

Abstract
Ribozymes of hepatitis delta virus have been proposed to use an active-site cytosine as an acid-base catalyst in the self-cleavage reaction. In this study, we have examined the role of cytosine in more detail with the antigenomic ribozyme. Evidence that proton transfer in the rate-determining step involved cytosine 76 (C76) was obtained from examining cleavage activity of the wild-type and imidazole buffer-rescued C76-deleted (C76Δ) ribozymes in D2O and H2O. In both reactions, a similar kinetic isotope effect and shift in the apparent pKa indicate that the buffer is functionally substituting for the side chain in proton transfer. Proton inventory of the wild-type reaction supported a mechanism of a single proton transfer at the transition state. This proton transfer step was further characterized by exogenous base rescue of a C76Δ mutant with cytosine and imidazole analogues. For the imidazole analogues that rescued activity, the apparent pKa of the rescue reaction, measured under kcat/KM conditions, correlated with the pKa of the base. From these data a Brønsted coefficient (β) of 0.51 was determined for the base-rescued reaction of C76Δ. This value is consistent with that expected for proton transfer in the transition state. Together, these data provide strong support for a mechanism where an RNA side chain participates directly in general acid or general base catalysis of the wild-type ribozyme to facilitate RNA cleavage.
General acid-base catalysis† is common in enzyme-catalyzed systems where proton transfer occurs in the transition state of the chemical step. General acid-base catalysis can accelerate chemical-catalyzed reactions 10- to 100-fold in solution (1); in enzyme systems, results from mutagenesis data indicate that the rate acceleration can be as high as 106-fold (2–4). With protein enzymes, general acid-base catalysis is commonly achieved by amino acid side chains with pKa values near neutral pH. However, RNA side chains have pKa values significantly higher or lower than neutral pH and, thus, would not generally be well suited for general acid-base catalysis. For an RNA side chain to participate directly in general acid-base catalysis, a significant shift in its pKa would appear to be required. Because most ribozymes require divalent cations for optimal activity, it has been proposed that hydrated divalent metal ions may act as a general acid or base in ribozyme catalysis (5–7). Divalent metal ions also can facilitate proton transfer through direct coordination to the attacking nucleophile (8–11). Thus, while there is precedent for the role of metal ions in catalyzing proton transfer in ribozymes, there is very little information on the direct involvement of RNA side chains in general acid-base catalysis.
Hepatitis delta virus (HDV) ribozymes, like other small ribozymes, catalyze a transesterification reaction of a phosphodiester linkage, generating a 2′,3′-cyclic phosphate and a 5′-hydroxyl. The proposed mechanism involves the adjacent 2′-hydroxyl group as an intramolecular nucleophile, a penta-coordinated dianionic transition state, and the 5′-oxyanion as a leaving group. In this mechanism, activation (deprotonation) of the 2′-hydroxyl nucleophile and stabilization (protonation) of the 5′-oxyanion could be achieved by general acid-base catalysis. The investigation of HDV ribozyme catalysis has been greatly facilitated by crystallographic studies of the genomic 3′ cleavage product (12). Although lacking the scissile phosphate, this structure is thought to closely resemble the precursor because most of the catalytic domain resides in the sequence 3′ to the cleavage site (13). An important residue, C75 in the genomic sequence (γC75), but no divalent metal ion, was observed within the active site of this structure; thus, a model of general base catalysis by an RNA side chain was suggested (12). A contribution of γC75 [or cytosine 76 in the antigenomic ribozyme (C76)] to HDV catalysis was consistent with mutagenesis studies (14, 15); a C to U (or G) mutation at this position resulted in a reduction of cleavage activity by >106. Only an adenine substitution for γC75/C76 retained detectable cleavage activity, but it was 103-fold less active than wild type (14, 16, 17). This result was consistent with a model where the ring nitrogen N1 of adenine fulfills a catalytic function similar to N3 of cytosine. General acid-base catalysis involving C76 also was supported by chemical rescue of cleavage of a C76u mutant with imidazole buffer (16). The imidazole rescue reaction followed a rate-law definition of general base catalysis and was consistent with a cleavage mechanism where C76 accepts a proton from the 2′-hydroxyl group. Nakano et al. (17) have proposed a kinetically equivalent mechanism, consistent with both biochemical and structural data, in which γC75 serves as a general acid by donating a proton to the 5′ oxyanion in the cleavage reaction.
Very recently a catalytic role for a conserved adenosine residue in 23S RNA of the large subunit of ribosome was proposed based on results from crystallographic studies and chemical modification experiments (18–20). The ring nitrogen N3 of the adenosine is positioned toward the reactive carbonyl carbon for peptide bond formation and is proposed to serve as both general base and general acid in the peptidyl transfer reaction. Because of the similarity in proton transfer involving a ring nitrogen of a nucleobase, results from characterization of the HDV ribozyme cleavage mechanism could have useful implications for understanding the catalytic mechanism of the ribosome.
In this study, general acid-base catalysis by cytosine is further explored by using the chemical rescue approach with exogenous bases and a C76-deleted mutant (C76Δ) of the HDV antigenomic ribozyme. The kinetic solvent isotope effect (KSIE) determined for self-cleavage of the wild type and chemical rescue of C76Δ ribozyme provides evidence for a single proton transfer at the transition state. In addition, a linear free energy relationship of log(rate constants) with pKa of exogenous bases was observed. The Brønsted coefficient, β, determined for the ribozyme-catalyzed phosphodiester bond cleavage was consistent with those obtained from model systems. These data support an involvement of a cytosine side chain, or an exogenous base, in general acid-base catalysis, and also provide a measurement of the approximate extent of proton transfer by general acid-base catalysis in the transition state of phosphodiester cleavage in an enzymatic system.
