

Abstract
Background
Alterations on glucose consumption and biosynthetic activity of amino acids, lipids and nucleotides are metabolic changes for sustaining cell proliferation in cancer cells. Irrevocable evidence of this fact is the Warburg effect which establishes that cancer cells prefers glycolysis over oxidative phosphorylation to generate ATP. Regulatory action over metabolic enzymes has opened a new window for designing more effective anti-cancer treatments. This enterprise is not trivial and the development of computational models that contribute to identifying potential enzymes for breaking the robustness of cancer cells is a priority.
Methodology/Principal Findings
This work presents a constraint-base modeling of the most experimentally studied metabolic pathways supporting cancer cells: glycolysis, TCA cycle, pentose phosphate, glutaminolysis and oxidative phosphorylation. To evaluate its predictive capacities, a growth kinetics study for Hela cell lines was accomplished and qualitatively compared with in silico predictions. Furthermore, based on pure computational criteria, we concluded that a set of enzymes (such as lactate dehydrogenase and pyruvate dehydrogenase) perform a pivotal role in cancer cell growth, findings supported by an experimental counterpart.
Conclusions/Significance
Alterations on metabolic activity are crucial to initiate and sustain cancer phenotype. In this work, we analyzed the phenotype capacities emerged from a constructed metabolic network conformed by the most experimentally studied pathways sustaining cancer cell growth. Remarkably, in silico model was able to resemble the physiological conditions in cancer cells and successfully identified some enzymes currently studied by its therapeutic effect. Overall, we supplied evidence that constraint-based modeling constitutes a promising computational platform to: 1) integrate high throughput technology and establish a crosstalk between experimental validation and in silico prediction in cancer cell phenotype; 2) explore the fundamental metabolic mechanism that confers robustness in cancer; and 3) suggest new metabolic targets for anticancer treatments. All these issues being central to explore cancer cell metabolism from a systems biology perspective.
Introduction
In recent years we have witnessed significative advances for identifying and understanding the role that individual genes have in genesis, development and progression on cancer [1]. However, despite significant advances in genomic sciences in identifying oncogenes and tumor suppressors, a systemic explanation of how these genes deregulate the normal function of genetic circuits and how its control may be used to design effective drugs against cancer still remains a great challenge in systems biology [2], [3], [4], [5], [6].
In conjunction with this molecular view of cancer, detailed studies monitoring the metabolic alterations in cells are a promising avenue for understanding and controlling cell proliferation in cancer cells [2], [7], [8]. For instance, researchers have extensively studied the p53 tumor suppressor's ability to trigger DNA repair, cell cycle arrest and apoptosis, but recently p53's capacity to influence mitochondrial respiration and energy metabolism have been elucidated [9], [10]. Similarly, enhanced effect on glycolysis, lactate (lac) production and control of fatty acids oxidation originated by Hypoxia inducible factors (HIF) and LKB1 tumor suppressor are clear examples linking genes expression, metabolism and cancer phenotype [3].
In this contextual scheme, development of computational procedures capable of surveying the physiological responses on cancer cells in terms of its metabolic topology and genetic information constitutes an attractive strategy for understanding, characterizing, designing and improving effectiveness of cancer drugs [11]. In this work we present a constraint-based analysis of a metabolic network integrated by a core of metabolic pathways participating in cancer cell growth: glycolysis, TCA cycle, pentose phosphate pathways (PPP) and oxidative phosphorylation. Constraint-based modeling has proven to be a successful paradigm in systems biology for describing and exploring the phenotype capacities for a variety of organisms based on its particular genome sequences and metabolic topology [11], [12], [13], [14], [15].
This paper's central aim is twofold: 1) the construction of a model simulating metabolisms that serves as a computational framework auxiliary to describe and understand physiological behavior in cancer cells; and 2) the identification of potential metabolic targets to induce a reduced phenotype on cancer cell growth. To qualitatively assess the in silico results obtained from our metabolic reconstruction with those experimentally observed, we accomplished a study of growth kinetics for Hela cell line. Furthermore, based on computational criteria we identified some enzymes with a relevant influence on cell growth and compared them with those considered as potential therapeutic targets in the literature.
Overall, we supply evidence that constraint-based modeling can be used as a platform for unraveling the biochemical mechanism underlying cancer cell growth and potentially contribute toward designing strategies for clinical treatments in cancer.
Results
Core Metabolism in cancer cells
Ever since the pioneering observation that aerobic glycolysis in cancer [7] is preferred over oxidative phosphorylation as a mechanism to generate ATP from glucose, numerous experiments have supported and extended the significant role that metabolisms have on transformation, proliferation, angiogenesis and metastasis in cancer [16], [17], [18]. Thus, scanning human tumors with positron emission tomography (PET) [17] has verified that a high uptake rate of glucose constitutes a hallmark in cancer cells, presumably required to confer adaptive advantages when facing acidic and hypoxic environments [19].
In light of these observations, an explanation of why energy production relies on glycolysis instead of on the more effective pathway driven by oxidative phosphorylation in mitochondria [2], [16] requires computational models capable of taking into account not only both pathways but a robust metabolic network containing its metabolic interconnectivity.
