1 cells that had reached 90% visual confluence. Replicate cultures were confirmed to express PC markers by immunostaining. Cells were rinsed in cold PBS and total RNA isolated using the RNEasy mini kit per the manufacturer’s instructions, including DNA-cleanup (Qiagen). Total RNA was used to generate labeled cRNA, which was purified using an Illumina TotalPrep RNA Amplification kit. Samples of cRNA were hybridized to Illumina Human 6 v3 Beadchip Arrays in accordance with the manufacturer’s recommendations (including appropriate controls). Briefly, samples were hybridized to the array platform for 16 hours in a hybridization oven, serially washed, stained with streptavidin-Cy3, washed again and dried. The array was scanned using an Illumina BeadArray reader and analyzed using the Beadstudio software (Illumina). Mean signal intensities were generated by background subtraction of negative controls. Detection signal p-values were used to infer whether a gene was highly expressed (p-value in all samples≤ 0.01), moderately expressed (p-value ≤ 0.05 in all three samples but ≥ 0.01 in at least one sample), or not expressed (p-value ≥ 0.05 in any sample). The entire array dataset can be found online at the NCBI GEO website.
Implantation of pericytes in vivo
PC were expanded as described above and, where indicated, retrovirally transduced to express EGFP. EGFP cDNA was inserted in the LZRSpBMN vector (provided by Dr. G.P. Nolan, Stanford University, Palo Alto, CA, USA). The retroviral vector DNA containing the EGFP coding sequence was directly transfected into the Phoenix packaging cell line by Lipofectamine 2000 (Invitrogen) and selected using 10 μg/ml puromycin (Calbiochem). Supernatants were collected in M199 medium, filtered using a 0.45 μm syringe filter (BD Biosciences), and used to transduce cultured cells in medium supplemented with 8 μg/ml polybrene (Sigma-Aldrich). Transduction efficiency, which routinely reached 55–65%, was assessed by flow cytometry.
Grafts for implantation were prepared by suspending HUVEC (3.0 × 106cells/ml final concentration) and/or PC (1.5 × 106cells/ml final concentration) in rat tail type I collagen (final concentration 1.5 mg/ml, BD Pharmingen) with 1X M199 (diluted from 10X, Gibco), 75 μg/ml fibronectin, 2.5% 1M HEPES, 2.2 mg/ml HCO3−, and pH neutralized to 7.4 with 1M NaOH. The cell-collagen mixture was injected into the interstices of a 8 mm × 8 mm square piece of non-woven PGA scaffold (Concordia Fibers), and the saturated scaffold placed into one well of a 24 well plate containing 300 μl of cell-collagen mixture. An additional 300 μl (600 μl total volume) of cell-collagen mixture was pipetted slowly into the well over the scaffold, such that the scaffold was completely immersed in collagen. Constructs were placed at 37°C for 15 minutes to allow for gel polymerization, after which time 500 μl 20%FBS/M199 was added to the well.
All procedures involving mice were conducted under protocols approved by the Yale University Institutional Animal Care and Use Committee following methods previously published by our group[12]. For subcutaneous implantation the PC and/or EC constructs were carefully liberated from the tissue culture well using forceps and bisected. Six to 10-week-old female C.B-17 SCID/bg mice were anesthetized with inhaled isoflurane, a right lateral 1.5cm incision was made, and a subcutaneous pocket created by blunt dissection. The grafts were then placed in the subcutaneous position on the abdominal wall and the incision closed using skin staples (Precise). Grafts were harvested at 5, 10, 15, 30 and 60 days, as indicated in the text.
Intravital microscopy
Anesthetized mice were given fluorescein isothiocyanate (FITC)-conjugated Ulex Europaeus Agglutinin 1 (Uea-1, Ulex, Vector) by intravenous administration via the jugular vein. FITC-Ulex was allowed to circulate for 30 minutes at which time the mice were euthanized and the skin overlying the implant was carefully dissected free, exposing the implant and underlying abdominal wall. Microvascular structures within the grafts were then imaged with a Nikon Microphot-FXA microscope fitted with a DAGE-MTI CCD-100 camera utilizing ATI multimedia imaging software.
Morphological analyses
For histological analyses the grafts, overlying skin, and underlying abdominal muscle were harvested and fixed in formalin for 12 to 24 hours. The harvested grafts were then paraffin embedded and cut into six μm sections for staining with hematoxylin and eosin (H&E) or relevant antibodies. Primary antibodies reactive with EGFP or α-SMA were used for enumeration and characterization vascular profiles (α-SMA, Novocastra, GFP, Abcam). FITC-Ulex was used for detection of human EC (Vector). A species-appropriate fluorochrome-conjugated secondary antibody (AlexaFluor 598, Invitrogen, AlexaFluor 588, JacksonImmuno) was used to detect the primary antibody. Vascular profiles were characterized by positive binding of FITC-Ulex in structures with an identifiable vascular lumen greater than 5μm.