Materials and Methods
Reagents and Chemicals.
T7 RNA polymerase was purified from an overexpressing clone (21). Restriction endonucleases, nucleotides, 32P-labeled nucleotides, chemicals, reagents, and other enzymes were purchased from commercial suppliers.
Preparation of RNA.
Self-cleaving ribozymes, PEX1 and its mutants, were derived from the HDV antigenomic sequence (16, 22). RNA was transcribed and isolated as described (16).
Self-Cleavage Reactions.
RNA was stored at −20°C, and aliquots were heated to 95°C in 25 mM Tris⋅HCl (pH 7.5), 5 mM EDTA, and 1 mM spermidine for 1 min immediately before use and preincubated at 37°C for 10 min. The reactions were adjusted to the final pH with buffer systems containing 25 mM acetic acid/25 mM Mes/50 mM Tris (pH 4.0–8.0) or 50 mM Mes/25 mM Tris/25 mM 2-amino-2-methyl-1-propanol (pH 7.0–10.0). Self-cleavage reactions at 25°C were initiated by addition of divalent metal ion; chemical rescue reactions were at 37°C and initiated by addition of both divalent metal ion and exogenous base. Aliquots were quenched with an equal amount of stop mix containing 50% formamide and 50 mM EDTA and fractionated on denaturing polyacrylamide gels. Results were quantified by using a PhosphorImager (Molecular Dynamics).
Solvent Isotope Experiments.
All solutions and buffers were prepared in 99.9 atom % D deuterium oxide (Sigma). The final pD of each reaction was determined with a standard glass electrode by adding 0.4 to the reading (23). The proton inventory experiments were done in nine compositions (23) of H2O/D2O at pL = 8.0 (plateau pH for both H2O and D2O reactions).
Data Analysis.
Time courses of cleavage reactions were fit to a first-order
single-exponential by using kaleidagraph data
analysis/graphics application (Synergy Software, Reading, PA):
Ft =
F∞ × (1 −
exp−kobst),
where Ft and
F∞ are the fractions cleaved at time
t and at the end point, respectively, and
kobs is the first-order rate constant.
For the cleavage reactions of the C76 mutant ribozymes in the presence
of exogenous base, the apparent second-order rate constant
(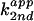 ) with respect to base concentration
is defined as
) with respect to base concentration
is defined as 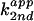 =
(kobs −
kuncat)/[base]total,
in which kuncat is the uncatalyzed
first-order rate constant in the presence of divalent metal ion but
absence of base. The second-order rate constant
(kb) with respect to the free base was
determined as the slope from the plot of
(kobs −
kuncat) vs.
[base]free, by measuring
kobs at various total concentrations
and pH of buffers.
=
(kobs −
kuncat)/[base]total,
in which kuncat is the uncatalyzed
first-order rate constant in the presence of divalent metal ion but
absence of base. The second-order rate constant
(kb) with respect to the free base was
determined as the slope from the plot of
(kobs −
kuncat) vs.
[base]free, by measuring
kobs at various total concentrations
and pH of buffers.
Calculations of the Molecular Volume of the Bases.
The molecular volumes of exogenous bases were calculated with advol, developed by J. M. Word (Duke University), using coordinates generated by insight ii (Molecular Simulations, San Diego).
Results
Rescue of Cleavage of C76 Mutants by Imidazole Buffer.
Ribozymes used in this study were derived from the HDV antigenomic
self-cleaving sequence (Fig. 1). The
wild-type ribozyme, PEX1, cleaves with a maximal first-order rate
constant of 13.4 min-1 in 11 mM
MgCl2 and 1 mM EDTA at 25°C. Changing C76 to u
(C76u) or deleting C76 (C76Δ) reduced cleavage activity
≈106-fold from wild type in 11 mM
MgCl2 and 1 mM EDTA. Consistent with what was
seen with C76u (16), cleavage activity of C76Δ was not stimulated by
a higher concentration (50 mM) of divalent metal ions, but could be
rescued by a combination of imidazole buffer and divalent metal ion
(Mg2+ or Ca2+). Also, a
second-order plot of the cleavage rate constants
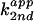 vs. fraction of imidazole
free base showed that C76Δ cleavage in imidazole buffer followed the
rate-law definition of general base catalysis (Fig.
2A).
vs. fraction of imidazole
free base showed that C76Δ cleavage in imidazole buffer followed the
rate-law definition of general base catalysis (Fig.
2A).
Figure 1.

Sequence and secondary structure of the HDV antigenomic ribozyme PEX1 with C76 emphasized. Numbering is from the cleavage site (indicated by an arrow) such that cleavage occurs between positions −1 and 1.
Figure 2.

Kinetic solvent isotope effect for wild-type and imidazole-rescue
reactions of C76Δ. (A) Second-order plot of C76Δ
cleavage by imidazole free base in H2O. The apparent
second-order rate constant (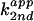 ) was
plotted against fraction of free base. For wild type (B)
and C76Δ (C), first-order
(kobs) and apparent second-order
(
) was
plotted against fraction of free base. For wild type (B)
and C76Δ (C), first-order
(kobs) and apparent second-order
(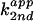 ) rate constants in
H2O (○) and D2O (□) were
plotted against pL (L = H, D). A ΔpKa ≈ 0.6 was
consistent with the expected value of 0.6 for an amino group.