Keeping in mind this systemic view, we constructed a metabolic network with those metabolic pathways that have a pivotal role in cancer cell growth: glycolysis, TCA cycle, pentose phosphate, glutaminolysis and oxidative phosphorylation [3], [16]. According to reconstruction protocols, our network was based on published knowledge about metabolism in cancer cells, basic thermodynamics and compartmentalization information associated with each metabolic reaction inside the cell, see Table S1. Thus, for example, studies on C13 NMR spectroscopy have demonstrated that glutaminolysis constitutes an active metabolic pathway in human glioblastoma cell lines [8], and consequently, a demand reaction of α-ketoglutarate representing an intermediary compound along the conversion of glutamine to lactate was included in the reconstruction. In addition, the reconstruction was complemented by transport reactions to resemble the physiological conditions prevailing in cancer cells, particularly those associated with glucose consumption, lactate production and hypoxia conditions, see Table S1. Overall, our reconstruction integrates 66 metabolites participating in 80 metabolic reactions representing glycolysis, pentose phosphate, TCA cycle, oxidative phosphorylation and glutaminolysis, as well as transport reactions of essential metabolites for cellular proliferation, specifically oxygen, hydrogen, carbon dioxide and water, see Table S1 in supplementary material. Figure 1 depicts the metabolic network used in this study. Mathematical representation of this set of reactions, through the stoichiometric matrix, constitutes our central platform for exploring and estimating the metabolic capacities potentially driving cancer cells [3], [16].
Figure 1. Metabolic pathways with a significant role in cancer cells.

As a result of a bibliography search, we have selected those metabolic pathways that potentially can constitute a metabolic core on most cancer cells. Orange, red and green dashed lines indicate metabolites that participate in other biosynthetic pathways, metabolites that can be transported from cytoplasm to mitochondrion and metabolites that can be transported from mitochondrion to cytoplasm, respectively. Compartment information has been denoted by external environment [e], cytoplasm [c] and mitochondria [m]. The set of reactions that integrates this reconstruction are listed in Table S1.
Dynamic Constraint-based Modeling and its experimental assessment
Experimental assessment of the results and hypothesis inferred from computational modeling is needed to ensure a high-quality metabolic reconstruction with a real scope for explaining and predicting cell behavior. Given that self-sufficiency in growth signals and mechanisms for evading apoptosis [20] in cancer cells contribute to uncontrolled cell proliferation, the feasibility of our model to simulate cancer cell growth constituted a principal issue to evaluate. Therefore, dynamic constraint-based modeling was applied to the metabolic reconstruction depicted in Figure 1 . According to this formalism, growth rate is calculated by assuming the existence of a characteristic time scale, at which a steady state condition for metabolite concentrations is a plausible assumption. Thus, hypothesizing that physiological growth rate at each time scale obeys optimization principles, linear programming was applied to identify the metabolic flux profile that maximized a function associated with growth rate [21], see methods section.
Malignant progression requires proper metabolic cell machinery in order to supply the energy and biosynthetic demand required for cancer cell growth. To quantify the cancer cell growth in terms of its metabolic networks and to link the topology of the reconstruction with the cancer cell physiology, we proceeded to construct an objective function that mathematically represents the metabolic demands required for successful cell growth [11], [13], [22], [23].
The proper selection of an objective function is crucial for reducing the steady-state stoichiometrically feasible solution to an optimal solution space [22], [24]. In this work the objective function was created by taking into account the expected metabolites supporting cancer cell proliferation [25]. Thus, based on a review of literature and considering the set of metabolites integrating our reconstruction, we suggest an objective function consisting of lactate (lac), ATP, ribose 5-phosphate (r5p), oxaloacetate (oaa) and citrate(cit) production, having been selected according to their fundamental roles as 1) precursors required for energy production, 2) precursors of amino acids and nucleotides and 3) intermediates in maintaining glycolysis and the reductive power required for biosynthesis of other cellular compounds [25], [26]:
where c, e and m denote the compartments utilized in the reconstruction (cytoplasm, external environment and mitochondria respectively).
In addition, proper computational representation of environmental conditions is essential to obtain reliable results and interpretations from in silico procedures [24]. Hence, sink and demand reactions were included to define proper metabolic boundaries to mimic the physiological conditions prevailing around cancer cells, see Table S1. Through sink reactions (which serve to introduce those metabolites that are produced or consumed by nonmetabolic cellular processes), we represent NADH, NAD, CO2, biphosphate, hydrogen, water, carbon dioxide, coenzyme A, FAD and FADH2. In turn, through demand reactions (which are unbalanced reactions that permit the accumulation of a compound otherwise not allowed in steady-state models because of mass-balancing requirements), we were able to include a source of ACCOA, ADP and oxygen.
Furthermore, plasma, an abundant source of glucose and glutamine in cancer cells, was represented by two demand reactions in the reconstruction, see Figure 1 and Table S1. In order to simulate glucose consumption, a simple transport of glucose was included in the metabolic reconstruction, while consumption on glutamine was represented through an external source of 2-oxoglutarate, one of the intermediary products of the glutaminolysis pathway in cancer cells, see Figure 1 [8].
Finally, consistent with hypoxia conditions governing cancer cell environment, all the simulations were constrained to low rates of oxygen uptake [27], see details in Table S1.
Constraint-base modeling assessment : Evaluating Objective Function
To evaluate the physiological significance of the proposed objective function, we decided to explore the extent to which growth rate derived from dynamic constraint-based modeling coincided with that obtained from a kinetic growth study of Hela cell lines. Therefore, the in silico temporal profile of growth rate was calculated by defining an initial cellular density, an initial available glucose concentration and a proper time scale for assuming steady-state condition, see methods section. Meanwhile, Hela cancer cell lines were cultivated in solution and a growth kinetic study was accomplished. As described in the methods section, experimental measurements of cellular density on Hela cells were made with six replicates for estimated experimental reproducibility and by monitoring the process every 24 hours for five days, see also Figure 2 (B).