Assessment of vessel permeability
Permeability of the microvessels within implanted grafts was determined using administration of Evans blue and quantitation of dye extravasation. Anesthetized mice were administered Evans blue (30 mg/kg body weight; approximately 150 μl per mouse) by tail vein injection. Evans blue was allowed to circulate for approximately 30 minutes, at which point the mice were euthanized by cervical dislocation. The thorax was rapidly opened and the heart exposed. The left ventricular apex was cannulated with a blunt 21G needle attached to polyethylene tubing and the right atrium cut. The animal was then perfused with 20 ml of 37oC PBS containing 300U/kg heparin. Visualization of hepatic blanching was used to confirm sufficient exsanguination and tissue perfusion. Grafts were excised and stored at −80C until analysis. Excised grafts were then desiccated at 55oC for 12 to 18 hours and dry tissue weight recorded. Evans blue was extracted from the desiccated tissue by immersion in formamide for 12 to 18 hours and quantified by absorbance at 610 nm (Abs610) compared to a standard curve of Evans blue Abs610.
Statistics
In vivo experiments assessing microvessel branching, number, and stability were conducted with at least 6 animals in each group at each time point, and similar results were seen in three independent experiments. Statistical analyses of microvessel branch points, densities, and permeabilities were performed with paired, two-tailed t-tests with Bonferroni post-hoc correction. Statistical analysis of mouse CD45 infiltration was performed using one-way ANOVA with both Kruskal-Walls and Dunn’s Multiple Comparison post-hoc test.
Results
Human microvessel fragments, collected by sieving enzymatically digested placental tissue, readily attach to tissue culture plastic. Outgrowth cell populations are visible by 5 to 7 days (fig. 1a). Extensive PC outgrowth is observed by day 10 and confluent monolayers are typically achieved between 14 to 21 days (fig. 1b). Outgrowth cells in primary culture express the contractile protein calponin (fig. 1c), the surface proteoglycan NG2 (fig. 1d), and Thy-1 (CD90) (fig. 1e), but do not express the vascular endothelial cell marker CD31 (fig. 1f). As many as 1–2 × 107 cells can be obtained in primary culture and these cells may be serially subcultured, for a minimum of 8 passages, yielding more than 1 × 109 cells per isolation. Primary cells that are serially passaged continue to express a characteristic PC phenotype that is distinct from other vascular cell types. Specifically, cultured PC express the surface molecules NG2, Thy-1, platelet-derived growth factor receptor-β (PDGFR-β ), and CD146 (fig. 2a) and the intracellular proteins α-smooth muscle actin (α-SMA) and calponin (fig. 2b). PC can be distinguished from smooth muscle cells (SMC) by lack of expression of smooth muscle 22-α (SM22-α ) and smooth muscle myosin heavy chain (SMMHC) (fig. 2b), markers of terminally differentiated SMC that are highly expressed by umbilical artery SMC (data not shown). Despite the lack of a positive selection step in our procedure, the resultant PC populations do not contain detectable contaminating CD31 or CD34 (EC markers), or CD45 or CD14 (leukocyte markers) expressing cells (fig. 2c).
Figure 1.
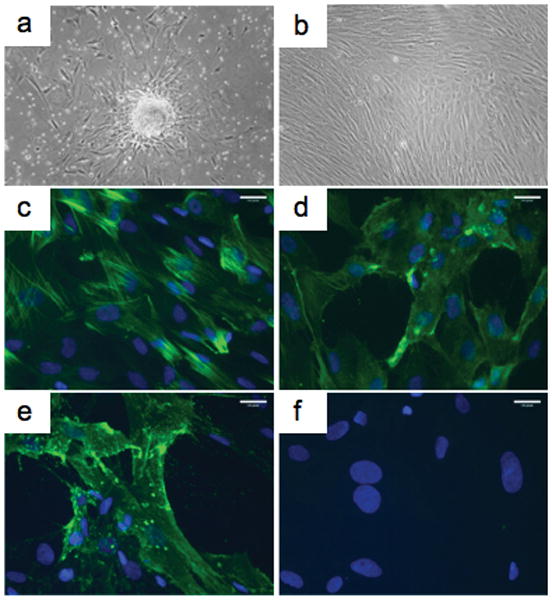
Characterization of primary outgrowth cells from human placental microvessels. Following enzymatic digestion, placental microvessel fragments readily attach to tissue culture plastic, with PC outgrowth evident by 7 days (a). Extensive PC outgrowth normally occurs after 10 to 14 days (b). PC express the contractile protein calponin (c), the surface proteoglycan NG2 (d) and Thy-1 (CD90) (e). Isolates were negative for CD31 (f). Specific antibody staining is shown in green and nuclei counterstaining with DAPI is blue (c–f). Scale bars represent 10 μm. One of eight experiments with similar results.
Figure 2.