(D) Proton inventory of the wild-type PEX1 reaction. The
first-order rate constants measured in varied solvent isotopic
composition were normalized to the rate constant in H2O
(
) rate constants in
H2O (○) and D2O (□) were
plotted against pL (L = H, D). A ΔpKa ≈ 0.6 was
consistent with the expected value of 0.6 for an amino group.
(D) Proton inventory of the wild-type PEX1 reaction. The
first-order rate constants measured in varied solvent isotopic
composition were normalized to the rate constant in H2O
(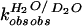 )
and plotted against molar ratio of D2O
(nD2O). The data were fit to a linear
dependence (n = 9; r = 0.99).
)
and plotted against molar ratio of D2O
(nD2O). The data were fit to a linear
dependence (n = 9; r = 0.99).
Kinetic Solvent Isotope Experiments Revealed a Single Proton Transfer.
To further establish that the ring nitrogen of C76 in the wild-type
cleavage reaction and that of the imidazole base in C76Δ cleavage is
involved in proton transfer at the transition state, the kinetic
solvent isotope effect for both reactions was determined. The
first-order rate constants
(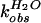 and
and
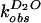 )
for wild-type self-cleavage were measured over a pL range of 5.0 to 9.0
in H2O and D2O (Fig.
2B). For the C76Δ ribozyme, the apparent
second-order rate constants
(
)
for wild-type self-cleavage were measured over a pL range of 5.0 to 9.0
in H2O and D2O (Fig.
2B). For the C76Δ ribozyme, the apparent
second-order rate constants
(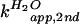 and
and
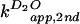 )
were measured at a constant concentration of 0.4 M imidazole over a pH
range of 6 to 9 (Fig. 2C). The KSIE was determined from the
ratio of the two rate constants
[k(H2O)/k(D2O)]
in the plateau region of the pH profile. KSIE for the wild-type
cleavage and chemical rescue C76Δ cleavage was 2.3 (ΔpKa =
0.7) and 2.7 (ΔpKa = 0.5), respectively (Fig. 2 B and
C). A ΔpKa ≈ 0.6 in both cases is the same as that
predicted for an amino group involved in proton transfer (23). A
similar KSIE seen with both the wild-type and the imidazole-rescued
C76Δ reactions, with an apparent reaction pKa corresponding to the
base, strongly supported the hypothesis that proton transfer at the
chemistry step is directly involved with the cytosine side chain or
exogenous imidazole buffer.
)
were measured at a constant concentration of 0.4 M imidazole over a pH
range of 6 to 9 (Fig. 2C). The KSIE was determined from the
ratio of the two rate constants
[k(H2O)/k(D2O)]
in the plateau region of the pH profile. KSIE for the wild-type
cleavage and chemical rescue C76Δ cleavage was 2.3 (ΔpKa =
0.7) and 2.7 (ΔpKa = 0.5), respectively (Fig. 2 B and
C). A ΔpKa ≈ 0.6 in both cases is the same as that
predicted for an amino group involved in proton transfer (23). A
similar KSIE seen with both the wild-type and the imidazole-rescued
C76Δ reactions, with an apparent reaction pKa corresponding to the
base, strongly supported the hypothesis that proton transfer at the
chemistry step is directly involved with the cytosine side chain or
exogenous imidazole buffer.
The partial KSIE (proton inventory) was determined for the
wild-type cleavage reaction (23). The first-order rate constants,
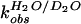 ,
were measured with varied compositions of H2O and
D2O, and normalized to the rate constant in
H2O
(
,
were measured with varied compositions of H2O and
D2O, and normalized to the rate constant in
H2O
(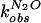 ).
A linear relationship (n = 9, r =
0.996) was obtained when the normalized rate constants
(
).
A linear relationship (n = 9, r =
0.996) was obtained when the normalized rate constants
(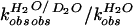 )
were plotted against solvent isotopic composition
nD2O
[mol.(D2O)/(mol.(H2O) +
mol.(D2O))] (Fig. 2D). The lack of
significant curvature in the plot suggested a single proton transfer at
the transition state.
)
were plotted against solvent isotopic composition
nD2O
[mol.(D2O)/(mol.(H2O) +
mol.(D2O))] (Fig. 2D). The lack of
significant curvature in the plot suggested a single proton transfer at
the transition state.
Chemical Rescue of C76 Mutants by Imidazole Analogues.
To better define the participation of imidazole in HDV catalysis, several imidazole analogues were tested in cleavage of the C76 mutants. The ability of imidazole analogues to rescue cleavage of the C76 mutants correlated strongly with the position of substituents on the imidazole ring. All 1-, 2-, or 1,2-substituted imidazole analogues failed to rescue activity (Table 1). A subset of 4-substituted imidazole analogues were able to rescue activity of the C76 mutants and yielded ≈5 to 1,000-fold enhancement over the background rate (≈1 × 10-5 min-1).
Table 1.