Figure 2. Temporal profile of kinetic cell growth.

(A) Comparative analysis between the growth rate obtained experimentally and in silico. (B) Average and standard deviation obtained in the kinetics measurements for Hela cell lines. As described in methods, growth rate was monitored every 24 hours for five days, and six replicates were obtained for each absorbance measurement. Statistical properties characterizing the kinetic growth on Hela cell lines are shown in Figure 2(B), while the temporal behavior of glucose uptake rate and external concentration predicted by in silico procedures are depicted in (C) and (D), respectively. Coefficients of variation obtained at each measurement are reported by the red points in (B).
The contribution of metabolic entities in the objective function has been assumed to carry equal weight on growth rate, in such a way that instead of using a quantitative criteria to evaluate the crosstalk between experiment and modeling, a qualitative procedure based on the normalization of cellular density profile was implemented. Thus, proceeding as described in the methods section, we found that our modeling was capable of obtaining a normalized temporal growth profile comparable with that associated with Hela cell lines, see Figure 2.
In light of this result, we postulate that the objective function associated with the metabolic reconstruction depicted in Figure 1 is potentially able to elucidate the metabolic flux activity required for supplying the metabolic demand for cancer cell growth. This is a crucial contribution in this study and constitutes the backbone for exploring the relationships among gene activity, metabolism and phenotype in cancer.
In silico simulations
Computational models on biological systems have two general purposes: 1) to reproduce what is physiologically observed and understand their biological principles, and 2) to create a platform capable of predicting the cellular phenotype when metabolic alterations are induced in the system. Having verified that in silico phenotype qualitatively reproduces the growth rate of Hela cell lines, we proceeded to survey the metabolic mechanisms supporting cell proliferation through Flux Balance Analysis (FBA), an in silico formalism that has been useful in exploring the genotype-phenotype relation for a variety of organisms [11], [12], [13], [22], [23], [28]. Specifically, we have used our metabolic reconstruction for identifying those biochemical reactions that have a strong influence on controlling cancer cell growth, a worthy issue when one desires to identify metabolic targets with effective results in cancer treatments [6]. For this purpose, metabolic targets with a central role in cancer cell growth were identified by two constraints: low flux variability and high enzymatic essentiality for cancer cell growth. Together, these constraints constitute computational criteria for selecting those reactions that ensure a low redundancy on metabolite synthesis with a maximal effect for decreasing its phenotype. Thus, this computational criteria lead us to identify a set of target enzymes whose metabolic activity may has a direct effect on cancer cell growth, see Figure 3.
Figure 3. Flux Variability and Enzyme essentiality on metabolic reactions.

To identify those reactions that could have a pivotal role in cancer growth rate, flux variability and enzyme essentiality analysis were accomplished over all the reactions included in the reconstruction. In panel (A), the metabolic reactions whose deletion produces a significant reduction on growth rate are highlighted in red. Those reactions that ensure a low variability and high essentiality constitute 27% of the complete metabolic reconstruction and these appear in red in panel (B). Exchange and sink reactions were excluded from this analysis. Abbreviation code: Enolase (ENO), glyceraldehyde-3-phosphate dehydrogenase(GAPD), phosphoglucomutase (PGMT), pyruvate kinase (PYK), triose-phosphate isomerase (TPI), lactate dehydrogenase (LDH), ribose-5-phosphate isomerase (RPI), pyruvate dehydrogenase (PDHm), 2-oxoglutarate dehydrogenase (AKGDm), cytrate synthase (CSm), Fumarate hydratase (FUMm), malate dehydrogenase (MDHm), succinate dehydrogenase (SUCD1m), succinyl-CoA synthetase (SUCOAS).
The robustness of this set of target enzymes in terms of the ratios among the objective function's components was subsequently verified: We repeatedly applied the in silico analysis to a set of objective functions whose equimolar contributions on objective function's components were not assumed. With this in mind, 1,000 objective functions (with components selected from a random uniform distribution ranging from 0 to 1 around numerical values estimated for other organisms [22]) were reconstructed, and enzymes with low flux variability and high enzymatic essentiality were identified in each realization. Despite growth rates highly dependent on the ratios of the objective function's components, we identified a set of enzymes that in 99% of all the realizations obeyed selection criteria, see Figure 4 . Among the target enzymes identified in silico, we determined that some participate in glycolysis, such as phosphoglucomutase (PGMT), enolase (ENO), glyceraldehyde-3-phosphate dehydrogenase (GAPD), pyruvate kinase (PYK) and lactate dehydrogenase (LDH). Consistent with this result, development of drugs mainly targeting glucose transport and phosphorylation steps in glycolytic pathways have shown to be a latent therapeutic strategy for reducing cancer phenotype [19], [27], [29].
Figure 4. Enzymes with high essentiality and low variability that are robust to the ratio of objective function's components.

Reactions with high essentiality and low variability were identified through a set of 1000 objective functions with nonequivalent ratios among function components. As panel (A) shows, reactions obeying both criteria (red on black regions) were plotted over the 1000 realizations. In each realization, those enzymes that obey the in silico criteria were denoted in black; all others in white. The percentage of times reactions obeyed the computational criteria are depicted in panel (B). Robust enzymes relevant to this study (excluding transporters, exchange and demand reactions) were labeled in red. EX, DM and Sink denote exchange, demand and sink reactions in the cytoplasm [c] and mitochondria [m] compartments.