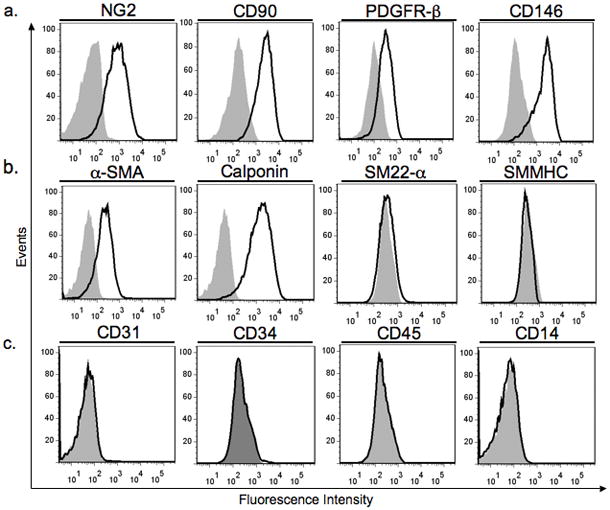
Flow cytometric characterization of cultured PC isolated from human placental microvessels. PC at subculture 4 uniformly express the cell surface molecules NG2, CD90, PDGFR-β , and CD146 (a) and the intracellular proteins α-SMA and calponin (b). PC do not express the intracellular proteins SM22-α or SMMHC (b), markers expressed by terminally differentiated umbilical artery SMC. PC isolates at subculture 1 do not contain cells expressing CD31 or CD34 (endothelial makers), or CD45 or CD14 (leukocyte markers) (c). Specific staining is shown by the black line (bold); isotype matched control staining is shown in grey (shaded). Similar results were observed in eight independent isolations.
PC have been identified as a tissue source of mesenchymal stromal cells (MSC) [3]. We therefore examined the relationship of our outgrowth populations to MSC by analysis of global gene expression profiles. Others have characterized human bone marrow derived MSC by gene expression arrays and identified those genes with differentially increased expression in undifferentiated and dedifferentiated MSC as compared to differentiated MSC, allowing for comparison with our own array [13]. Of the 74 genes previously identified, 65 were included on our Illumina platform. We found that our cells do share strong expression of a number of genes highly expressed by undifferentiated/dedifferentiated MSC (32/65, shown in Table IA). However, they fail to express 21 other strongly expressed MSC genes (Table IC). We conclude that our outgrowth population has not reverted to a more primitive MSC-like cell type.
Table 1.
Pericyte expression of 65 transcripts highly expressed in undifferentiated Mesenchymal Stromal Cells
| Probe ID | Gene Symbol | Description | Mean Signal Intensity ± SEM | Entrez Gene ID |
|---|---|---|---|---|
| A. HIGHLY DETECTED IN PC(32 TOTAL) | ||||
| ILMN_2360710 | TPM1 | Tropomyosin 1 | 26408.2 ± 2722.5 | 7168 |
| ILMN_2206746 | BGN | Biglycan | 15589.6 ± 505.5 | 633 |
| ILMN_1779228 | CDH2 | N-cadherin | 10852.9 ± 2166.7 | 1000 |
| ILMN_1744381 | SERPINE1 | Serpin peptidase inhibitor clade E member 1 | 9653.9 ± 1840.8 | 5054 |
| ILMN_1686116 | THBS1 | Thrombospondin 1 | 8538.7 ± 3575.5 | 7057 |
| ILMN_1680738 | C5orf13 | Chromosome 5 open reading frame 13 | 6713.7 ± 119.6 | 9315 |
| ILMN_1757387 | UCHL1 | Ubiquitin carboxyl-terminal esterase L1 | 6601.1 ± 1602.7 | 7345 |
| ILMN_1720048 | CCL2 | Chemokine (C-C motif) ligand 2 | 6508.5 ± 2607.3 | 6347 |
| ILMN_1779875 | THY1 | Thy-1 cell surface antigen, CD90 | 6003.5 ± 2542.6 | 7070 |
| ILMN_1699651 | IL6 | Interleukin 6 | 4967.8 ± 1428.4 | 3569 |
| ILMN_1764228 | DAB2 | Disabled homolog 2 mitogen-responsive phosphoprotein | 4137.6 ± 383.8 | 1601 |
| ILMN_2129927 | EXT1 | Exostoses 1 | 3856.0 ± 851.2 | 2131 |
| ILMN_1697220 | NT5E | 5'-nucleotidase ecto (CD73) | 3698.1 ± 271.8 | 4907 |
| ILMN_1770338 | TM4SF1 | Transmembrane 4 L six family member 1 | 3025.6 ± 446.48 | 4071 |
| ILMN_1758067 | RGS4 | Regulator of G-protein signalling 4 | 2169.0 ± 581.5 | 5999 |
| ILMN_2350634 | EFEMP1 | EGF-containing fibulin-like extracellular matrix protein 1 | 1994.2 ± 1682.1 | 2202 |
| ILMN_2146418 | CRIM1 | Cysteine rich transmembrane BMP regulator 1 | 1961.4 ± 208.7 | 51232 |
| ILMN_1724686 | CLDN1 | Claudin 1 | 1730.2 ± 1428.8 | 9076 |
| ILMN_2406084 | ITGA11 | Integrin alpha 11 | 1505.1 ± 849.0 | 22801 |
| ILMN_1676663 | TNFRSF11B | Osteoprotegerin | 1444.0 ± 304.6 | 4982 |
| ILMN_1797822 | SEL1L3 | Sel-1 suppressor of lin-12-like 3, KIAA0746 | 1377.0 ± 244.4 | 23231 |
| ILMN_1756439 | SCRN1 | Secernin 1 | 1024.0 ± 100.6 | 9805 |
| ILMN_1736939 | UGCG | UDP-glucose ceramide glucosyltransferase | 830.7 ± 21.76 | 7357 |
| ILMN_1707652 | KRTAP1-5 | Keratin associated protein 1-5 | 682.4 ± 653.3 | 83895 |
| ILMN_1741404 | MSC | Musculin | 674.2 ± 121.7 | 9242 |
| ILMN_1656335 | RIT1 | Ras-like without CAAX 1 | 591.6 ± 43.13 | 6016 |