Bases tested in rescue experiments and the apparent
second-order rate constants
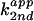 * for 76u and 76D
cleavage at pH 7
* for 76u and 76D
cleavage at pH 7
| 76u | 76Δ | |
|---|---|---|
| Cytosine analogues | ||
| Cytosine (C) | 50 | 120 |
| Isocytosine | 12 | 33 |
| 5-Methylcytosine | —† | — |
| 5-Fluorocytosine | — | — |
| Pyridine | 0.008 | 0.21 |
| Purine | ||
| Adenosine | — | — |
| Imidazole analogues | ||
| Imidazole | 32 | 15 |
| 1-Methylimidazole | — | — |
| 2-Methylimidazole | — | — |
| 1,2-Dimethylimidazole | — | — |
| 4-Methylimidazole | 4.5 | 10 |
| 4-Hydroxymethylimidazole | 3.6 | 10 |
| Histamine | 0.069 | 2.5 |
| Pyrazole | — | — |
| Histidine | 0.049 | 0.82 |
| 3-Amino-1,2,4,-triazole | 1.3 | 5.1 |
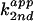 (×
103 min−1⋅M−1) was
determined with reactions at pH 7 in 26 mM MgCl2, with
respect to the total base concentration, not to the free base
component.
(×
103 min−1⋅M−1) was
determined with reactions at pH 7 in 26 mM MgCl2, with
respect to the total base concentration, not to the free base
component.
— indicates no detectable rescue activity.
The concentration and pH dependence for cleavage of C76 mutants with
4-substituted imidazole analogues was similar to that of the
imidazole-rescue reaction. For each of these analogues, the first-order
rate constant (kobs) showed a linear
dependence on total base concentration up to 600 mM. The dissociation
constant was estimated at >1.2 M (data not shown). Thus, all
rates were determined under
kcat/KM
conditions with respect to imidazole analogue concentrations, and the
apparent pKa of the C76u and C76Δ cleavage reactions corresponded to
the pKa of the buffer (Table 2 and Fig.
3A). The second-order plot of
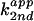 against fraction of free base
exhibited a linear dependence with a y-intercept close to 0
(data not shown). These data suggested that rescue of C76u and C76Δ
cleavage activity with 4-substituted imidazole analogues followed the
rate-law definition of general base catalysis, with no acid catalysis
from the buffer observed.
against fraction of free base
exhibited a linear dependence with a y-intercept close to 0
(data not shown). These data suggested that rescue of C76u and C76Δ
cleavage activity with 4-substituted imidazole analogues followed the
rate-law definition of general base catalysis, with no acid catalysis
from the buffer observed.
Table 2.
Reaction pKa and second-order rate constant kb for 76u and 76D cleavage in the presence of 4-substituted imidazole analogues
| pKa | m.v.* | C76u
|
C76Δ
|
|||
|---|---|---|---|---|---|---|
| pKa | kb | pKa | kb | |||
| 4-Methylimidazole | 7.8 | 79 | 7.9 | 33 | 7.8 | 76 |
| Imidazole | 7.0 | 62 | 7.0 | 63 | 7.1 | 30 |
| 4-Hydroxymethylimidazole | 6.5 | 84 | 6.6 | 5.0 | 6.6 | 14 |
| 3-Amino-1,2,4-triazole | 5.5 | 67 | 5.4 | 1.3 | 5.5 | 5.3 |
| Histamine | 6.0 | 104 | — | — | 6.7 | 3.7 |
The second-order rate constants kb (× 103 min−1⋅M−1) were determined from a plot of first-order rate constants against free base concentration. — indicates the numbers were not determined.
Molecular volume of each base (m.v., Å3).
Figure 3.

Brønsted analysis of C76Δ cleavage activity with imidazole
analogues. (A) The apparent second-order rate constant
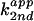 as a function of pH. The
apparent pKa of each reaction is labeled for 4-methylimidazole
(squares), imidazole (circles), 4-hydroxymethylimidazole (diamonds),
and 3-amino-1,2,4-triazole (triangles). (B) The
Brønsted plot of C76Δ cleavage in the buffers shown in
A; Brønsted coefficient β of 0.51 is determined from
the slope (n = 4; r = 0.96).
as a function of pH. The
apparent pKa of each reaction is labeled for 4-methylimidazole
(squares), imidazole (circles), 4-hydroxymethylimidazole (diamonds),
and 3-amino-1,2,4-triazole (triangles). (B) The
Brønsted plot of C76Δ cleavage in the buffers shown in
A; Brønsted coefficient β of 0.51 is determined from
the slope (n = 4; r = 0.96).
Brønsted Coefficient for C76 Mutants.
In general acid-base catalysis, the efficiency of catalysis correlates with the strength of the Brønsted acid or base; that is, the second-order rate constant shows a log-linear relationship with the pKa of the Brønsted acid or base. For C76Δ cleavage in the presence of imidazole and imidazole analogues, where catalysis is contributed by the free base of the buffer, the second-order rate constant can be described as
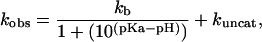 |
1 |
in which kobs and kuncat are the first-order rate constants in the presence and absence of exogenous bases, respectively; kb is the second-order rate constant with respect to the free imidazole base; pKa is the measured buffer pKa. The second-order rate constant, kb, for each base was determined at various pH and total base concentrations and is listed in Table 2. Chemical rescue by imidazole and imidazole analogues (4-methylimidazole, 4-hydroxymethylimidazole, and 3-amino-1,2,4-triazole) was analyzed with a Brønsted plot based on the following equation
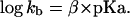 |
2 |
The Brønsted coefficient, β, determined for cleavage of the C76Δ ribozyme was 0.51 (n = 4, r = 0.99) (Fig. 3B). It is worth noting that the Brønsted coefficient reported here was not corrected for molecular volume of the bases (Table 2).‡
Chemical rescue of C76u cleavage by imidazole analogues showed several similarities to that of the C76Δ ribozyme. For each 4-substituted imidazole derivative the apparent reaction pKa corresponded to the buffer pKa (Table 2), and the second-order rate constant, kb, was slightly lower than, but of the same order as, that of C76Δ (Table 2). However, for imidazole rescue of C76u, kb was higher than that of C76Δ. When the imidazole data were omitted, the Brønsted coefficient determined for C76u, using the remaining points, was 0.53 (n = 3; r = 0.99), consistent with that determined for the C76Δ mutant. This result suggested that, with respect to buffer accessibility or binding, the active site of C76u might be more crowded than that of C76Δ.