Furthermore, constraint-based modeling suggests that lactate dehydrogenase can be used as a metabolic control point over phenotype behavior in agreement with previous studies, see Figure 5 [2], [3]. Specifically, there has been experimental evidence that inhibition of lactate dehydrogenase induces a decreased activity of some glycolytic enzymes and consequently reduces growth rate in cancer cells [30]. Motivated by this fact and with the purpose of further assessing our computational interpretation, we evaluated to what extent a reduction of enzymatic capacity of lactate dehydrogenase influences the metabolic activity on enzymes participating in glycolysis, pentose phosphate and TCA cycle. As Figure 5 shows (panel A, B and C), flux balance analysis exhibits that an increment on enzyme activity for lactate dehydrogenase is followed by an increased metabolic activity over glycolysis and some enzymes participating in TCA cycle and pentose phosphate pathway. Consistent with this in silico observation, an increase of lactate production has been proposed to be a necessary condition supporting tumor cell transformation through the Warburg effect [31]. To confirm that this property is a consequence of the geometry of the flux steady-state solution space and not of the particular selections of ratios in the objective function's components, a nonbiased Monte Carlo sampling method was applied for characterizing the solution spaces [23], see methods section. As Figure 5 (D) shows, a significant correlation emerged between metabolic activity of lactate dehydrogenase (LDH) and the first enzyme in glycolysis: phosphoglucomutase (PDGM). A decrease of LDH tends to be related with a decrease on glucose metabolism through PDGM, hence, our in silico analysis suggests LDH as a control point in cancer cell metabolism.
Figure 5. Lactate dehydrogenase and its influence on central pathways.

Lactate dehydrogenase (LDH) has been suggested as a pivotal metabolic control on cancer cell growth with a significant role in the Warburg effect. Panels (A), (B) and (C) show the effects that variations of LDH activity have on some enzymes participating in glycolysis, TCA cycle and pentose phosphate, respectively. Metabolic activity of LDH increases from bottom to top. Panel (D) shows the correlation between flux activity of LDH and phosphoglucomutase (PGMT) obtained through sampling the null space of the stoichiometric matrix. Phenotype phase plane for glucose-6-phosphate dehydrogenase (G6PDH) and transketolase (TKT1), enzymes quantifying the activity of the oxidative and non-oxidative branches of pentose phosphate, is depicted in panel (E). White arrows indicate the direction at which the metabolic flux increases.
On the other hand, our computational platform suggests that pyruvate dehydrogenase (PDHm) can perform a central role in driving cell proliferation due to its low flux variability and high enzymatic essentiality for metabolism in cancer cell growth, see Figure 4 (A). Consistent with this finding, there is evidence that metabolic inhibition of PDHm contributes to Warburg metabolism and enhances malignant phenotype in human neck and head squamous carcinomas [26], [32]. This observation could make sense in light of additional regulatory components integrating this metabolic puzzle. First, hypoxia condition in tumors induces the activation of HIF (Hypoxia Inducible Factor), which in turn activates pyruvate dehydrogenase kinase 1, an enzyme that negatively regulates the catalytic activity of PDHm. In addition, aerobic glycolysis is enhanced by the fact that HIF induces the overproduction of enzymes participating in the glycolytic pathway and lactate production [31]. Overall, enhancement of the Warburg effect and diminishing activity of PDHm seems to be a metabolic response that confers selective advantage for survival and cell proliferation.
With the purpose of surveying how growth in cancer cells may vary when changing the metabolic activities on both PDHm and glucose transport, we have accomplished phenotypic phase plane analysis, a computational procedure to visually explore how objective function behaves when flux variations over two independent metabolic reactions occurs [21], [23]. Remarkably, as Figure 6 (B) shows, our analysis suggests that at fix glucose uptake rate a decrease on PDHm enzymatic activity may improve the phenotype growth rate in cancer cell lines, arrow in region I. Despite the fact that this result is in agreement with some experimental reports, our computational model predicts the existence of a threshold on PDHm whose reduced activity could be beneficial to arrest cancer cell growth (region II), a result that requires posterior experimental verification.
Figure 6. Phenotype phase plane for glycolytic and TCA cycle enzymes.

Panel (A) is a three-dimensional representation of how metabolic activity of succinate dehydrogenase and glucose uptake rate influence growth. As Panels (B) and (C) show, in silico modeling leads us to identify some regions where variations on pyruvate dehydrogenase and fumarate hydratase, both associated with tumor suppressor activity, can result in different phenotypes. White lines indicate the direction at which the metabolic fluxes increase; black lines, the direction at which they decrease. The potential effect that pyruvate kinase activity can produce on cancer cell growth is depicted in (D). In panel (D) objective function components were selected as follows: cATP = 12.47, cLactate = 0.13, cNADPH = 0.93, cR5P = 0.6, cNAD = 0.89, cOAA = 0.75, cATP[m] = 17.09 and cCitrate = 0.55. The threshold flux activity is denoted by a red line.