| ILMN_2129234 | TMEM47 | Transmembrane protein 47 | 582.5 ± 587.1 | 83604 |
| ILMN_1765796 | ENO2 | Enolase 2 | 447.2 ± 88.43 | 2026 |
| ILMN_1758548 | NEK7 | NIMA (never in mitosis gene a)-related kinase 7 | 346.6 ± 31.22 | 140609 |
| ILMN_1691376 | JAG1 | Jagged 1 | 274.1 ± 37.83 | 182 |
| ILMN_1709026 | C6orf145 | Chromosome 6 open reading frame 145 | 243.2 ± 27.71 | 221749 |
| ILMN_1684620 | SPAG4 | Sperm associated antigen 4 | 241.3 ± 26.54 | 6676 |
| B.MODERATELY DETECTED IN PC(12 TOTAL) | ||||
| ILMN_1768940 | COL15A1 | Collagen type XV alpha 1 | 982.4 ± 877.6 | 1306 |
| ILMN_1722845 | RAB3B | RAS oncogene family member | 432.4 ± 319.7 | 5865 |
| ILMN_2131861 | SOCS2 | Suppressor of cytokine signaling 2 | 297.0 ± 127.5 | 8835 |
| ILMN_1665686 | FAM38B | Family with sequence similarity 38 member B | 236.7 ± 61.00 | 63895 |
| ILMN_1732197 | MN1 | Meningioma 1 | 207.2 ± 29.90 | 4330 |
| ILMN_1795275 | USP53 | Ubiquitin specific peptidase 53 | 185.1 ± 30.76 | 54532 |
| ILMN_2215119 | SYNJ2 | Synaptojanin 2 | 182.7 ± 4.80 | 8871 |
| ILMN_2350970 | SOCS5 | Suppressor of cytokine signaling 5 | 177.6 ± 7.88 | 9655 |
| ILMN_1838885 | KIAA1632 | KIAA1632 | 170.7 ± 1.753 | 57724 |
| ILMN_2240221 | SYTL2 | Synaptotagmin-like 2 | 159.8 ± 10.31 | 54843 |
| ILMN_1675429 | SMURF2 | SMAD specific E3 ubiquitin protein ligase 2 | 151.1 ± 8.87 | 64750 |
| ILMN_1725896 | AMIGO2 | Adhesion molecule with Ig-like domain 2 | 135.6 ± 3.31 | 347902 |
| C.NOT DETECTED IN PC(21TOTAL) | ||||
| ILMN_2113490 | NTN4 | Netrin 4 | 200.6 ± 54.91 | 59277 |
| ILMN_2048477 | SLC22A3 | Solute carrier family 22 member 3 | 172.7 ± 37.18 | 6581 |
| ILMN_2118832 | RBM24 | RNA binding motif protein 24 | 169.8 ± 30.08 | 221662 |
| ILMN_1672350 | JAM2 | Junctional adhesion molecule 2 | 167.1 ± 30.41 | 58494 |
| ILMN_1695041 | GATA6 | GATA binding protein 6 | 158.9 ± 19.31 | 2627 |
| ILMN_1710523 | ATP8B1 | ATPase class I type 8B member 1 | 132.5 ± 6.34 | 5205 |
| ILMN_1729409 | DLC1 | Deleted in liver cancer 1 | 124.4 ± 6.43 | 10395 |
| ILMN_1719547 | INHBA | Inhibin beta A | 123.9 ± 5.61 | 3624 |
| ILMN_2263086 | NTM | Neurotrimin | 121.2 ± 2.37 | 50863 |
| ILMN_1787548 | HSPG2 | Heparan sulfate proteoglycan 2 | 119.1 ± 4.85 | 3339 |
| ILMN_1810093 | TSPAN2 | Tetraspanin 2 | 116.5 ± 3.75 | 10100 |
| ILMN_1699520 | SLC8A1 | Solute carrier family 8 member 1 | 114.1 ± 5.05 | 6546 |
| ILMN_1723921 | C10orf30 | Chromosome 10 open reading frame 30 | 113.9 ± 1.58 | 222389 |
| ILMN_1742307 | MEST | Mesoderm specific transcript homolog | 112.9 ± 3.48 | 4232 |
| ILMN_1740696 | MAP2K3 | Mitogen-activated protein kinase kinase 3 | 112.5 ± 4.46 | 5606 |
| ILMN_2250344 | DACT1 | Dapper homolog 1 | 109.9 ± 2.77 | 51339 |
| ILMN_1803318 | PTPRF | Protein tyrosine phosphatase receptor type f | 107.3 ± 5.16 | 5792 |
| ILMN_1719820 | BDNF | Brain-derived neurotrophic factor | 106.4 ± 6.83 | 627 |
| ILMN_1775830 | TM4SF20 | Transmembrane 4 L six family member 20 | 100.6 ± 2.14 | 79853 |
| ILMN_1660424 | AFAP1 | Actin filament associated protein 1 | 97.5 ± 1.62 | 60312 |
| ILMN_1675092 | VLDLR | Very low density lipoprotein receptor | 94.5 ± 2.95 | 7436 |
Gene expression profiles of human placental PC populations from three isolates at subculture 1 as described in the Methods. Mean Signal Intensities (MSI) ± SEM are background subtracted expression measurements as determined by Illumina Microarray BeadChips. The 65 transcripts analyzed here were all highly expressed in undifferentiated or poorly differentiated MSC based on ref 13. PC transcripts for which the signal intensity detection p-value is ≤ 0.01 in all three isolates are considered highly detected, whereas transcripts for which the signal intensity detection p-value is ≤ 0.05 in all three isolates but is ≥ 0.01 in at least one isolate are considered moderately detected, and transcripts for which the signal intensity detection p-value is ≥ 0.05 in any of the three isolates are considered not detected.