Chemical Rescue of C76 Mutants by Exogenous Cytosine Bases and Analogues.
To link catalysis of C76 mutants in imidazole buffer to wild-type self-cleavage, exogenous cytosine base was tested for rescue activity with the C76 mutants. In contrast to rescue with imidazole and its analogues, cleavage activity of C76Δ showed a saturating dependence on the cytosine concentration, suggesting a possible binding site for exogenous cytosine with a Kd of ≈40 mM at pH 7. The apparent pKa determined from the pH profile of the cleavage reactions with 50 mM cytosine was 5.2, which suggested a shift in pKa of about ≈+0.8 from the measured pKa for cytosine in solution (24). Cleavage of C76u in the presence of cytosine exhibited the same pH and concentration dependence, but at slightly lower efficiency for rescue (data not shown).
Several other cytosine analogues were tested with the C76 mutants (Table 1). Only isocytosine, a close structural analogue of cytosine, was able to rescue cleavage activity. However, it was less effective than cytosine. The concentration and pH dependence of isocytosine-rescued cleavage of C76Δ was similar to that of cytosine-rescued cleavage, giving an apparent binding constant of ≈40 mM and an apparent pKa of 5.0 in 50 mM isocytosine. Similar results were seen for C76u cleavage (data not shown). Cleavage activity of C76 mutants was not rescued to a significant extent in the presence of pyridine, 5-methylcytosine, or 5-fluorocytosine (Table 1). Moreover, these bases inhibited cleavage activity of C76 mutants in the presence of cytosine or imidazole with a Ki of 20–30 mM (data not shown), suggesting that these bases might bind at the active site of the ribozyme, but were not able to support catalysis.
Discussion
The data presented here provide strong evidence that the cytosine side chain is involved in proton transfer at the transition state in the HDV ribozyme cleavage reaction. Previous studies have suggested that the chemistry step is rate-limiting for PEX1 self-cleavage.§ Kinetic solvent isotope and proton inventory experiments indicate that, in the presence of 10 mM divalent metal ion, the rate-limiting step of wild-type antigenomic PEX1 ribozyme cleavage involves a single proton transfer at the transition state. The KSIE of ≈2.7 and ΔpKa of 0.7 observed with PEX1 is consistent with what is seen with the wild-type genomic ribozyme under similar conditions (17). However, these experiments alone do not identify the functional group in the ribozyme responsible for this proton transfer. Similar KSIE seen in the chemical rescue of C76Δ, but with an apparent pKa corresponding to the buffer pKa, strongly implicate γC75/C76 in proton transfer in the wild-type reactions. In addition, chemical rescue of C76Δ demonstrates a linear free energy relationship with the strength of the base (pKa). The Brønsted coefficient for the general base of 0.51 is consistent with a model, in which the ring nitrogen of imidazole is involved in proton transfer at the transition state. Moreover, cleavage rescued with exogenous cytosine in place of imidazole supports the proposal that chemical catalysis by imidazole buffer in the C76 mutants is analogous to the catalysis by the cytosine sidechain in wild-type cleavage.