Optimization of the objective function leads us to conclude that glutaminolysis, starting at glutamine uptake rate and ending with lactate production, is an active pathway during cancer cell growth. From a functional and biological point of view, glutaminolysis performs a fundamental role in replenishing TCA cycle and generating additional reductive power required for fatty acids biosynthesis. Furthermore, our in silico analysis suggests that fumarate hydratase (FUMm) and succinate dehydrogenase (SUCD1m) can be independently used as metabolic targets for regulating cell proliferation, see Figure 6A and C. Phenotype phase plane accomplished over these enzymes allows us to conclude that when activity of FUMm (SUCD1M) is reduced different regions separated by a threshold value are identified. As can be appreciated in Figure 6 (A) and (B), when metabolic activity on FUMm or SUCD1M is decreased, phenotype growth rate in region I is enhanced while in region III is reduced. Interestingly, phenotype behavior observed in region I is in accordance with the fact that FUMm or SUCD1m can participate as a tumor suppressor when its enzymatic activity is deficient [33]. Even though the model can sense the influence that the enzymatic activity of FUMm or SUCD1m has on cancer growth rate, further analysis is required to assess if in silico interpretation on region II and III has a biological meaning.
We highlight that in our simulations mitochondria-derived citrate constitutes a fundamental metabolite to be optimized for supporting cell proliferation. As a result, low citrate transport from mitochondria toward cytoplasm induces a decreased effect on in silico growth rate. Consistent with published findings, inhibition of ATP citrate lyase participating in conversion of mitochondria-derived citrate into acetyl-coenzyme A in cytoplasm prevents cancer cell proliferation and tumor growth due to its central role as a precursor for lipids [2], [34]. Even though inhibition of ATP citrate lyase and low citrate transport have the final effect of reducing acetyl-coenzyme A, a more detailed analysis should be considered in future reconstructions. Specifically, fatty acids metabolism should be included in the reconstruction in order to evaluate if cytoplasm acetyl-coenzyme A could be a more appropriate component than cytoplasm citrate to simulate computationally cancer cell growth.
The pharmaceutical design of drugs targeting the pentose phosphate pathway (PPP) seems to be an appealing strategy to reduce growth rate in cancer cells due to its essential role in synthesizing ribose-5-phosphate (r5p), essential for biosynthesis of nucleotides and nucleic acids [35]. To quantify the contribution of PPP to cancer cell growth, we analyzed in silico how transketolase (TKT1) and glucose-6-phosphate dehydrogenase (G6PDH), representing the nonoxidative and oxidative branches in PPP respectively, affect cell growth phenotype, see Figure 5E. As Figure 3A shows, in silico mutation of TKT or G6PDH predicts its nonessentiality in cancer cell growth, this due to the fact that non-oxidative or oxidative branches in PPP can both produce ribose-5 phosphate (r5p). Converse to this harmless effect of simple mutations, however, in silico modeling suggests that simultaneous mutation is lethal for phenotype in cancer cell growth, a finding that agrees with already published reports. Thus, enzymatic down-regulation of glucose-6-phosphate dehydrogenase (G6PDH) and TKT1 is required to effectively arrest growth rate for cancer cells in animals [6], [35]. Furthermore, phenotype phase plane done with G6PDH and TKT1 allows us to infer not only that a simultaneous reduction of TKT1 and G6PDH metabolic activity clearly predicts a drop in growth rate but also that, although both branches of PPP can produce r5p, the oxidative branch can be a limiting factor for growth rate in cancer: it means if TKT1 flux is null an increment of G6PDH metabolic activity can produce phenotype, however the opposite situation does not occurs, see Figure 5(E) , a hypothesis that needs to be verified with more precise experimental measurements [35].
All together, the complete set of benchmarks used for supporting this reconstruction are shown in Figure 7 . In light of these results, our constraint-based modeling represents an effort to construct a computational platform that serves as a guide for accomplished descriptive and predictive analysis about cancer metabolisms, thereby creating a dialogue with an experimental counterpart.
Figure 7. Physiological assessment of in silico interpretations.

A proper assessment of in silico results is required to ensure proper reconstruction. Ten proofs were used here to evaluate the physiological consistency of the results from constraint-based modeling. In sequential order, columns indicate the metabolic property computationally analyzed, in silico predictions and its consistency with some representative references. The blue text in column 1 represents metabolic pathways, while red lines indicate the effects that enzyme mutation has on cancer cell growth.
High throughput technology and cancer variability
Previous sections were devoted to constructing a metabolic network, simulating growth rate and evaluating its predictive scope based on published findings about cancer cells. From a conceptual view, metabolic activity resulting from our constraint-based analysis in nature represents an average behavior obtained from a cancer cell population whose variability and stochasticity on phenotype have been neglected. However, it is well known that the cancer cell population in tumors is heterogeneous, and a subsequent question is, then, to what extent cancer cell diversity in tumors can display heterogeneous metabolic activity [18]. A significant contribution to this issue was published regarding a cervical uterine cancer study. It concluded that significant variability within tumor regions mainly is concentrated on genes participating in transcriptional regulatory and metabolic mechanisms [18]. Thus, with the purpose of estimating the extent of the cellular heterogeneity inherent in tumors could affect the metabolic activity of our reconstruction, we have done a statistical analysis of more than 33 samples of expression profiling stored in the GEO repository at NCBI (accession GSE5787) obtained from several regions of 16 patients with cervical cancer [18]. In contrast to the methodology used by the original report [18], our variability analysis was quantified through classical statistics over the expression data obtained from affymetrix technology, see methods section. In agreement with the original findings that the expression of genes acting on signal transduction, regulatory mechanism and metabolic pathways was significantly varied, we obtained that genes participating in oxidative phosphorylation potentially induce a variability on enzymatic activity within the population, see Table S2 in supplementary material.