To further characterize PC isolated from placental microvessels we examined the ability of these cells to stabilize human EC-lined microvessels in vivo. To do so, we co-engrafted placental PC and/or human umbilical vein EC (HUVEC) into poly(glycolic acid) (PGA)-supported collagen/fibronectin gels and implanted the gels subcutaneously into the abdominal walls of immunodeficient C.B-17 SCID/bg mice as previously described [12]. PGA support was required as PC, like SMC, will otherwise contract the gel resulting in loss of cell viability. All implants containing EC formed erythrocyte-containing microvessel-like structures. Intravital fluorescence microscopy of implants at 30 days revealed a more highly arborized microvessel architecture in gels containing EC plus PC as compared to gels containing EC alone, as evidenced by an increased number of branch points in microvessels within implants (fig. 3a–b).
Figure 3.
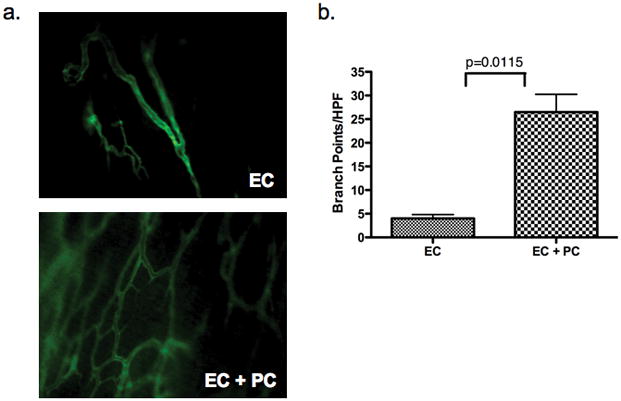
Co-engraftment of PC with EC increases microvascular network formation in vivo. Intravital microscopy with the use of intravenous injection of FITC-labeled Ulex was used to evaluate the microvascular network of human EC-lined vessels present in protein gel implants containing EC alone or EC plus PC after 30D (a), 10X magnification. Quantification of branch points per high power field (HPF) reveals increased vessel arborization in implants containing EC plus PC versus EC-only implants (b). Values represent average branch point counts in twelve grafts (six per condition). One of three experiments with similar results.
We next assessed PC investment of EC-lined microvessels at various time points by immunohistochemical analysis of paraffin-embedded sections from EC/PC implants. To aid in the identification of human PC and distinguish these cells from host-derived (mouse) stromal cells, enhanced green fluorescent protein (EGFP)-transduced PC were used in these studies. At 15 days post-implantation, EGFP-positive cells surrounded the walls of human EC-lined microvessels (fig. 4a and, at higher magnification, in 4b), confirming that these cells were engrafted human PC and not host-derived mesenchymal cells. Engrafted EGFP-positive PC remained present and in association with the microvessel walls at 30 days post-implantation (fig. 4c). Implants were noted to contain many cells that were neither Ulex nor GFP-positive (fig. 4a, c). Staining with mouse CD45 revealed many positive inflammatory cells infiltrating the EC/PC grafts as well as EC-only grafts (Supplemental fig. 1), a finding consistent with our previous observation that PGA induces an inflammatory response [12]. However, the presence of PC did not appear to influence the extent of leukocytic infiltration, as EC-only and EC/PC grafts contained significantly indistinguishable numbers of mouse CD45+ cells (Supplemental fig. 1).
Figure 4.