An important objective of a mechanistic study is to assign catalytic roles to specific residues. Self-cleavage of the wild-type and C76a mutant ribozymes both demonstrated an apparent pH dependence that would be consistent with a model where the unprotonated ring nitrogen (N3 for C and N1 for A) accepts a proton and thus acts as a general base. A pH profile suggestive of base catalysis also is seen in the rescue of the C76 mutants by imidazole and cytosine. Furthermore, the second-order plot for catalysis in each imidazole buffer exhibits a linear dependence with respect to the free base component of the buffer, but no acid catalysis from the protonated form of the buffer. From the proton inventory of the wild-type reaction, the linear dependence of the rate on solvent isotopic composition indicates that there is only one proton “in flight” in the transition state. These observations all would be consistent with a mechanism of general-base catalysis in which the unsaturated ring nitrogen of cytosine (or imidazole) base accepts a proton from the 2′-OH group at the transition state to facilitate nucleophilic attack (Fig. 4A). However, these data also can be explained by a kinetically equivalent cleavage mechanism involving specific base and general acid catalysis. In this model, the 2′-OH nucleophile is deprotonated at pre-equilibrium (specific base catalysis) followed by donation of a proton to the 5′-leaving oxyanion by a protonated cytosine side chain or imidazolium at the transition state (general acid catalysis) (Fig. 4B). Specific base catalysis might be achieved by hydroxide anion accompanied by direct metal coordination to the 2′-oxygen, or by a hydrated metal hydroxide. These two mechanisms, general base catalysis and specific base/general acid catalysis, are indistinguishable with the kinetic data presented here. However, it has been argued that the rate-determining step for cleavage of a RNA phosphodiester linkage is not formation of the 2′O-P but breaking of the P-5′O bond (see ref. 25 and references therein). Thus, general acid catalysis by the protonated cytosine or imidazolium to stabilize the 5′-leaving group in the transition state is an attractive model. Biochemical data in support of general acid catalysis was suggested from self-cleavage reactions of the wild-type genomic ribozyme in the presence of high concentrations of monovalent cation with no added divalent metal ion (17). Under these conditions, self-cleavage demonstrated a pH profile essentially reverse of that of the “standard” cleavage reactions containing divalent metal ions. The interpretation for this observation was that in the absence of base catalysis involving a hydrated metal hydroxide, the general acid mechanism of γC75 is unmasked. The apparent pKa of 5.7 determined from this reverse pH profile corresponds well to the apparent pKa of 6.1 determined under standard reactions conditions. However, whereas a γC75a mutant is active under standard conditions, there was no detectable activity with γC75a in the absence of added divalent metal ion. Therefore, it has not been possible to more definitively link the apparent pKa for cleavage activity in the absence of the added divalent metal ion to γC75. Wadkins et al. (26) recently have found that cleavage with a reverse pH profile in high concentrations of monovalent cation requires the presence of a specific trinucleotide sequence, γC41-A42-A43. This sequence is present in the wild-type genomic ribozyme but absent in the antigenomic ribozyme. Thus, the reverse pH profile alone does not justify assignment of a role of general acid to γC75/C76. However, a persuasive argument for a role of γC75/C76 as proton donor comes from the crystal structure of the 3′ cleavage product of the genomic ribozyme; in the structure, the 5′-OH leaving group is within hydrogen bonding distance of N3 of the γC75 residue (12). Assuming that there is no major conformational change between structures of the transition state and product, this geometrical arrangement is strongly suggestive of a mechanism of acid catalysis involving γC75/C76 (17).
Figure 4.

Two kinetically equivalent mechanisms for general acid-base catalysis by RNA side chains or imidazole buffer. (A) In the C76u and C76Δ mutants, the unsaturated ring nitrogen of imidazole base accepts the proton from the nucleophilic group for general-base catalysis; in the wild type, cytosine serves a similar role. An unidentified functional group stabilizes the leaving group. (B) Imidazolium or protonated cytosine sidechain acts as a general acid by donating a proton to the leaving group. A specific base deprotonates the 2′-OH nucleophilic group at pre-equilibrium.
Difference in the concentration dependence of cytosine and imidazole analogues in the rescue reactions may reflect features of the active site. With cytosine and isocytosine, rescue of the C76 mutants demonstrated saturating concentration dependence. Moreover, the cytosine analogues that cannot rescue inhibit the cytosine-dependent reactions. These results would be consistent with a binding pocket for cytosine in these mutants. A higher second-order rate constant kb for cytosine than imidazole (Tables 1 and 2) suggested that interactions of cytosine (or isocytosine) with other functional groups in the putative binding pocket might allow a more favorable orientation for the exogenous cytosine and hence increases efficiency of the chemical rescue. Interactions in the binding pocket for cytosine also could lead to better discrimination against cytosine analogues and might explain the observation that 5-methylcytosine, which has a pKa close to neutral pH, cannot rescue the C76 mutants.
With cytosine rescue of C76Δ, the reaction pKa (5.2) was higher than the pKa of cytosine in solution (4.4), but lower than the apparent pKa of the intact antigenomic ribozyme reaction (6.1) (16). In the crystal structure of the genomic 3′ cleavage product, the N4-amino group of γC75 is involved in a hydrogen bonding triad with the 2′-O of γU20 and the nonbridging phosphate oxygen of γC22 (12). If this interaction forms at the transition state, it would increase electronegativity of the cytosine ring and contribute to a pKa shift of γC75. A very similar structure is likely to be found in the antigenomic ribozyme and would be consistent with a shift in the reaction pKa in the rescue reaction with exogenous cytosine. If, in the cytosine-rescue reaction, the apparent pKa reflects the pKa of bound cytosine, the shift in reaction pKa would be greatest when the binding site is saturated (kcat conditions), whereas the apparent pKa should equal the solution pKa of cytosine under kcat/KM conditions. The cytosine concentration used in this study (50 mM) was at the practical upper limit under these conditions, thus these reactions were carried out at a concentration of cytosine that was subsaturating but exceeding kcat/KM conditions (KM ≈40 mM).
The self-cleavage reaction of the HDV ribozyme, with formation of a 2′,3′-cyclic phosphate and a 5′-hydroxyl, is very similar to the first step of RNase A-catalyzed cleavage. In both cases the adjacent 2′-OH group serves as an intramolecular nucleophile. The classical mechanism for RNase A cleavage is concerted general acid/general base catalysis involving two histidine side chains (His-119 and His-12) that catalyze protonation of the 5′-oxyanion leaving group and deprotonation of the 2′-OH group. Based on this mechanism, a dianionic penta-coordinated phosphorane is proposed for the transition state, but mechanisms involving protonation of a nonbridging phosphate oxygen also have been proposed¶ (27–29). Consistent with the concerted mechanism proposed for RNase A, proton inventory of the RNase A reaction showed a quadratic dependence on isotope composition, indicating that two protons are transferred in the transition state (30). In contrast, the linear dependence on isotopic composition for the HDV ribozyme reaction suggests that a single proton is transferred in the transition state. This result would be inconsistent with a mechanism of concerted general acid/general base catalysis, but, given other data in this paper, would be consistent with a kinetically indistinguishable mechanisms of general base catalysis or sequential specific base/general acid catalysis. The latter mechanism would be very similar to the mechanism of general acid/general base catalysis proposed for the genomic ribozyme by Nakano et al. (17), except that deprotonation of the 2′-OH group would occur at pre-equilibrium, thus accounting for the proton inventory data.