Guided by this result, we conclude that even though some genes involved in metabolism can be expressed quite variably in tumors, expression profiling reported for cervical cancer may suggest that most of the metabolic pathways included in this reconstruction are uniformly expressed at a population level, being the exception oxidative phosphorylation. Computational and experimental assessment for verifying this heterogeneity and exploring its biological consequences will be an issue to address in future.
Discussion
Metabolic alterations constitute a hallmark in cancer cells: They are required for driving transformation, progression and spreading cancer in tissues [16]. Consequently, development of computational procedures for identifying those enzymes with an influential role on cancer cell growth constitutes an active line of research essential to establishing the bases of a rationalized design of drugs with the desired therapeutic effects [16]. In our study, a metabolic reconstruction integrating the best-known metabolic pathways participating in cancer development was accomplished: glycolysis, pentose phosphate pathway, oxidative phosphorylation, glutaminolysis and TCA cycle.
To assess the practical scope of the metabolic reconstruction, four criteria were evaluated: 1) the model's ability to simulate growth rate in Hela cells, 2) its ability to identify the global metabolic activity during cell proliferation; 3) the expected phenotype on growth rate when variations on enzymatic activity are induced; and 4) the effect that cancer heterogeneity has on metabolic reconstruction. Qualitative assessment of points 1 to 3, summarized by Figure 2 and 6, were successfully evaluated in silico and have led us to suggest that constraint-based modeling can be used as a descriptive and predictive framework to study the metabolic alterations supporting cancer in cells.
Although this computational study constitutes a good start in genome scale modeling in cancer metabolisms, our reconstruction and modeling is far from complete [36], and improvements, additional tests and more specific studies should be undertaken in future. Between the most relevant issues we name the follows:
It has been reported that the expression of M2-PK, a specific-tumor pyruvate kinase, may enhance cell biosynthetic capabilities by decreasing the transformation of phosphoenolpyruvate (PEP) to pyruvate and shunting the intermediary substrates upstream of PEP toward biosynthetic pathways required for cancer cell growth [1], [24]. This qualitative behavior was reproduced by constraint-based modeling only when a proper ratio among the objective function's components is tune by one that may quantify a more reliable conversion in cancer cell growth, see Figure 6 D . According with previous studies, an isoform of pyruvate kinase PKM1, PKM2, is highly represented during cancer cell growth such that it induces a reduced metabolic activity over pyruvate kinase producing a beneficial effects on cancer cell growth [27]. Consistently with this fact, our model can mirror this fact by identifying a region in phase plane whose decreased activity of pyruvate kinase can induce an increment on cancer growth rate, such as occurs with the switching between PKM1 and PKM2 during cell transformation. An inverse effect on cancer cell growth is observed at a threshold activity of PKM1, red line in Figure 6(D), a result that requires of additional experimental studies. This result makes evident the need of determining proper coefficients in the objective function's components to obtain meaningful biological interpretations, specially when exploring the finest of details in metabolic behavior.
In this study, we have selected a partial metabolic reconstruction in order to permit a comparison between in silico predictions and published findings in literature, such that, this approach facilitated a reliable assessment of the in silico results, see Figure 7 . This primer modeling functions as a benchmarks that will allow us to sequentially introduce other pathways into the description. The reconstructed human metabolic network [36] constitutes an excellent source for extending our studies and potentially evaluate the role that new pathways have to sustain cancer phenotype.
From a genetic point of view, over expression of oncogenes and/or dysfunction of tumor repressors induce changes in genetic circuits that trigger the temporal transition between normal and cancer cell phenotype. Additional analysis will be required to explore how regulatory networks will constrain metabolic phenotype [37], [38] and to what extent the development of dynamic formalism can contribute to unraveling the main principles behind the robustness of cancer cells [39], [40]. In our opinion, both issues are relevant to distinguish the biological mechanism governing proliferation in cancer and normal functioning cell from a systems biology point of view.
Methods
Flux Balance Analysis
Constraint-based modeling is a computational frame for analyzing and exploring the phenotype space of metabolic networks obeying mass conservation and steady-state assumption [21], [22]. Thus, beginning from metabolic reconstruction of cancer cells, we applied linear programming to identify those metabolic fluxes (νj, j = 1,2…n) that maximize biomass production when constrained by thermodynamics and mass balance principles;
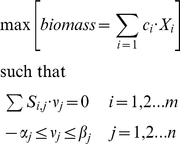 |
where Si,j represents the stoichiometric coefficient of metabolite i participating in the j reaction. In addition, biomass is represented by a linear combination of metabolites essential to cell growth (Xi) and whose contribution to biomass production are weighted by coefficients (ci). Irreversible constraints and enzyme capacities for each metabolic reaction are quantified by parameters αj and βj.