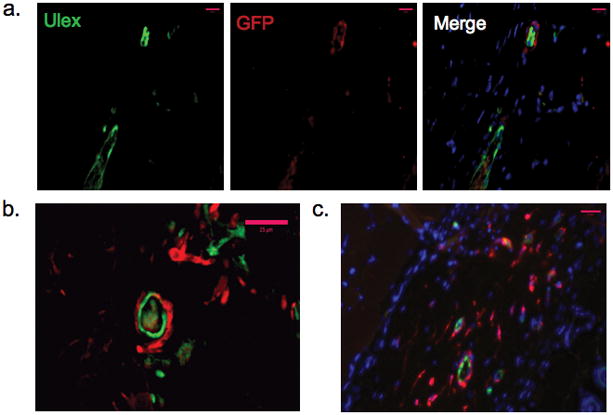
Investment of the microvascular wall by engrafted PC. EGFP-positive PC (stained red), co-engrafted with EC, could be found investing the wall of human EC-lined (green, Ulex-positive) microvessels after 15D (a–b). Panels show single color immunoflourescence and then a color merge in a. Engrafted EGFP-positive PC (stained red) remained present in the wall of human EC-lined microvessels at 30D post-implantation (c). Pink scale bars represent 25 μm. One of three experiments with similar results.
We then compared implants containing or lacking PC for vessel density, fragility, and permeability. Histologic analysis of grafts containing PC alone showed sparse host vessels at 15 days that disappeared by 30 days, despite persistence of engrafted PC within the constructs at 15 and 30 days, suggesting that engrafted human PC cannot support a host (mouse)-derived angiogenic response (fig. 5a–d). Comparison of EC-only and EC/PC grafts harvested at 5, 10, and 15 days post-implantation revealed that engrafted PC decrease microvessel fragility, as indicated by a reduction in extravascular erythrocytes at day 10 and an absence at day 15 (fig. 6a–f). Quantification of microvessel density revealed a statistically significant increase in the number of microvessels at 5D and 10D in grafts containing PC and EC versus grafts containing EC-alone (fig. 7). To test the functional maturity of developing microvessels we measured vessel permeability using the albumin-binding dye Evans blue. Inclusion of PC in the implants results in a statistically significant reduction in Evans blue extravasation at day 10 (fig. 8). This may actually underestimate the effect of PC since PC-containing implants have a greater number of vessels. To correct for this, we also normalized the amount of Evans blue extravasation to the vessel density (fig 8). We therefore conclude that the cultured placental outgrowth cells isolated by our method behave as PC in vivo by investing, stabilizing, and maturing developing microvessels.
Figure 5.
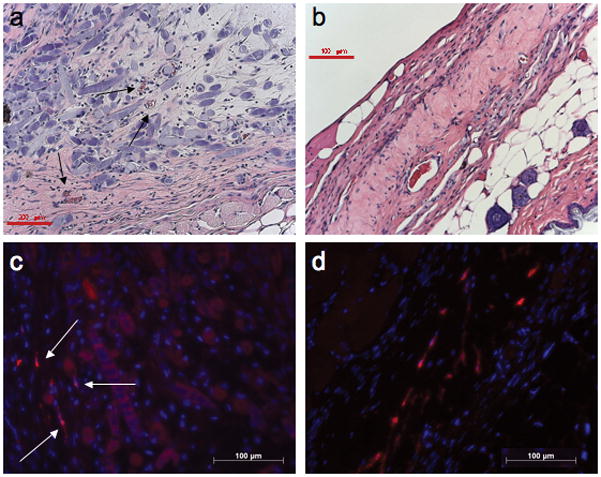
Gel implants containing PC alone do not become vascularized. Limited vessels were present after 15D (a, arrows) with still fewer microvessels found at 30D (b) in PGA-supported collagen gels containing only PC. Red scale bar represents 200 μm in panel a and 100 μm in panel b. Despite a lack of microvessel development, PC could still be identified within the implanted constructs at 15D (c, arrows) and 30D (d) by immunofluorescent detection of EGFP (stained red). White scale bars represent 100 μm in panels c and d. One of three experiments with similar results.
Figure 6.

PC co-engraftment with EC increases microvessel stability. Sections harvested after 5D reveal few microvessels present in EC-only implants (a) but more robust vascularization in EC/PC implants (b). Microvessels in EC-only grafts remain fragile EC-lined tubes with extravascular erythrocytes present at 10D (c), but co-engraftment with PC leads to PC investment of microvessel walls (arrows) and increased vessel number (d). After 15 days numerous, simple EC-lined microvessels are present in EC-only grafts (e), while EC/PC grafts have well-differentiated microvessels (arrows) and reduced numbers of extravascular erythrocytes (f). Scale bars represent 100 μm. One of three experiments with similar results.
Figure 7.