Numerous studies have been directed at elucidating the details of general acid-base catalysis and the nature of the transition state for the class of reactions typified by RNase A cleavage. Charge distribution of the transition state in both enzymatic and nonenzymatic systems has been probed by using a linear free energy relationship. For a poor leaving group, such as the 5′-oxyanion in RNA cleavage, several model compounds yielded a highly negative βleaving group (βlg) of −0.9 to −1.3 (25, 31), suggesting that the rate-determining step for this cleavage pathway would be breakdown of the penta-coordinated structure. Consistent with this idea, a βlg of −0.54 to −0.59 was determined for an aryl phosphate diester that bears a good leaving group (32). It has been shown that the βlg for a series of aryl-phosphate diester compounds is even less negative (βlg = −0.2) in the active site of RNase A, suggesting a considerable contribution of electrophilic catalysis in the active site (33). Brønsted analysis using aryl-phosphate diester compounds gave a Brønsted coefficient, β, of 0.67 (α of 0.33) (32, 34). In the study presented here, a Brønsted coefficient was obtained by using a natural RNA phosphodiester as the substrate: for a mechanism of general base catalysis, β is 0.51, or for the kinetically equivalent mechanism, specific base/general acid catalysis, α is 0.49. These numbers suggest a proton transfer in the transition state with approximately half of the charge shared between the donor and acceptor atoms. This value agrees well with the value determined in the nonenzymatic system and is consistent with a model where the proton is “in flight” at the transition state (35). The β value for the HDV ribozyme is also comparable to that of other reactions in enzymatic systems with general acid-base catalysis by histidine and/or lysine side chains (β = 0.3–0.8) (2–4).
Chemical rescue experiments presented here suggested a linear free energy relationship between the cleavage rate and the strength of bases involved in general acid-base catalysis. Although additional work is required for more detailed understanding of the catalytic role of γC75/C76 in the HDV ribozymes, Brønsted analysis and proton inventory strongly support the hypothesis that the cytosine side chain is involved in the single proton transferred in the transition state of the wild-type cleavage. Using the linear dependence between the second-order rate constants of 4-substituted imidazole analogues and buffer pKa and the apparent reaction pKa of cytosine, the effective molarity of the C76 side chain in the antigenomic HDV active site is estimated to be ≈103 M. This is comparable to 103–104 M for the intramolecular general acid catalysis observed in a model compound (36). In addition, Brønsted analysis for phosphodiester bond cleavage in RNA in the HDV ribozyme lends itself to detailed understanding of the structure of the penta-coordinated transition state in an enzyme-catalyzed system.
Acknowledgments
We thank T. S. Wadkins and A. T. Perrotta for helpful comments on the manuscript, D. Herschlag for encouragement and skepticism, and Dr. S. C. Lovell for help in calculating the molecular volumes. Special thanks to a reviewer for insightful comments and points of emphasis. This work was supported by National Institutes of Health Grant GM47233.
Abbreviations
- HDV
hepatitis delta virus
- KSIE
kinetic solvent isotope effect
- C76
cytosine 76 in the antigenomic ribozyme
- γC75
cytosine 75 in the genomic ribozyme
- C76u
C76 to u mutant
- C76Δ
C76 deleted mutant
Footnotes
The term general acid-base catalysis is used in this paper to specifically refer to catalysis involving proton transfer at the transition state, but not to catalysis involving pre-equilibrium proton transfer. The latter is referred to as specific acid-base catalysis regardless of the proton donor/acceptor (37).
Brønsted analysis in an enzymatic system can be complicated by the molecular volume (m.v.) of the exogenous bases. The relationship of kb vs. the pKa of the bases can be corrected for the m.v. of the bases using the following equation: log kb = β × (pKa) + V × (m.v.), in which, the value of the coefficient V is negative. For the set of 4-substituted imidazole analogues used in this study, the m.v. varied less than 25%. Assuming a V of −0.02 (2–4) the Brønsted coefficient β, obtained by using the equation above, is 0.45 with r = 0.87.
The behavior of the antigenomic self-cleaving ribozyme PEX1 used in these studies suggests that chemistry, rather than a folding step, is rate determining for cleavage. The evidence is as follows: (i) when properly prefolded, it cleaves to near completion (>95%) with a first-order single exponential kinetics and the observed rate constants are log-linear with pH values below the apparent pKa (16), (ii) no increase in activity is seen with this form of the ribozyme upon addition of denaturants to the reactions (22), and (iii) a modification of the linkage at the scissile phosphate slows the rate of the reaction (38).