Dynamic Flux Balance Modeling
To model the temporal growth rate, we have assumed that along the time line, it is possible to identify a time scale, Δt, where the steady-state condition can be applicable. Briefly, assuming that at the moment of initiation, the cell environment has a glucose concentration, soc and a cell density X0, the algorithm underlying dynamic constraint-based modeling consists of an iterative process among the following steps [21]. 1) Glucose concentrations sc available for the cell must be identified. 2) The glucose concentration then should be scaled to define quantity available per unit of biomass and per unit of time:
where sav denotes the initial concentration of glucose available in the environment. 3) Assuming a steady-state condition for metabolism, maximal growth rate, μ, at the interval of time Δt is calculated through the linear programming algorithm [21], [23] and consequently growth rate and the new substrate glucose uptake rate Su are updated. 4) At the next described time step (t+Δt), glucose concentration is obtained through the solution of the classic set of differential equations:
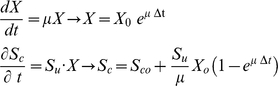 |
where X is the cell density, Su is the available concentration of glucose and μ represents the growth rate. In our simulation, the algorithm was repeated until null concentration of glucose was reached. The algorithm was implemented using Cobra Toolbox package in Matlab software [28].
Flux Variability and Enzyme Essentiality analysis
Flux Variability analysis was applied to explore metabolic redundancy in optimal phenotype. Thus, this procedure identifies the range of numerical values for each flux over the reconstruction, while still satisfying the given constraints and optimizing a selected objective function [23]. Enzyme essentiality analysis was used to quantify to what extent the deletion of an enzymatic reaction affects the optimization of selected objective function. In order to identify those metabolic reactions obeying low flux variability and high essentiality we selected two threshold values. First, we defined as essential those metabolic reactions whose deletion reduced by 50% or more the original objective function. Second, reactions with low flux variability were those whose absolute values on flux variability were less or equal to 50% of the maximal flux variation along the entire distribution. Those reactions intersecting both sets constituted the target enzymes reported in Figures 3 and 4. Both computational analyses were accomplished in a Matlab environment and COBRA toolbox.
Sampling null space of the stoichiometric matrix
The identification of metabolic fluxes (ν) obeying the steady-state condition, 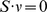 ,with S as the stoichiometric matrix, was accomplished by sampling the null space of S by an Artificial Center Hit and Run Algorithm (ACHR) [41], [42]. Basically, this algorithm defines an initial point along the null space of S. Once this point is defined, the algorithm calculates “warm-up” points by an iterative procedure. These warm-up points are stored in a matrix W by which a centroid xc is calculated. Finally the sample points are calculated by selecting one point yn in the W matrix and moving in the direction vector given by (xc−y). The new vector yn+1 is substituted by the previous point yn in W. The centroid is recalculated, and this process continues iteratively until a desired number of sample points are reached. ACHR was done using the COBRA tool box [28]. Overall, 10,000 sampled points belonging to the null space of the stoichiometric matrix were included in the analysis.
,with S as the stoichiometric matrix, was accomplished by sampling the null space of S by an Artificial Center Hit and Run Algorithm (ACHR) [41], [42]. Basically, this algorithm defines an initial point along the null space of S. Once this point is defined, the algorithm calculates “warm-up” points by an iterative procedure. These warm-up points are stored in a matrix W by which a centroid xc is calculated. Finally the sample points are calculated by selecting one point yn in the W matrix and moving in the direction vector given by (xc−y). The new vector yn+1 is substituted by the previous point yn in W. The centroid is recalculated, and this process continues iteratively until a desired number of sample points are reached. ACHR was done using the COBRA tool box [28]. Overall, 10,000 sampled points belonging to the null space of the stoichiometric matrix were included in the analysis.
Comparative analysis between in silico and experimental growth kinetics
To verify that the kinetic growth curve obtained in silico can exhibit a comparable behavior with the experimentally obtained growth curve from Hela cell lines, we have applied a normalization procedure in both curves. We have denoted G as a cell growth matrix whose entry g i,j represents the j replicate of cellular density measured at time i, with i = 1….5 (five days) and j = 1…6 (six replicates per day). In addition, <A> represents the average growth vector whose entries Ai indicate the average over rows of G. The normalization applied on both curves, theoretical and experimental, was obtained through this transformation
where Ai represents the average cellular density at sample i, and min and max represent the minimal and maximal numerical value of the i row of <A>. In other words, above normalization permits selection of a framework in which the minimal value of AiNorm is equal to zero and a maximal value equal to a unit.
Microarray analysis
In order to have an idea of how cancer heterogeneity can influence metabolic phenotype, we accomplished a statistical analysis of microarray data on 33 samples of cervical uterine cancer among 16 women patients. The analysis used previously published microarray data [18] stored in GEO (repository at NCBI under accession GSE5787) and analyzed in R with affy package in bioconductors, http://www.bioconductor.org/. To identify those genes that significantly vary with in the 33 samples, a t-statistics was accomplished selecting those genes with a log-ratio higher than 1.2 and a p-value less than 0.01. The set of genes identified and its corresponding information (such as symbol, description, reference chromosome location, gene ontology, and participation in metabolic pathways) are shown in Table S2.
Cell Culture Conditions and Growth Kinetics
Growth rate were obtained through standard technique of crystal violet procedure. The cell line HeLa was obtained from the Unidad de Diferenciacion Celular y Cáncer at FES-Zaragoza UNAM. Cancer cell line was cultured in RPMI-advanced 1640 serum-free media (Gibco BRL, USA) with red phenol and antibiotic-antimycotic solution (10,000 units penicillin, 10 mg streptomycin, and 25 µg amphotericin B per mL). The cells were incubated in 5% of CO2 and humidity saturation at 37°C. Cells were cultured in 1mL of RPMI-Advanced medium in 24-well cell culture plates (BD Falcon, USA) starting from an estimated cellular population of 105 cells. Every day the medium for 6 different wells was removed and 100uL of glutaraldehyde at 1.1% were added, until you have enough samples to complete the kinetics. After this the glutaraldehyde is removed from each well and the plate is left at room temperature until dryness is reached. Violet crystal at 0.1% is added during 20 minutes and the wells are then washed with bidistillate water. The water is removed and then citric acid at 10% was added and shaken during 20 minutes. The absorbance at 600 nm was obtained for the supernadant in each well.