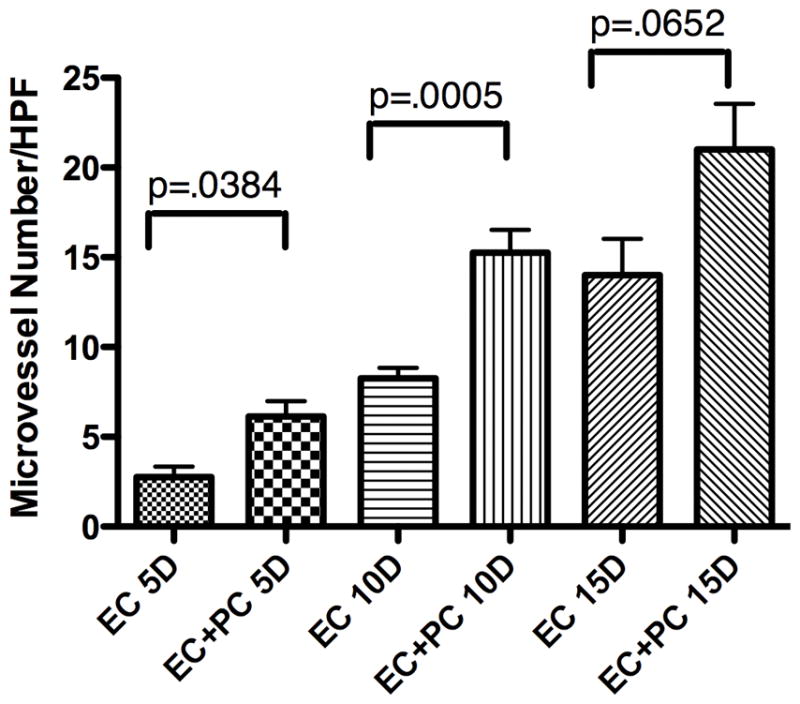
PC co-engraftment with EC increases microvessel number. Quantification of microvessel density demonstrates a significant increase in vessel number in grafts containing EC plus PC versus EC alone at 5 and 10 days post-implantation. Values reflect the average number of vessels per five high power fields (HPF) analyzed in six different grafts per condition. One of three experiments with similar results.
Figure 8.
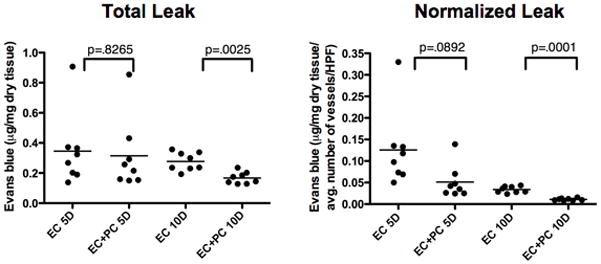
Pericytes decrease human microvessel permeability in vivo. Tissue content of Evans blue was quantified in gel implants at 5 and 10 days post-implantation, demonstrating decreased permeability in gels containing EC plus PC versus EC alone at 10D. Graphs show quantification of total Evans blue as well as after normalization to microvessel number. One of three experiments with similar results.
Discussion
Here we report a simple method for culturing human PC from placental microvessels, without a need for positive cell selection and resulting in a homogenous primary culture population from which a large number of expanded cells can be generated. In our protocol, PC outgrowth is preferentially encouraged by the choice of medium and optimization of conditions for microvessel attachment, a prerequisite for efficient outgrowth. Key features of our method that differ from previously published reports include 1. collagenase digestion of placenta in a balanced salt solution, instead of medium, for only 2 hours with rigorous agitation; 2. two sequential sieving steps to remove larger tissue fragments and then single cells, collecting only appropriately sized microvessel fragments (40–100 microns) from the digested suspension; 3. minimized plating volume and delayed washing and medium change to increase microvessel attachment; 4. thorough phenotypic characterization of primary outgrowth cells in vitro; and 5. extensive characterization of the expanded cells including demonstration that they behave functionally as PC in vivo. The technical aspects and tools required for our protocol are simple, thereby making the collection and culture of these cells feasible for any cell biology laboratory.
A major shortcoming of previously reported PC isolation techniques is the presence of contaminating cell populations and the inability to distinguish, by phenotype, some of these contaminating cells from PC. This fact required investigators to either positively sort cells after primary tissue digestion or serially propagate resultant populations prior to use in assays so that contaminating populations would die off. We found that PC isolated by our method uniformly express characteristic surface and intracellular proteins and lack both EC and SMC markers, as early as the first passage in culture. Furthermore, subculture one isolates are free from leukocytes, as indicated by absence of cells expressing CD14 or CD45, and hematopoietic stem cells as indicated by absence of cells expressing CD34. While our PC do, as expected, express the SMC markers α-SMA, Thy-1, and calponin, there is minimal to no expression of SMMHC and SM22-α , markers of terminally differentiated SMC that are expressed by SMC isolated from umbilical artery segments in primary and early subcultures. The cells obtained by our protocol continue to maintain a high proliferative capacity and preserve their characteristic phenotype over many passages in culture, as well as after cryopreservation. In addition, microarray gene expression analysis suggests that our outgrowth cell population has not dedifferentiated to a more primitive MSC phenotype in culture [13].