References
- 1.Jencks W P. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 2.Toney M D, Kirsch J F. Science. 1989;243:1485–1488. doi: 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- 3.Huang S, Tu S C. Biochemistry. 1997;36:14609–14615. doi: 10.1021/bi9722554. [DOI] [PubMed] [Google Scholar]
- 4.Newmyer S L, de Montellano P R O. J Biol Chem. 1996;271:14891–14896. doi: 10.1074/jbc.271.25.14891. [DOI] [PubMed] [Google Scholar]
- 5.Brown R S, Dewan J C, Klug A. Biochemistry. 1985;24:4785–4801. doi: 10.1021/bi00339a012. [DOI] [PubMed] [Google Scholar]
- 6.Dahm S C, Derrick W B, Uhlenbeck O C. Biochemistry. 1993;32:13040–13045. doi: 10.1021/bi00211a013. [DOI] [PubMed] [Google Scholar]
- 7.Dahm S C, Uhlenbeck O C. Biochemistry. 1991;30:9464–9469. doi: 10.1021/bi00103a011. [DOI] [PubMed] [Google Scholar]
- 8.Shan S, Yoshida A, Sun S, Piccirilli J A, Herschlag D. Proc Natl Acad Sci USA. 1999;96:12299–12304. doi: 10.1073/pnas.96.22.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinstein L B, Jones B C N M, Cosstick R, Cech T R. Nature (London) 1997;388:805–808. doi: 10.1038/42076. [DOI] [PubMed] [Google Scholar]
- 10.Piccirilli J A, Vyle J S, Caruthers M H, Cech T R. Nature (London) 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- 11.Narlikar G J, Herschlag D. Annu Rev Biochem. 1997;66:19–59. doi: 10.1146/annurev.biochem.66.1.19. [DOI] [PubMed] [Google Scholar]
- 12.Ferré-D'Amaré A R, Zhou K, Doudna J A. Nature (London) 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 13.Perrotta A T, Been M D. Biochemistry. 1992;31:16–21. doi: 10.1021/bi00116a004. [DOI] [PubMed] [Google Scholar]
- 14.Perrotta A T, Been M D. Nucleic Acids Res. 1996;24:1314–1321. doi: 10.1093/nar/24.7.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanner N K, Schaff S, Thill G, Petit-Koskas E, Crain-Denoyelle A-M, Westhof E. Curr Biol. 1994;4:488–497. doi: 10.1016/s0960-9822(00)00109-3. [DOI] [PubMed] [Google Scholar]
- 16.Perrotta A T, Shih I-h, Been M D. Science. 1999;286:123–126. doi: 10.1126/science.286.5437.123. [DOI] [PubMed] [Google Scholar]
- 17.Nakano S-I, Chadalavada D M, Bevilacqua P C. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 18.Ban N, Nissen P, Hansen J, Moore P B, Steitz T A. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 19.Nissen P, Hansen J, Ban N, Moore P B, Steitz T A. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 20.Muth G W, Ortoleva-Donnelly L, Strobel S A. Science. 2000;289:947–950. doi: 10.1126/science.289.5481.947. [DOI] [PubMed] [Google Scholar]
- 21.Davanloo P, Rosenberg A H, Dunn J J, Studier F W. Proc Natl Acad Sci USA. 1984;81:2035–2039. doi: 10.1073/pnas.81.7.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perrotta A T, Been M D. J Mol Biol. 1998;279:361–373. doi: 10.1006/jmbi.1998.1798. [DOI] [PubMed] [Google Scholar]
- 23.Schowen K B, Schowen R L. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 24.Dawson R M C, Elliott D E, Elliott W H, Jones K M. Data for Biochemical Research. Oxford: Clarendon; 1986. [Google Scholar]
- 25.Oivanen M, Schnell R, Pfleiderer W, Lonnberg H. J Org Chem. 1991;56:3623–3628. [Google Scholar]
- 26.Wadkins T S, Shih I-h, Perrotta A T, Been M D. J Mol Biol. 2001;305:1045–1055. doi: 10.1006/jmbi.2000.4368. [DOI] [PubMed] [Google Scholar]
- 27.Breslow R. Acc Chem Res. 1991;24:317–324. [Google Scholar]
- 28.Breslow R, Xu R. J Am Chem Soc. 1993;115:10705–10713. [Google Scholar]
- 29.Breslow R, Chapman W H., Jr Proc Natl Acad Sci USA. 1996;93:10018–10021. doi: 10.1073/pnas.93.19.10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matta M S, Vo D T. J Am Chem Soc. 1986;108:5316–5318. [Google Scholar]
- 31.Perreault D M, Anslyn E V. Angew Chem Int Ed Engl. 1997;36:432–450. [Google Scholar]
- 32.Davis A M, Hall A D, Williams A. J Am Chem Soc. 1988;110:5105–5108. [Google Scholar]
- 33.Davis A M, Regan A C, Williams A. Biochemistry. 1988;27:9042–9047. doi: 10.1021/bi00425a024. [DOI] [PubMed] [Google Scholar]
- 34.Dalby K N, Kirby A J, Hollfelder F. J Chem Soc Perkin Trans. 1993;2:1269–1281. [Google Scholar]
- 35.Jencks W P. Acc Chem Res. 1976;9:425–432. [Google Scholar]
- 36.Brown C J, Kirby A J. J Chem Soc Perkin Trans. 1997;2:1081–1093. [Google Scholar]
- 37.Jencks W P. Catalysis in Chemistry and Enzymology. New York: McGraw–Hill; 1969. [Google Scholar]
- 38.Shih I-h, Been M D. RNA. 1999;5:1140–1148. doi: 10.1017/s1355838299990763. [DOI] [PMC free article] [PubMed] [Google Scholar]