Supporting Information
Metabolic reconstruction for central metabolism in cancer.
(0.03 MB XLS)
Genes with a significative variation on gene expression in cervical cancer.
(0.51 MB XLS)
Acknowledgments
OR-A. thanks discussions and suggestions from Prof. Pamela Silver and her systems biology group during a research stay at Harvard Medical School. The authors are grateful for comments and suggestions from two anonymous referees during the review process of this paper.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was partially supported by National Council of Science and Technology (CONACyT-Mexico: grant 83461) and Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica-Universidad Nacional Autonoma de Mexico (PAPIIT: grant IN203809-3). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Weinberg RA. The biology of Cancer. Garland Sciences, Taylor & Francis Group, LLC; 2007. [Google Scholar]
- 2.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 3.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Kreeger PK, Lauffenburger DA. Cancer Systems Biology: A Network Modeling Perspective. Carcinogenesis; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolkenhauer O, Auffray C, Baltrusch S, Bluthgen N, Byrne H, et al. Systems biologists seek fuller integration of systems biology approaches in new cancer research programs. Cancer Res. 70:12–13. doi: 10.1158/0008-5472.CAN-09-2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cascante M, Boros LG, Comin-Anduix B, de Atauri P, Centelles JJ, et al. Metabolic control analysis in drug discovery and disease. Nat Biotechnol. 2002;20:243–249. doi: 10.1038/nbt0302-243. [DOI] [PubMed] [Google Scholar]
- 7.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 8.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 10.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 11.Palsson B. Metabolic systems biology. FEBS Lett. 2009 doi: 10.1016/j.febslet.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Resendis-Antonio O, Reed JL, Encarnacion S, Collado-Vides J, Palsson BO. Metabolic reconstruction and modeling of nitrogen fixation in Rhizobium etli. PLoS Comput Biol. 2007;3:1887–1895. doi: 10.1371/journal.pcbi.0030192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BO. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol. 2009;7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feist AM, Palsson BO. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol. 2008;26:659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Thiele I, Weekes D, Li Z, Jaroszewski L, et al. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science. 2009;325:1544–1549. doi: 10.1126/science.1174671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garber K. Energy deregulation: licensing tumors to grow. Science. 2006;312:1158–1159. doi: 10.1126/science.312.5777.1158. [DOI] [PubMed] [Google Scholar]
- 18.Bachtiary B, Boutros PC, Pintilie M, Shi W, Bastianutto C, et al. Gene expression profiling in cervical cancer: an exploration of intratumor heterogeneity. Clin Cancer Res. 2006;12:5632–5640. doi: 10.1158/1078-0432.CCR-06-0357. [DOI] [PubMed] [Google Scholar]
- 19.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 21.Varma A, Palsson B. Stoichiometric Flux Balance Models Quantitatively predict growth and metabolic Byproduct secretion in Wild-type Escherichia coli W3110. Applied and Environmental Microbiology. 1994;60:3724–3731. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palsson B. Systems Biology: Properties of Reconstructed Networks. New York: Cambridge University Press; 2006. [Google Scholar]
- 23.Price ND, Reed JL, Palsson BO. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat Rev Microbiol. 2004;2:886–897. doi: 10.1038/nrmicro1023. [DOI] [PubMed] [Google Scholar]
- 24.Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 28.Becker SA, Feist AM, Mo ML, Hannum G, Palsson BO, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc. 2007;2:727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- 29.Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- 30.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 31.Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72. doi: 10.1016/s0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 32.McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283:22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- 34.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Ramos-Montoya A, Lee WN, Bassilian S, Lim S, Trebukhina RV, et al. Pentose phosphate cycle oxidative and nonoxidative balance: A new vulnerable target for overcoming drug resistance in cancer. Int J Cancer. 2006;119:2733–2741. doi: 10.1002/ijc.22227. [DOI] [PubMed] [Google Scholar]
- 36.Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci U S A. 2007;104:1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- 38.Covert MW, Palsson BO. Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J Biol Chem. 2002;277:28058–28064. doi: 10.1074/jbc.M201691200. [DOI] [PubMed] [Google Scholar]
- 39.Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 40.Resendis-Antonio O. Filling kinetic gaps: dynamic modeling of metabolism where detailed kinetic information is lacking. PLoS One. 2009;4:e4967. doi: 10.1371/journal.pone.0004967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price ND, Schellenberger J, Palsson BO. Uniform sampling of steady-state flux spaces: means to design experiments and to interpret enzymopathies. Biophys J. 2004;87:2172–2186. doi: 10.1529/biophysj.104.043000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiback SJ, Famili I, Greenberg HJ, Palsson BO. Monte Carlo sampling can be used to determine the size and shape of the steady-state flux space. J Theor Biol. 2004;228:437–447. doi: 10.1016/j.jtbi.2004.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Metabolic reconstruction for central metabolism in cancer.
(0.03 MB XLS)
Genes with a significative variation on gene expression in cervical cancer.
(0.51 MB XLS)