This is the first report, to our knowledge, demonstrating that human PC acquired by any method can act functionally as PC in vivo, evidenced in our study by the ability to invest and stabilize developing human microvessels. Cultured placental PC co-engrafted with human EC into collagen gels accelerate microvessel formation and stability when implanted subcutaneously over the abdominal wall of immunodeficient mice. A previous report from our group demonstrated increased vessel arteriolization and development of larger calibur vessels when cultured SMC were co-engrafted with EC in a similar model [12]. This is distinct from what we find here when PC are co-engrafted with EC, where PC support the development of a highly arborized microvasculature with improved small vessel patterning. This in vivo ability is appropriate given the normal physiologic role of PC in facilitating capillary network stabilization during new vessel formation and vessel remodeling. Not only did PC associate with and invest EC-lined human microvessels in our model, but their presence also correlated with decreased extravasation of erythrocytes and decreased microvessel permeability, both indicators of vessel stability and maturation.
As with many cell types, PC isolated from different tissues may behave differently in culture and in different research models. While this variability and plasticity are attractive for PC application in fields such as tissue engineering, there exists a real need to define a standard PC population to improve experimental rigor and to offer a comparative reference point against which other perivascular cells are described. Even within the tissue engineering field “cell sourcing and cell/tissue characterization” was among the top three concepts identified as an area of priority in order to achieve clinical success within the next two decades [9]. The utilization of what seems to be a limitless human tissue, placenta, as the tissue source for such a standardized population should provide all interested investigators the ability to study these cells in a variety of experimental systems. An additional advantage of using placenta as the starting material is that EC and SMC can be expanded from the umbilical cord of the same donor for comparison and for studies of vascular cell interactions.
In conclusion, we describe an efficient and simple method for obtaining well differentiated human PC that may be used to better define the functions that this cell type serves in the microcirculation.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Cathy Shanahan for sharing her experiences related to pericyte isolation and biology that provided the basis from which our optimized protocol was developed. We greatly appreciate the help of Aiping (Amy) Lin and the Keck Foundation Biotechnology Resource Laboratory at Yale for technical and biostatistical assistance with microarray acquistion and interpretation. We also thank Gwendolyn Arrington, Louise Benson, and Lisa Gras for assistance in placenta acquisition and endothelial cell culture. This work was funded by NIH grants R01-HL051014, R01-HL085416 and T32-GM-007205.
This work was funded by NIH grants R01-HL051014, R01-HL085416 and T32-GM-007205.
References
- 1.Bryan BA, D’Amore PA. Pericyte isolation and use in endothelial/pericyte coculture models. Meth Enzymol. 2008;443:315–331. doi: 10.1016/S0076-6879(08)02016-8. [DOI] [PubMed] [Google Scholar]
- 2.Churchman AT, Siow RCM. Isolation, culture, and characterization of vascular smooth muscle cells. Methods Mol Biol. 2009;467:127–138. doi: 10.1007/978-1-59745-241-0_7. [DOI] [PubMed] [Google Scholar]
- 3.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, Norotte C, Teng PN, Traas J, Schugar R, Deasy BM, Badylak S, Buhring HJ, Giacobino JP, Lazzari L, Huard J, Peault B. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E, Huang J, Scheppke L, Stockmann C, Johnson RS, Angle N, Cheresh DA. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456:809–813. doi: 10.1038/nature07424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammes HP. Pericytes and the pathogenesis of diabetic retinopathy. Horm Metab Res. 2005;37:39–43. doi: 10.1055/s-2005-861361. [DOI] [PubMed] [Google Scholar]
- 6.Hirschi KK, D’Amore PA. Pericytes in the microvasculature. Cardiovasc Res. 1996;32:687–698. [PubMed] [Google Scholar]
- 7.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 9.Johnson PC, Mikos AG, Fisher JP, Jansen JA. Strategic Directions in Tissue Engineering. Tissue Engineering. 2007;13:2827–2837. doi: 10.1089/ten.2007.0335. [DOI] [PubMed] [Google Scholar]
- 10.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 11.Proudfoot D, Skepper JN, Shanahan CM, Weissberg PL. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of Osteopontin expression. Arterioscler Thomb Vasc Biol. 1998;18:379–388. doi: 10.1161/01.atv.18.3.379. [DOI] [PubMed] [Google Scholar]
- 12.Shepherd BR, Jay SM, Saltzman WM, Tellides G, Pober JS. Human aortic smooth muscle cells promote arteriole formation by coengrafted endothelial cells. Tissue Eng Part A. 2009;15:165–173. doi: 10.1089/ten.tea.2008.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song L, Webb NE, Song Y, Tuan RS. Identification and Functional Analysis of Candidate Genes Regulating Mesenchymal Stem Cell Self-Renewal and Multipotency. Stem Cells. 2006;24:1707–1718. doi: 10.1634/stemcells.2005-0604. [DOI] [PubMed] [Google Scholar]
- 14.Sundberg C, Kowanetz M, Brown LF, Detmar M, Dvorak HF. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Laboratory Investigation. 2002;82:387–401. doi: 10.1038/labinvest.3780433. [DOI] [PubMed] [Google Scholar]
- 15.Thornton SC, Mueller SN, Levine EM. Human endothelial cells: use of heparin in cloning and long-term serial cultivation. Science. 1983;222:623–625. doi: 10.1126/science.6635659. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.